

Abstract
Previous studies in rodents have shown that after a moderate traumatic brain injury (TBI) with a controlled cortical impact (CCI) device, the adult-born immature granular neurons in the dentate gyrus are the most vulnerable cell type in the hippocampus. There is no effective approach for preventing immature neuron death following TBI. We found that tyrosine-related kinase B (TrkB), a receptor of brain-derived neurotrophic factor (BDNF), is highly expressed in adult-born immature neurons. We determined that the small molecule imitating BDNF, 7, 8-dihydroxyflavone (DHF), increased phosphorylation of TrkB in immature neurons both in vitro and in vivo. Pre-treatment with DHF protected immature neurons from excitotoxicity-mediated death in vitro, and systemic administration of DHF before moderate CCI injury reduced the death of adult-born immature neurons in the hippocampus 24 hours following injury. By contrast, inhibiting BDNF signaling using the TrkB antagonist ANA12 attenuated the neuroprotective effects of DHF. These data indicate that DHF may be a promising chemical compound that promotes immature neuron survival after TBI through activation of the BDNF signaling pathway.
Keywords: 7, 8-dihydroxyflavone; Brain-derived neurotrophic factor; Cell death; Newborn neuron; Neuroprotection; Traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI) causes a mechanical injury that can induce a primary injury of the immediate tissue (1, 2), and a secondary injury of the surviving cells that is triggered by the primary event (1, 3). The secondary injury occurs in many brain areas, including the cortex and hippocampus in both humans (4–9) and experimental animals (1, 10, 11). This injury contributes to further cognitive, sensory, and motor dysfunction (1, 12). Currently, there is no FDA-approved therapeutic treatment for these disorders that occur following TBI.
The hippocampus is one of the regions of the adult brain that can support neurogenesis throughout life, as demonstrated in rodents and primates, including humans (13–17). New neurons are continuously generated from neural stem/progenitor cells (NSCs) in the subgranular zone of the hippocampal dentate gyrus (HDG) (18, 19). These newborn neurons integrate into the neural circuitry and are believed to play a critical role in learning and memory (18–20). We previously showed that adult-born immature neurons in the HDG are the most vulnerable cell type to a moderate controlled cortical impact (CCI) TBI (21–23). Most of the neuron death occurs within 24 hours (23). Because the hippocampus is critical for higher cognitive function (24, 25) and is frequently associated with post-traumatic seizure generation (26), disturbances in hippocampal neurogenesis may play a significant role in the pathogenesis of TBI-related injuries. Thus, in addition to other mechanisms of neuroprotection, it is important to explore approaches to prevent immature neuron death after TBI.
One approach is to explore the use of neurotrophins that exist naturally in the brain. Brain derived neurotrophic factor (BDNF) is a member of the neurotrophin family of growth factors. BDNF is active broadly in the adult brain and is highly active in the hippocampus (27). It regulates diverse and important functions of neurons, i.e. helping mature (28) and immature neurons to survive (29–31). BDNF is the most abundant neurotrophin in the hippocampal formation of cortex in both adult rodents and humans (2, 32).
We found that reduction of BDNF expression in the hippocampus by conditional knockout resulted in exacerbated death of both immature and mature neurons after TBI (30). This suggested that BDNF is involved in regulating the neuronal survival in the adult brain, and potentially might be used to protect immature neurons from death following TBI. Although direct injection of BDNF into the hippocampus decreased immature neuron death in a rodent TBI model (unpublished data), direct injection of BDNF into the hippocampus is an invasive treatment. Furthermore, because BDNF is a polypeptide growth factor, direct administration of BDNF into the brain may also cause an immune response and a further increase in inflammation following TBI. Therefore, an alternative molecule that can imitate BDNF function and can be used non-invasively is essential. One small molecule that imitates BDNF is 7,8-dihydroxyflavone (DHF), which protects neurons from death (33). Treatment with DHF improves neurological functions in rodent models of stress (34), depression (35), aging (36), and Alzheimer disease (37). In this study, we tested the role of DHF in immature neuron survival following TBI in a pre-treatment paradigm.
