

The optimal approach to desmoid-type fibromatosis has not been established as of today.
Considering the unpredictable behaviour and the heterogeneity of this disease, we propose a treatment algorithm, based on a front-line wait and see (WS) approach and subsequent therapy in case of progression.
A careful counseling at a referral center is mandatory and should be offered to all patients affected by sporadic DF from the time of their diagnosis.
Keywords: desmoid tumor, aggressive fibromatosis, wait and see approach, surgery, radiation therapy, medical therapy, outcome
Abstract
Desmoid-type fibromatosis (DF) is a rare locally aggressive monoclonal proliferation of myofibroblasts lacking metastatic capacity. It may be observed in nearly every part of the body. Considering the variable clinical presentations, anatomic locations, and biologic behaviors, an individualized treatment approach is required. The pathogenesis of DF is not completely understood even if a high prevalence (∼85%) of CTNNB1 mutations discovered in sporadic DF underlies the importance of the Wnt/β-catenin pathway. No established and evidence-based approach for the treatment of this neoplasm is available as of today. Considering the unpredictable behavior and the heterogeneity of this disease, we propose a treatment algorithm approved by the French and the Italian Sarcoma Group, based on a front-line wait and see approach and subsequent therapy in the case of progression. A careful counseling at a referral center is mandatory and should be offered to all patients affected by sporadic DF from the time of their diagnosis.
introduction
Desmoid-type fibromatosis (DF) is a rare tumor characterized by a monoclonal proliferation of myofibroblasts in muscles, tendons, and ligaments. According to WHO (2013), it belongs to the group of locally aggressive, non-metastasizing mesenchymal tumors. It accounts for 0.03% of all neoplasms and 3% of all soft tissue tumors [1], but the true frequency may be underestimated as patients with small indolent tumors are not seen in tertiary centers. The tumor never metastasizes but recurs frequently after surgery and can be multifocal [1]. It usually occurs sporadically but in ∼5%–10% of patients it is associated with familial adenomatous polyposis (FAP) [2]. Sporadic DF predominantly affects young adults, especially females, and although it may be observed in nearly every part of the body, it often involves the extremities (including pelvic and shoulder girdles), the trunk, and the abdominal cavity (mostly within the mesentery or the pelvis) [3]. Given the variable clinical presentations, anatomic locations, and biologic behavior of this entity, also termed ‘desmoid tumor’ (DT), an individualized treatment approach is required [1]. In this perspective, sporadic DF should be clearly differentiated from FAP-associated DF and also from the so-called retroperitoneal fibrosis which is now generally referred to as IgG4-related pseudo-inflammatory tumor [4]. Therefore, in abdominal DF, it might be helpful to perform a colonoscopy to exclude signs of polyposis, while in retroperitoneal ones dosing IgG classes may help in the differential diagnosis. Given the rarity of the disease, a diagnosis of DF has to be confirmed by an expert soft tissue pathologist.
The pathogenesis of DF is not completely understood: the cell of origin is not known, precursor lesions are not described, and a substantial lack of knowledge still exists especially regarding potential risk factors. A possible derivation from mesenchymal stromal cell has recently been suggested [5]. Several pathogenetic mechanisms have been proposed. Increased incidence of DF during and shortly after pregnancy, reports of spontaneous regression and stabilization of disease using hormonal agents support the hypothesis of a possible role of estrogens in the genesis and maintenance of this disease [6]. A previous history of surgical trauma may be also elicited, wound healing bringing about growth factors, which may be crucial in predisposed patients. Since DF is a fibroblast-like proliferation with abundant collagen, they resemble an ‘uncontrolled’ wound healing process [7]. A possible correlation with tissue repair has also been sustained by the demonstration of transiently elevation of β-catenin in fibroblasts during tissue repair and by the fact that forced β-catenin overexpression results in the formation of hypertrophic scars in mice [8]. A large number of growth factors and cytokines such as platelet derived growth factor and transforming growth factor are secreted from platelets at the site of injury and induce β-catenin signaling in fibroblast in a paracrine manner [9]. Altogether, the phenomenon that β-catenin plays a physiological role in wound healing is consistent with the hypothesis that the deregulation of this pathway is of functional relevance in DF. The potential role of the APC/β-catenin pathway in DF was first provided by studies of FAP-associated DF [10]. While somatic APC mutations were identified in a small subset of sporadic DF, a high prevalence (∼85%) of CTNNB1 (the gene coding for β-catenin) mutations was discovered in sporadic DF [11]. The result of such mutation is the accumulation of non-phosphorylated β-catenin in the cytoplasm and in its consequent translocation into the nucleus where it promotes the transcription of specific target genes such as c-MYC, c-JUN, MMP7, Nr-CAM, cyclin D1, and COX-2 [12].