MATERIALS AND METHODS
Animals
Male C57BL/6 mice (Harlan Laboratories, Indianapolis, IN) were group-housed with a 12/12-hour light/dark cycle and had access to food and water ad libitum. They were used in experiments at the age of 8 to 10 weeks. All procedures were performed under protocols approved by the Animal Care and Use Committee of Indiana University.
Hippocampal Cells In Vitro
Brains of C57BL/6 mice (post-natal day 0) were dissected, the meninges were removed, and the hippocampi were isolated. Hippocampal tissue was digested with papain, and single-cells were collected, plated onto polylysine-coated coverslips in plates, and maintained in a B27 supplemented Neurobasal serum-free medium (38).
DHF Treatment In Vitro
The hippocampal cells were cultured in vitro for 5 days (day-in-vitro 5 [DIV 5]) and were treated with either DHF (10, 100, 500, 1,000, or 5,000 nMol/L) or dimethyl sulfoxide (DMSO) as control. Thirty minutes after DHF or DMSO treatment, the cells were then exposed to 100 μMol/L glutamate with 20 μMol/L glycine for 60 minutes to create an excitotoxicity model in vitro. The cells were then washed with phosphate-buffered saline (PBS) and cultured with a B27-supplemented Neurobasal serum-free medium in the incubator at 37°C.
Propidium Iodide Staining
Propidium iodide (PI) staining was performed to assess cell death at 24 hours after glutamate and glycine treatment (39). Briefly, PI (10 μg/mL) was applied to cell culture. After 30 minutes at 37°C in a humid incubator, the cells were washed briefly with PBS and then fixed with 2% paraformaldehyde (PFA) in PBS. Fixed cells were checked by phase contrast microscopy. Five random fields on each coverslip were chosen for images of both brightfield and PI staining. The total number of cells and the number of PI-positive cells were counted in each field and the percentages of PI-positive cells were calculated. Triplicate slides were included in each group.
TBI Model
C57BL/6 male mice (n = 131), 8 to 10 weeks old, were randomly subjected to either moderate CCI injury or sham surgery, as previously described (40, 41), using an electromagnetic model (42). Briefly, the mice were anesthetized with avertin and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA) prior to TBI. Using sterile procedures, the skin was retracted and a 4-mm craniotomy centered between the lambda and bregma sutures was performed. A point was identified midway between the lambda and bregma sutures and midway between the central suture and the temporalis muscle laterally. The skullcap was carefully removed without disruption of the underlying dura. Prior to the injury, the impacting tip was angled perpendicularly to the exposed cortical surface. The moderate severity brain injury was then introduced with the depth of deformation set at 1.0 mm and the piston velocity controlled at 3.0 m/sec. Sham (non-injured) animals (n = 18) received the craniotomy but no CCI injury. During all surgical procedures and recovery, the core body temperature of the animals was maintained at 36°–37°C. Animals did not receive analgesics after surgery.
DHF Administration
One hour prior to surgery, the animals received intraperitoneal injection of either DHF once (5 mg/kg in 17% DMSO) or vehicle (17% DMSO). Twenty-four hours after surgery, the animals were perfused and brains were collected for assessments.
ANA12 Administration
One hour prior to surgery, animals received an intraperitoneal injection of 0.5 mg/kg the tyrosine-related kinase B (TrkB) antagonist ANA12 (Fisher Science, Waltham, MA) (43, 44). Right after ANA12 injection, the animal received another intraperitoneal injection of DHF (5 mg/kg in 17% DMSO) or vehicle (17% DMSO).
Tissue Processing
Animals were deeply anesthetized with an overdose of avertin and then perfused transcardially with 0.9% saline, followed by an ice-cold fixative containing 4% PFA in PBS. The brains were removed and post-fixed in 4% PFA overnight, then cryoprotected with 30% sucrose for 48 hours. Serial coronal sections (30 μm thick) were cut using a cryostat (Leica CM 1950) and stored at −20°C.