The challenge is finding a suitable treatment, given the peculiar natural history of the disease. In fact, surgery (alone or in association with radiation therapy) has been considered the mainstay of treatment until few years ago, but it is nevertheless associated with morbidity and high recurrence rates even after apparently adequate local treatment [13–15]. Besides, several medical approaches have been used to treat patients with DF. These include antiestrogen therapy such as tamoxifen (Fidia, Italy) or toremifene (Orion Pharma, Finland), non-steroidal anti-inflammatory drugs such as indomethacin (Biofutura Pharma, Italy), chemotherapy (mainly methotrexate and vinblastine [Teva, Israel]/vinorelbine [Pierre Fabre Pharma, France] or doxorubicin-based [Pfizer, United States] combinations) [16]. However, there is no established or evidence-based approach for the treatment of this neoplasm as of today. In a recent retrospective series, antiestrogens and anthracycline-containing regimens appeared to be associated with a higher radiological response rate compared with other treatments [17]. The initial management of patients with DF remains unclear: it is often adapted to disease evolution and it is highly dependent on the treating physician's expertise. Recently, a front-line wait and see (W&S) policy with medical treatment at progression has been proposed by several authors with encouraging results, i.e. a prolonged progression-free survival in a substantial proportion of patients [18, 19]. Since spontaneous stabilizations or regressions are regularly noticed, an initial ‘observation’ period could be considered as a reasonable option [20]. Unfortunately, there are no data from randomized trials available to assess which treatment modality is most effective or less harmful. Because of the rarity of this disease and the heterogeneity of its behavior, clinical trials have been problematic and published series are mainly based on retrospective data analyses. Recently, a stepwise approach for the treatment of DF patients has been proposed: a W&S policy at the beginning to discriminate indolent and aggressive forms, since >50% of patients have a slow growing or potentially regressive disease, with treatment reserved to progressing cases [20].
We propose hereafter an algorithm of treatment approved by the members of the French and the Italian Sarcoma Group. It renders the treatment approach selected by institutions belonging to these two groups and is offered to the medical community as a contribution for further discussion and possibly collection of prospective case series treated homogeneously. These data would help to validate the approach outlined hereafter.
surgery and/or radiotherapy: switching the previous standard to later lines
Before 2000, surgery with negative margins had been considered the standard of care for patients affected by DF reflecting the same approach to extremity soft tissue sarcomas [21].
Due to the specific pattern of infiltrative growth, the resection needed to achieve negative margins in DF could be often larger than for a same-sized sarcoma leading to a potential impairment of function and cosmetic alterations in patients affected by a disease that can be indolent in as many as half cases [18–21].
Nevertheless, unlike soft tissue sarcomas, where positive margins are consistently a predictive factor for local failure [22–24], the prognostic significance of positive margins after excision of DF has been debated for long without achieving a worldwide accepted consensus. Few large series with more than 100 patients were unable to establish a predictive role of positive margins. In fact, the Massachussets General Hospital (MGH) [14], the earlier MD Anderson Cancer Center (MDACC) [25] and the Institute Gustave Roussy (IGR) [18] studies suggested that margin positivity was a important prognostic factor for local recurrence-free survival (19% negative versus 39% positive in the MGH and 27% versus 54% in the MDACC). On the opposite, the Memorial Sloan Kettering Cancer Center (MSKCC) [15] and the Istituto Nazionale Tumori (INT) [26] series did not demonstrate such an impact (22% versus 24% and 21% versus 18%, respectively). Moreover, a more recent analysis of MDACC did not reproduce their previous results [27]. Finally, a recent French multi-institutional retrospective analysis confirmed the absence of any impact of positive microscopic margin on patient outcome [28].
This heterogeneous behavior and the benign nature of the disease paved the way to more conservative resections, with accepted marginal and microscopic positive surgical margins where function preservation was the aim.
In parallel with these observations, it became evident over the years that DF is not a unique disease but is made by at least two different entities, marked by either an indolent or an aggressive behavior, which, however, is hard to predict. It is then likely that ‘indolent tumors’ will not recur regardless of positive margins due to their natural tendency to regress or remain stable, whereas ‘aggressive lesions’ do recur despite the adequacy of surgical resection and, of course more commonly, after positive margins resections. Today, in those cases in which surgery is felt to be needed, one should be aware at least whether the disease is indolent or aggressive, in order to adjust the extent of resection and plan possible adjuvant therapies. In the lack of prognostic factors, this is the main rationale for a W&S policy.
Radiation therapy had also been used in order to reduce the rate of local recurrence especially for the extra-abdominal DF. It was preferably used alone for unresectable tumors (surgery not feasible or associated with a significant loss of function), for palliative intent (pain management) or in combination with surgery in the adjuvant setting, mostly postoperatively after microscopic positive margins. The benefit of radiotherapy has been claimed in several reports. In particular, a review including more than 20 retrospective studies focusing on the role of the combination (surgery and radiotherapy) showed that surgery plus radiotherapy or radiotherapy alone could obtain a better local control rate (75% and 78%, respectively) compared with surgery alone (61%) [29]. The advantage of radiotherapy in combination or alone compared with surgery alone was suggested in both negative and positive margins. The fact that radiotherapy and surgery achieved the same local control than radiotherapy alone suggested that radiotherapy alone could have been preferable for tumors located at sites where surgery would have been marginal and followed by major side-effects, such as head and neck, girdles, and pelvis.
More recently, Guadagnolo et al. published a series of patients treated with radiotherapy alone or in association with surgery, showing that radiation doses over 56 Gy did not significantly improve local control but were associated with an increased risk of complications, especially in patients ≤30 years. Median time to radiation-related complications was 33 months and included fibrosis, soft-tissue necrosis, anesthesia/paresthesia, pathological fractures, edema, and rarely vascular complications requiring amputation or secondary malignancies [30]. Based on the published literature, the recommended dose of radiotherapy is 50–56 Gy in 2-Gy fractions. A phase 2 study on exclusive radiotherapy at moderate doses (50 Gy) has just been reported, with long-term tumor control rates close to 80% [31].
However, the role of radiotherapy in DF management remains controversial and extensively debated, given the fact that DF is a relatively benign condition and treatment complications (including secondary malignancies) may be severe especially in such a young, often healthy subgroup of patients, who have a long life expectancy. This is the reason why we believe it should be proposed only for documented progressive disease and in the lack of other alternatives.
why should we change our approach in treating patients? a new era: W&S approach as the first step
Considering the morbidity related to surgical and radiation treatments, the unpredictable behavior of the disease with described spontaneous regression and growth arrest in the absence of any treatments, some authors proposed an observational front-line approach for asymptomatic patients affected by DF before considering any further treatment [18–21]. This approach had been initially used in patients with recurrent but stable disease in order to avoid repeated surgical excisions and preserve function and cosmesis [32–35]. Considering the benefit and the safety of such approach (progression was generally relative slow), an observational period was then proposed in patients with primary unresectable disease for whom radiotherapy could have been an option but could carry serious side-effects or sequelae [32].
The IGR group was the first to propose watchful surveillance for primary resectable diseases [18]. The validity of this strategy was later confirmed by a combined retrospective French/Italian study [19], which showed a 50% progression-free survival at 5 years for patients managed with a front-line conservative approach. The vast majority of progressions were registered in the first 2 years of observation and almost all of them within the first 5 years. The increase in size was usually mild and in the case of progression medical therapy and eventually surgery/radiotherapy could be used. The observational approach was intended to select patients who did not need any treatment as opposed to those who needed to be treated in an aggressive manner because of their tendency to progress. Different patterns of growth were observed, also depending on the time point of the disease natural history during which patients were first evaluated (Figure 1).
Figure 1.
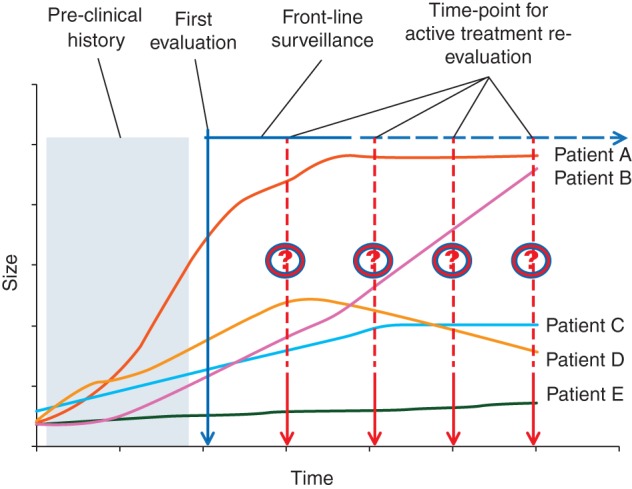
Different patterns of tumor growth according to the time point of first observation in the clinic.
More recently, a potential role of β-catenin mutational analysis as a predictor of behavior has been shown [11, 36]. In fact, a specific mutation (45F) in patients surgically treated was correlated with a worse outcome in terms of recurrence-free survival. Nevertheless, the potential value of CTNNB1 mutational analysis as a predictor of progression-free survival in patients ‘observed’ has not yet been evaluated and prospective trials are ongoing.
algorithm proposal
Since the approach to this rare and challenging disease has been changing in the last years, we decided to propose a decision algorithm that could guide clinicians in tailoring therapeutic decisions (Figure 2). The present algorithm is intended to be prospectively studied in the context of an observational study algorithm (ClinicalTrials.gov Identifier: NCT01801176).