Fluoro-Jade B Staining of Dying Neurons
The staining procedures were implemented as previously described (45, 46). Briefly, the sections mounted on slides were incubated in 0.06% potassium permanganate for 20 minutes, then rinsed in distilled water for 5 minutes. Thereafter, the sections were incubated in 0.0004% fluoro-Jade-B ([FJB], Histo-Chem, Inc., Jefferson, AR) for 20 minutes, and counterstained with 4′,6-diamidino-2-phenylindole ([DAPI], Sigma, St. Louis, MO) for 5 minutes. Finally, sections were rinsed in distilled water and air-dried overnight. The dry slides were mounted with DPX (Fluka).
Hippocampal Neuron Death Assessment
To determine the FJB-positive cell density across the entire hippocampus, 1 out of every 6 sections throughout the entire extent of the hippocampal formation was selected for assessment. The anatomical boundaries of each hippocampal subregion (CA1; CA3; granular cell layer [GCL]; molecular layer; hilus) were identified as previously described (47). The number of FJB-positive neurons in each subregion was determined by a blinded quantitative histological analysis through the Z-axis under a microscope system (Zeiss Axiovert 200 M, Carl Zeiss MicroImaging, Inc., Thornwood, NY), with a 40X objective. The subregion area (μm2) was measured with imaging software (AxioVision, v4.0, Carl Zeiss MicroImaging, Inc.). The results of FJB-positive cell density were calculated with the section thickness (30 μm) and presented as N/mm3.
Immunohistochemistry
Tissue sections were rinsed with PBS and then blocked with 10% donkey serum in PBS with 0.3% Triton for 2 hours at 4°C. The primary antibodies were sheep-anti-mouse p-TrkB (phospho Y816), (ab74841, Abcam, Cambridge, MA), diluted 1:20, mouse-anti-mouse NeuN, (MAB377, Millipore, Billerica, MA), diluted 1:100, and guinea pig-anti-mouse doublecortin (Dcx) (ab2253, Millipore) diluted 1:1000. The antibodies were applied and the sections incubated for 48 hours at 4°C. After they were rinsed with PBS, the sections were incubated with the appropriate secondary antibody (all from Invitrogen) for 1 hour at 4°C. The antibodies were donkey-anti-sheep Alexa488 IgG, 1:1000, (A11015), in 10% donkey serum in PBS, goat-anti-mouse cy3 IgG, 1:400, (A10521), or goat-anti-guinea pig cy5 IgG, 1:200, (A21450) in blocking buffer (1% bovine serum albumin, 5% normal goat serum in PBS). After rinsing, the sections were counterstained with DAPI for 2 minutes, rinsed with PBS, and then briefly with water. They were then mounted on slides with the mounting medium. Images were taken by using a Zeiss microscope.
Western Blot
Hippocampi of all groups (n = 3 mice/group) were dissected and protein samples were prepared for the Western blot. Equal amounts of protein were loaded into the wells of the SDS-PAGE gel, along with molecular weight markers. The gels were then run for 1 to 2 hours at 100V. Afterward, the proteins were transferred from the gel to the membrane and were incubated overnight. For antibody staining, the membrane was blocked for 1 hour at room temperature using 5% blocking solution. Then the membrane was incubated with appropriate dilutions of the above-described primary antibody overnight at 4°C. After washing in Tris-buffered saline with Tween (TBST) 3 times, the membrane was incubated with the recommended dilution of labeled secondary antibody for 1 hour at room temperature. The membrane was washed 3 times and rinsed in TBST. Finally, images were acquired using normal image scanning methods for colorimetric detection.
Statistical Analyses
All data are presented as mean ± SE, with the number of repetitive experiments or mice indicated. Data of different groups were statistically compared using one-way ANOVA or two-way ANOVA followed by post hoc tests.
RESULTS
DHF Protects Immature Hippocampal Neurons from Excitotoxic Injury In Vitro
Following experimental TBI, the interstitial concentration of glutamate may increase to a more than 100-fold higher than the normal level; glutamate-mediated excitotoxicity is well documented and leads to cell death right after injury (48). To imitate in vivo traumatic injury of immature neurons, hippocampi at neonatal day 0 were dissected, dissociated into single cells, and cultured in vitro. At DIV5, we assessed the cell types of the cultured cells using double immunostaining with an antibody against nestin, a marker for neural progenitors, and an antibody against Dcx, a marker for newborn neurons (49). Nine percent of the cells expressed only Nestin, 25% of the cells expressed both Nestin and Dcx, and 44% of the cells expressed only Dcx (Supplemental Fig. 1). These results indicated that 69% of the cells are either new neurons or are differentiating into new neurons at DIV 5 (50).
To assess the effect of DHF on neuronal survival following the glutamate/glycine-induced injury, the cells were pretreated with DHF in DMSO in a concentration of 500 nM or DMSO without DHF for 30 minutes before 1-hour exposure to glutamate (100 μM) and glycine (20 μM) to induce excitotoxicity. Twenty-four hours later, PI staining was performed to detect dead cells (Fig. 1a). In the vehicle (DMSO)-treated group there were 6% ± 1% of neurons labeled by PI (Fig. 1b–d, n). Glutamate/glycine treatment caused massive neuronal death (47% ± 3%, p < 0.001 vs. vehicle group; Fig. 1e–g, n). DHF treatment significantly reduced the PI-positive cells to 20% ± 1%, suggesting that DHF protected neurons from death induced by glutamate/glycine (Fig. 1h–j, n, p < 0.001 vs. glutamate/glycine treatment group). DHF treatment alone did not significantly affect the death (7% ± 1%) of healthy neurons without glutamate/glycine treatment (Fig. 1k–m, n). These results indicate that DHF protects neurons from death induced by glutamate/glycine-mediated excitotoxicity in vitro.
Figure 1.

7, 8-Dihydroxyflavone (DHF) protects immature hippocampal neurons from excitotoxic injury in vitro. (a) Experimental procedures. (b) Brightfield image of hippocampal granular neurons at the sixth day-in-vitro (DIV 6) treated with dimethyl sulfoxide (DMSO) as a control. (c) Propidium iodide (PI) staining to detect the dead cells. (d) Merged image of b and c. (e) Brightfield image of hippocampal granular neurons at DIV 6 treated with glutamate (Glu) (100 μM), glycine (Gly) (20 μM), and DMSO for 1 hour. (f) PI staining to detect the dead cells. (g) Merged image of e and f. (h) Brightfield image of hippocampal granular neurons at DIV 6 pretreated with DHF (500 nM) before treating with glutamate (100 μM), glycine (20 μM) for 1 hour. (i) PI staining to detect dead cells 25 hours after DHF treatment. (j) Merged image of h and i. (k) Brightfield image of hippocampal granular neurons at DIV 6 treated with DHF (500 nM). (l) PI staining to detect the dead cells 25 hours after treatment. (m) Merged image of k and l. (n) Quantification of PI-positive cells at different treatment conditions. o: Dose-response curve of DHF on neuroprotection. (%): Percentage of PI-positive cells of total number of cells. *p < 0.05; **p < 0.01; ***p < 0.001. N.S., not significant.
To determine an optimal dose of DHF for neuroprotection, the neurons from hippocampi (post-natal day 0) at DIV 5 were incubated with different concentrations of DHF from 0 to 5,000 nM 30 minutes before glutamate/glycine treatment (Fig. 1o). Twenty-four hours after glutamate/glycine treatment, PI staining was performed to detect the dead neurons. Cell death was induced by glutamate/glycine-mediated excitotoxicity without DHF treatment. When the cells were treated with DHF at a very low concentration of 10 nM, the cell glutamate/glycine-induced cell death was significantly reduced to 37% ± 1%, indicating that DHF showed a neuroprotective effect at the low concentration (p < 0.01). The neuroprotective effect of DHF was exhibited in a dose-dependent manner (Fig. 1o). The rate of cell death induced by glutamate/glycine-mediated excitotoxicity decreased to 23% ± 3% when treated with DHF at a concentration of 100 nM (F = 62.834, p < 0.001), and to 17% ± 2% at 500 nM (p < 0.05 vs. DHF at a concentration of 100nM). The neuroprotective effect reached a plateau after a concentration of 500 nM. The rate of cell death was 14% ± 2% when treated with DHF at a concentration of 1,000 nM, and was 13% ± 1% at 5,000 nm. The increase in DHF concentration to more than 500 nM did not further reduce the rate of cell death, suggesting that 500 nM is an optimal concentration for neuroprotection in vitro.