Figure 2.

Consensus algorithm of treatment of sporadic desmoid-type fibromatosis.
Indeed, when a patient with sporadic DT is first evaluated, we do not know at which time point of the natural history of the disease we are observing him/her (Figure 1). For this reason and considering that sporadic DT is often an indolent and slowly growing disease, we can safely propose to the vast majority of patients a W&S option. Besides, women of child-bearing age have to stop birth control medications. The only caveat is to carefully follow-up tumors located at critical sites, such as the head and neck, pelvis, and intra-abdominal cavity, since a loco-regional growth may become problematic or even life-threatening in exceptional cases. Of note, patients with important symptoms may sometimes skip the initial observation.
Essentially, ‘observation’ represents a way to understand the behavior of the disease and tailor next treatments.
When this conservative approach is chosen, then patients should be followed-up with contrast enhanced MRI (preferably) or, in the case of intolerance or intra-abdominal location, with CT scan, every month during the first 2 months, then every 3 months in the first year, then 6 monthly up to the 5th year, and yearly thereafter. Indeed, the frequency of imaging is fairly intense, especially in the first year, but it is intended to avoid missing the few rapidly progressing cases and will be studied prospectively. In the case of tumors ease to follow clinically and located in safe sites, follow-up could be adjusted. In the case of progression, alternative active treatment should be discussed with the patient, even if supplemental ‘observation’ at close intervals can be still an option, if safe. What progression means and which is the cut-off to decide that active treatment should be started has not been defined yet. Treatment is switched to a definitive treatment (surgery or radiotherapy) mainly in the case of obvious tumor progression. Pain can be managed with medical treatment. In fact, the pathogenetic mechanisms of pain in DF are certainly complex and multifold. Pain is not strictly correlated with the DF progression, some stable DF may be painful, while some progressive DF may not, and sometimes pain can be the consequence of previous loco-regional treatments rather than being caused by the disease itself. In other words, pain has to be treated independently of the DF course. Different factors should be taken into account to define the need of a specific treatment: the initial size, growth rate, anatomical site, risk of organs/vessels/nerves compression, and worsening of function, although also pain onset may anticipate an increase in size, especially in primary disease.
Indeed, patients with a significant pain syndrome or with tumors to threatening anatomical sites (i.e. head and neck or pelvis or intra-abdominal cavity) may sometimes receive a front-line medical therapy, skipping the initial observation period. The same medical therapy could be used for patients who substantially progress during the initial observation period. Different pharmacological options can be proposed. When pain is the main issue, anti-hormonal agents such as Toremifene or Tamoxifen should be used, alone or in combination with anti-COX2 as first-line medical treatment, because of their limited toxicity [17, 37–38]. In general, this strategy is well tolerated and devoid of major adverse events, but for the rare increased risk of thrombotic events which should always be taken into account. Of note, different doses have been reported, from 20 to 80 mg/day in the case of Tamoxifen and 60 to 180 mg/day in the case of Toremifene. The general feeling is that higher doses may be more active, although side-effects are more pronounced in young females. Higher doses may infact interfere more with their physiological hormonal status and accelerate the menopause. On the contrary, no major side-effects are seen in males. The response to anti-hormonal therapy is slow and in the vast majority of patients who respond objective response is observed after few months of treatment. Generally, when the drug is active, a stabilization of disease growth and an improvement in pain is promptly observed. Of note, response to anti-hormonal agents as well as to all other agents discussed below can be lasting even beyond treatment discontinuation.
When the relevant issue is critical anatomic site or in the case of hormonal/anti-COX2 therapy failure, chemotherapy should be then considered.
Different regimens can be used: the two commonest ones include the so-called ‘low-dose’ chemotherapy with methotrexate and/or vinblastine/vinorelbine [39–42] and the conventional chemotherapy with doxorubicin (including liposomal doxorubicin [Jansen Cilag, United States] with less cardiotoxicity)±dacarbazine (Sanofi Aventis, Switzerland) [43, 44]. Although administered for a longer time, ‘low-dose’ chemotherapy is usually the first choice, given the more limited toxicity profile and the absence of tumorigenic effect in the long run. However, patients' compliance with therapy may be an issue, since chemotherapy is usually administered for 1 year at least. Moreover, liver toxicity as well as peripheral neuropathy, although rare, may also prevent the administration of this regimen for the whole period and should always be monitored. When active, the response to chemotherapy in terms of pain relief is usually prompt, while radiological tumor attenuation and shrinkage may occur at a later stage and maintained beyond treatment discontinuation (similarly to hormonal therapies). Conventional chemotherapy is then reserved to patients who fail the ‘low-dose’ regimen. Toxicity is not different from what extensively reported in all other diseases in which these drugs are used. If shrinkage is an issue due to location, size, or morbidity caused by the tumor, conventional chemotherapy may be considered as the first choice. When active, response is more prompt and shrinkage possibly more substantial.
Loco-regional chemotherapy, such as isolated limb perfusion (ILP) with tumor necrosis factor alpha (Boehringer Ingelheim, Germany) and Melphalan (Laboratorio Farmacologico Milanese, Italy), can also be considered as an alternative in patients affected by tumors located in extremities, especially for those who have multifocal disease and in patients with DF of the hand or foot, although only few case reports are available so far [45]. After ILP, patients are not routinely operated because a stabilization of the disease or a slow regression is commonly observed. However, the addiction of radiotherapy or surgery can be discussed on an individualized basis.
Initial experiences with cryoablation have also been reported. This modality appears to be an effective alternative treatment for the achievement of local control of small and moderately sized extra-abdominal tumors, but it is likely of limited use in patients with larger tumors that have untreatable regions due to involvement of vital structures [46, 47].
Target therapy—in particular Imatinib (Novartis Farma, Switzerland)—has been used, initially with encouraging [48] results, but unfortunately they were not confirmed in prospective studies [49, 50]. As a result, Imatinib has a limited use, but other anti-tyrosine kinase therapies (i.e. Sorafenib [Bayer, Germany], Pazopanib [Glaxosmithkline, United Kingdom]) are under investigation with promising results in an uncontrolled trial with sorafenib [51]. These therapies could be offered when other options have failed, preferably within clinical studies.
In selected conditions, surgery could eventually be proposed if resection is feasible without major sequelae and if previous treatment failed to obtain a local control. The limited risk of post-surgical recurrence of abdominal wall [26–28, 52] and intra-abdominal sporadic tumors needs to be taken into account. On the contrary, radiotherapy should be discussed first in patients affected by tumors originating from critical sites (such as girdles, head and neck, and pelvis). In fact, when surgery is not safely feasible, exclusive radiation therapy can be an option and doses could be limited to 50–56 Gy [31].
When W&S is the initial approach, it must be emphasized that if surgery is then chosen for the patients who will have progressed, it will have to be eventually more aggressive than the one which would have been chosen upfront indiscriminately.
Site and symptoms as well as the tendency to grow or not are the main criteria selected to choose the more appropriate therapy in a stepwise fashion. Biological studies to identify potential molecular predictors are crucially warranted.
Of note, DF is not a contraindication to pregnancy, but these patients must be followed in tertiary centers and a closer follow-up is necessary while pregnant. When tumor is in place, there is a 50% risk of progression during pregnancy, which is followed in most cases by a subsequent spontaneous regression after delivery [53].
Careful counseling at a referral center is mandatory and should be offered to all patients affected by sporadic DF at the time of diagnosis. These patients should never be treated without a specific expertise, and an international prospective database should be funded. In such a rare and bizarre non-metastasizing, though locally aggressive, neoplasm, along with cross-institutional collaborations, this would be the only chance we have to validate tentative guidelines as those we outlined in this consensus paper from two national sarcoma cooperative research groups.
disclosure
The authors have declared no conflicts of interest.
acknowledgements
To the patients and patient advocacy groups (such as SOS-Desmoïde) that keep stimulating the Italian and the French Sarcoma Group.
references
- 1.Fletcher JA, Bridge JA, Hogendoorn PCW, et al. WHO Classification of Tumors of Soft Tissue and Bone. Lyon: IARC Press; 2013. Desmoid-type fibromatoses; pp. 72–73. [Google Scholar]
- 2.Clark SK, Phillips RKS. Desmoids in familial adenomatous polyposis. Brit J Surg. 1996;83:1494–1504. doi: 10.1002/bjs.1800831105. doi:10.1002/bjs.1800831105. [DOI] [PubMed] [Google Scholar]