DHF Treatment Reduced Neuron Death in the Hippocampus After TBI
TBI abruptly induces cell death, neuronal dysfunction, vascular damage, and glial reactivation (51). Cell death in the hippocampus following CCI peaks at 24 hours, drops sharply to a low level at 48 hours, and then maintains a minimal level for up to 2 weeks after TBI (23).
We assessed whether DHF can protect neurons from death in the hippocampus following TBI. One hour before inducing TBI, we administered DHF (5 mg/kg, i.p. injection) or DMSO as control. The DHF dose has been shown to protect neurons from apoptotic cell death (33). The mice were perfused 24 hours after TBI to evaluate cell death with FJB staining (52, 53). Although we did not observe any FJB-positive cells in the HDG of sham operated mice (Fig. 2 a), we observed a large number of FJB-positive dying neurons mainly located in the HDG of TBI-injured mice (Fig. 2b–d), which confirmed our prior report (23). Further quantification of FJB-positive cells in the HDG showed there were 25395 ± 1260/mm3 FJB-positive cells in the HDG of mice that received only a TBI (Fig. 2b). The number of FJB-positive cells was reduced to 19505 ± 600/mm3 in TBI-injured mice with DHF treatment (Fig. 2c), which is a 23.19% reduction in cell death; this was statistically significant (Fig. 2e, F=7.123, p < 0.01). In contrast, in TBI injury mice with DMSO treatment, the number of FJB-positive cells in the HDG (25426 ± 1717/mm3) was not different from the untreated TBI group (Fig. 2d). These data indicated that DHF treatment can protect neurons in the HDG from death following TBI.
Figure 2.

Systemic administration of 7, 8-dihydroxyflavone (DHF) reduced neuron death in hippocampus after subsequent traumatic brain injury (TBI). (a) Brain sections containing hippocampus from sham operated mice with Fluoro-Jade B (FJB) and 4′,6-diamidino-2-phenylindole (DAPI) staining (blue). No FJB-positive cells were detected in sham-operated mice. (b) FJB staining (green) to detect the dying cells in the hippocampus 24 hours after moderate TBI. The section was counterstained with DAPI (blue). (c) FJB staining (green) to detect the dying cells in the hippocampus 24 hours after moderate TBI and treated with DHF. The section was counterstained with DAPI (blue). (d) FJB staining (green) to detect the dying cells in the hippocampus 24 hours after moderate TBI and treated with DMSO as a control. The section was counterstained with DAPI (blue). (e) Quantification of FJB-positive cells in the different regions of the hippocampus with different treatments. (n = 5/group, **p < 0.01).
DHF Treatment Protects Immature Neurons From Death in the Hippocampus After TBI
We next assessed whether protecting cell death in the HDG with pre-injury DHF increases the number of immature neurons in this area after TBI using immunostaining with anti-Dcx antibody, a specific marker for newborn immature neurons (49). Immature neurons stained red by Dcx located in the inner one third of the GCL (Fig. 3a). In sham-operated mice there were 187±14 Dcx-positive immature neurons in the hippocampus (Fig. 3a, e). In tissue from mice that received only a TBI, the number of Dcx-positive cells was reduced to 76 ± 4 (Fig. 3b, e). Between the 2 groups, the reduction seen in the number of immature neurons at the epicenter sections was 59.36% within 24 hours after TBI (p < 0.001), confirming previous results (21).
Figure 3.