- 3.de Bree E, Keus R, Melissas J, et al. Desmoid tumors: need for an individualized approach. Expert Rev Anticancer Ther. 2009;9(4):525–535. doi: 10.1586/era.09.9. doi:10.1586/era.09.9. [DOI] [PubMed] [Google Scholar]
- 4.Stone JH, Zen Y, Deshpande V. Igg4 related disease. N Engl J Med. 2012;366(6):539–551. doi: 10.1056/NEJMra1104650. doi:10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 5.Carothers AM, Rizvi H, Hasson RM, et al. Mesenchymal stromal cell mutations and wound healing contribute to the etiology of desmoid tumors. Cancer Res. 2012;72(1):346–355. doi: 10.1158/0008-5472.CAN-11-2819. doi:10.1158/0008-5472.CAN-11-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deyrup AT, Tretiakova M, Montag AG. Estrogen receptor-β expression in extraabdominal fibromatoses—an analysis of 40 cases. Cancer. 2006;106:208–213. doi: 10.1002/cncr.21553. doi:10.1002/cncr.21553. [DOI] [PubMed] [Google Scholar]
- 7.Amini Nika S, Ebrahima RP, Van Dama K, et al. TGF-β modulates β-catenin stability and signaling in mesenchymal proliferations. Exp Cell Res. 2007;313:2887–2895. doi: 10.1016/j.yexcr.2007.05.024. doi:10.1016/j.yexcr.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 8.Cheon SS, Cheah AYL, Turley S, et al. β-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc Natl Acad Sci USA. 2002;10:6973–6978. doi: 10.1073/pnas.102657399. doi:10.1073/pnas.102657399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennet NT, Schultz GS. Growth factors and wound healing; biochemical properties of growth factors and their receptors. Am J Surg. 1993;165:728–737. doi: 10.1016/s0002-9610(05)80797-4. doi:10.1016/S0002-9610(05)80797-4. [DOI] [PubMed] [Google Scholar]
- 10.Alman BA, Li C, Pajerski ME, et al. Increased β-catenin and somatic mutations in sporadic aggressive fibromatosis (desmoids tumors) Am J Pathol. 1997;151:329–334. [PMC free article] [PubMed] [Google Scholar]
- 11.Lazar AJ, Tuvin D, Hajibashi S, et al. Specific mutations in β-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173:1518–1527. doi: 10.2353/ajpath.2008.080475. doi:10.2353/ajpath.2008.080475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotiligam D, Lazar AJ, Pollock RE, et al. Desmoid tumor: a disease opportune for molecular insights. Histol Histopathol. 2008;23:117–126. doi: 10.14670/HH-23.117. [DOI] [PubMed] [Google Scholar]
- 13.Reitamo JJ, Sheinin TM, Pekka H. The desmoid syndrome. New aspects in the cause, pathogenesis and treatment of the desmoid tumor. Am J Surg. 1986;15:230–237. doi: 10.1016/0002-9610(86)90076-0. doi:10.1016/0002-9610(86)90076-0. [DOI] [PubMed] [Google Scholar]
- 14.Spear MA, Jennings LC, Mankin HJ, et al. Individualizing management of aggressive fibromatoses. Int J Radiat Oncol Biol Phys. 1998;40(3):637–645. doi: 10.1016/s0360-3016(97)00845-6. doi:10.1016/S0360-3016(97)00845-6. [DOI] [PubMed] [Google Scholar]
- 15.Merchant NB, Lewis JJ, Woodruff JM, et al. Extremity and trunk desmoid tumors: a multifactorial analysis of outcome. Cancer. 1999;86(10):2045–2052. [PubMed] [Google Scholar]
- 16.Janinis J, Patriki M, Vini L, et al. The pharmacological treatment of aggressive fibromatosis: a systemic review. Ann Oncol. 2003;14:181–190. doi: 10.1093/annonc/mdg064. doi:10.1093/annonc/mdg064. [DOI] [PubMed] [Google Scholar]
- 17.de Camargo VP, Keohan ML, D'Adamo DR, et al. Clinical outcomes of systemic therapy for patients with deep fibromatosis (desmoid tumor) Cancer. 2010;116(9):2258–2265. doi: 10.1002/cncr.25089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonvalot S, Eldweny H, Haddad V, et al. Extra-abdominal primary fibromatosis: aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol. 2008;34(4):462–468. doi: 10.1016/j.ejso.2007.06.006. doi:10.1016/j.ejso.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Fiore M, Rimareix F, Mariani L, et al. Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol. 2009;16(9):2587–2593. doi: 10.1245/s10434-009-0586-2. doi:10.1245/s10434-009-0586-2. [DOI] [PubMed] [Google Scholar]
- 20.Bonvalot S, Desai A, Coppola S, et al. The treatment of desmoid tumors: a stepwise clinical approach. Ann Oncol. 2012;23(Suppl 10):x158–x166. doi: 10.1093/annonc/mds298. doi:10.1093/annonc/mds298. [DOI] [PubMed] [Google Scholar]
- 21.Colombo C, Gronchi A. Desmoid-type fibromatosis: what works best? Eur J Cancer. 2009;45(Suppl 1):466–467. doi: 10.1016/S0959-8049(09)70092-9. doi:10.1016/S0959-8049(09)70092-9. [DOI] [PubMed] [Google Scholar]
- 22.Stojadinovic A, Leung DH, Hoos A, et al. Analysis of prognostic significance of microscopic margins in 2084 localized primary adult soft tissue sarcomas. Ann Surg. 2002;235(3):424–434. doi: 10.1097/00000658-200203000-00015. doi:10.1097/00000658-200203000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft tissue- sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer. 2003;97(10):2530–2543. doi: 10.1002/cncr.11365. doi:10.1002/cncr.11365. [DOI] [PubMed] [Google Scholar]
- 24.Gronchi A, Casali PG, Mariani L, et al. Status of surgical margins and prognosis in adult soft tissue sarcomas of the extremities: a series of 911 consecutive patients treated at a single institution. J Clin Oncol. 2005;23(1):96–104. doi: 10.1200/JCO.2005.04.160. doi:10.1200/JCO.2005.04.160. [DOI] [PubMed] [Google Scholar]
- 25.Ballo MT, Zagars GK, Pollack A, et al. Desmoid tumor: prognostic factors and outcome after surgery, radiation therapy, or combined surgery and radiation therapy. J Clin Oncol. 1999;17:158–167. doi: 10.1200/JCO.1999.17.1.158. [DOI] [PubMed] [Google Scholar]
- 26.Gronchi A, Casali PG, Mariani L, et al. Quality of surgery and outcome in extra-abdominal aggressive fibromatosis: a series of patients surgically treated at a single institution. J Clin Oncol. 2003;21:1390–1397. doi: 10.1200/JCO.2003.05.150. doi:10.1200/JCO.2003.05.150. [DOI] [PubMed] [Google Scholar]
- 27.Lev D, Kotilingam D, Wei C, et al. Optimizing treatment of desmoid tumors. J Clin Oncol. 2007;25(13):1785–1791. doi: 10.1200/JCO.2006.10.5015. doi:10.1200/JCO.2006.10.5015. [DOI] [PubMed] [Google Scholar]
- 28.Salas S, Dufresne A, Bui B, et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. J Clin Oncol. 2011;29(26):3553–3558. doi: 10.1200/JCO.2010.33.5489. doi:10.1200/JCO.2010.33.5489. [DOI] [PubMed] [Google Scholar]
- 29.Nuyttens JJ, Rust PF, Thomas CR, et al. Surgery versus radiation therapy for patients with aggressive fibromatosis or desmoid tumors. A comparative review of 22 articles. Cancer. 2000;88:1517–1523. [PubMed] [Google Scholar]
- 30.Guadagnolo BA, Zagars GK, Ballo MT. Long-term outcomes for desmoid tumors treated with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;71:441–447. doi: 10.1016/j.ijrobp.2007.10.013. doi:10.1016/j.ijrobp.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 31.Keus R, Nout RA, Blay JY, et al. Results of a phase II pilot study of moderate dose radiotherapy for inoperable desmoid-type fibromatosis–an EORTC STBSG and ROG study (EORTC 62991–22998) Ann Oncol. 2013;24(10):2672–2676. doi: 10.1093/annonc/mdt254. [DOI] [PubMed] [Google Scholar]
- 32.Pignatti G, Barbanti-Brodano G, Ferrari D, et al. Extraabdominal desmoid tumor. A study of 83 cases. Orthop Relat Res. 2000;375:207–213. doi:10.1097/00003086-200006000-00025. [PubMed] [Google Scholar]
- 33.Rock MG, Pritchard DJ, Reiman HM, et al. Extra-abdominal desmoid tumors. J Bone Joint Surg Am. 1984;66:1369–1374. [PubMed] [Google Scholar]
- 34.Lewis JJ, Boland PJ, Leung DH, et al. The enigma of desmoid tumors. Ann Surg. 1999;229:866–872. doi: 10.1097/00000658-199906000-00014. doi:10.1097/00000658-199906000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips SR, A'Hern R, Thomas JM. Aggressive fibromatosis of the abdominal wall, limbs and limb girdles. Br J Surg. 2004;91:1624–1629. doi: 10.1002/bjs.4792. doi:10.1002/bjs.4792. [DOI] [PubMed] [Google Scholar]
- 36.Colombo C, Miceli R, Lazar A, et al. CTNNB1 45f mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence: an independent multicenter validation study. Cancer. 2013 doi: 10.1002/cncr.28271. doi:10.1002/cncr.28271. [DOI] [PubMed] [Google Scholar]
- 37.Skapek SX, Anderson JR, Hill DA, et al. Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: results of a children's oncology group (COG) phase II study. Pediatr Blood Cancer. 2013;60(7):1108–1112. doi: 10.1002/pbc.24457. doi:10.1002/pbc.24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiore M, Colombo C, Radaelli S, et al. Activity of toremifene in sporadic desmoid-type fibromatosis. J Clin Oncol. 2011;29(Suppl)) abstr 10033. [Google Scholar]
- 39.Azzarelli A, Gronchi A, Bertulli R, et al. Low-dose chemotherapy with methotrexate and vinblastine for patients with advanced aggressive fibromatosis. Cancer. 2001;92(5):1259–1264. doi: 10.1002/1097-0142(20010901)92:5<1259::aid-cncr1446>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 40.Weiss AJ, Horowitz S, Lackman RD. Therapy of desmoid tumors and fibromatosis using vinorelbine. Am J Clin Oncol. 1999;22(2):193–195. doi: 10.1097/00000421-199904000-00020. doi:10.1097/00000421-199904000-00020. [DOI] [PubMed] [Google Scholar]
- 41.Skapek SX, Ferguson WS, Granowetter L, et al. Vinblastine and methotrexate for desmoid fibromatosis in children: results of a pediatric oncology group phase II trial. J Clin Oncol. 2007;25(5):501–506. doi: 10.1200/JCO.2006.08.2966. doi:10.1200/JCO.2006.08.2966. [DOI] [PubMed] [Google Scholar]
- 42.Garbay D, Le Cesne A, Penel N, et al. Chemotherapy in patients with desmoid tumors: a study from the French Sarcoma Group (FSG) Ann Oncol. 2012;23(1):182–186. doi: 10.1093/annonc/mdr051. doi:10.1093/annonc/mdr051. [DOI] [PubMed] [Google Scholar]
- 43.Constantinidou A, Jones RL, Scurr M, et al. Pegylated liposomal doxorubicin, an effective, well-tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer. 2009;45(17):2930–2934. doi: 10.1016/j.ejca.2009.08.016. doi:10.1016/j.ejca.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 44.Patel SR, Benjamin RS. Desmoid tumors respond to chemotherapy: defying the dogma in oncology. J Clin Oncol. 2006;24(1):11–12. doi: 10.1200/JCO.2005.03.6566. doi:10.1200/JCO.2005.03.6566. [DOI] [PubMed] [Google Scholar]
- 45.Bonvalot S, Rimareix F, Causeret S, et al. Hyperthermic isolated limb perfusion in locally advanced soft tissue sarcoma and progressive desmoid-type fibromatosis with TNF 1 mg and melphalan (T1-M HILP) is safe and efficient. Ann Surg Oncol. 2009;16(12):3350–3357. doi: 10.1245/s10434-009-0733-9. doi:10.1245/s10434-009-0733-9. [DOI] [PubMed] [Google Scholar]
- 46.Kujak JL, Liu PT, Johnson GB, et al. Early experience with percutaneous cryoablation of extra-abdominal desmoid tumors. Skeletal Radiol. 2010;39:175–182. doi: 10.1007/s00256-009-0801-z. doi:10.1007/s00256-009-0801-z. [DOI] [PubMed] [Google Scholar]
- 47.Walczak BE, Rose PS. Desmoid: the role of local therapy in an era of systemic options. Curr Treat Options Oncol. 2013;14(3):465–473. doi: 10.1007/s11864-013-0235-7. doi:10.1007/s11864-013-0235-7. [DOI] [PubMed] [Google Scholar]
- 48.Heinrich MC, McArthur GA, Demetri GD, et al. Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor) J Clin Oncol. 2006;24:1195–1203. doi: 10.1200/JCO.2005.04.0717. doi:10.1200/JCO.2005.04.0717. [DOI] [PubMed] [Google Scholar]
- 49.Penel N, Le Cesne A, Bui BN, et al. Imatinib for progressive and recurrent aggressive fibromatosis (demoid tumors): an FNCLCC/French Sarcoma Group phase II trial with long-term follow up. Ann Oncol. 2011;22:452–457. doi: 10.1093/annonc/mdq341. doi:10.1093/annonc/mdq341. [DOI] [PubMed] [Google Scholar]
- 50.Chugh R, Wathen JK, Patel SR, et al. Efficacy of imatinib in aggressive fibromatosis: results of a phase II multicenter sarcoma alliance for research through collaboration (SARC) trial. Clin Cancer Res. 2010;16(19):4884–4891. doi: 10.1158/1078-0432.CCR-10-1177. doi:10.1158/1078-0432.CCR-10-1177. [DOI] [PubMed] [Google Scholar]
- 51.Gounder MM, Lefkowitz RA, Keohan ML, et al. Activity of sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res. 2011;17(12):4082–4090. doi: 10.1158/1078-0432.CCR-10-3322. doi:10.1158/1078-0432.CCR-10-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonvalot S, Ternes N, Fiore M, et al. Spontaneous regression of primary abdominal wall desmoids: more common than previously thought. Ann Surg Oncol. 2013 doi: 10.1245/s10434-013-3197-x. doi:10.1245/s10434-013-3197-x. [DOI] [PubMed] [Google Scholar]
- 53.Fiore M, Coppola S, Cannell AJ, et al. Desmoid-type fibromatosis and pregnancy. A multi-institutional analysis of recurrence and obstetric risk. Ann Surg. 2013 doi: 10.1097/SLA.0000000000000224. doi:10.1097/SLA.0000000000000224. [DOI] [PubMed] [Google Scholar]