7, 8-Dihydroxyflavone (DHF) treatment increases the number of immature neurons in hippocampus after subsequent traumatic brain injury (TBI). (a–d) Immunostaining with antibody against Doublecortin (Dcx, red) to show newborn neurons in the hippocampal dentate gyrus of a sham operated mouse (a), a TBI mouse 24 hours after surgery (b), a TBI-injured mouse 24 hours after receiving DHF treatment (c), or a TBI-injured mouse 24 hours after receiving DMSO treatment as a control (d). Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). (e) Quantifications of Dcx-positive newborn neurons after various treatments. (n = 5/group; *p < 0.05; **p < 0.01; ***p < 0.001)
The number of Dcx-positive immature neurons increased (142 ± 9) in the epicenter sections from TBI-injured mice with DHF pre-treatment, as compared to TBI-injured mice with DMSO (control) treatment (Fig. 3c, e, p < 0.01). After DHF treatment, only 24% of immature neurons were induced to death by TBI, indicating a 59.46% protection rate. In contrast, in the control group, DMSO treatment did not significantly change the number of newborn neurons (82 ± 3) following TBI (Fig. 3d, e). These data indicate that DHF effectively protects immature neurons from death following TBI.
TrkB Phosphorylation Is Reduced in the HDG Following TBI and DHF Treatment Restored TrkB Phosphorylation
Brain-derived neurotrophic factor and its receptor TrkB are broadly expressed in the developing and adult mammalian brain (54). TrkB tyrosine autophosphorylation in response to BDNF stimulates intracellular signaling that is critical for neuronal survival, morphogenesis, and plasticity. The level of phosphorylated TrkB (p-TrkB) is used as an indicator for the activity of BDNF signaling (55). We evaluated the total TrkB and p-TrkB level in the hippocampus 24 hours after TBI with Western blot and found that the level of phosphorylated TrkB was reduced after injury, although the total protein level of TrkB was not obviously changed (Fig. 4a). DHF treatment increased p-TrkB level in the hippocampus (Fig. 4a). Treatment with ANA12, which can specifically inhibit the phosphorylation of the TrkB receptors by occupying the binding site shared with BDNF and DHF (56), attenuated DHF-induced TrkB phosphorylation (Fig. 4a). These results suggest that the BDNF signaling activity is impacted by TBI and that DHF restored it through activating its receptor TrkB.
Figure 4.
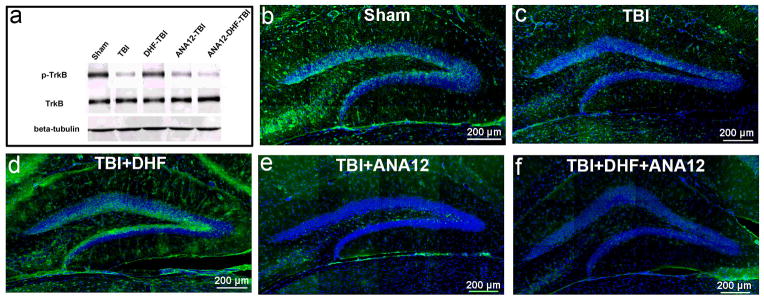
Phosphorylation of tyrosine-related kinase B (TrkB) receptor in the hippocampus following traumatic brain injury (TBI). (a) Western blot to assess the phosphorylated and un-phosphorylated TrkB receptor in the hippocampus. (b) Immunostaining to detect the phosphorylated TrkB receptor (p-TrkB) (green) in the hippocampus of mice 24 hours after sham operation. The section was counterstained with DAPI (blue). (c) Immunostaining to detect p-TrkB receptor (green) in the hippocampus of a mouse 24 hours after moderate TBI. The section was counterstained with DAPI (blue). (d) Immunostaining to detect the p-TrkB receptor (green) in the hippocampus of mice 24 hours after moderate TBI and 7, 8-dihydroxyflavone (DHF) treatment. The section was counterstained with DAPI (blue). (e) Immunostaining to detect the p-TrkB receptor (green) in the hippocampus of a mouse 24 hours after moderate TBI and treatment with ANA12. The section was counterstained with DAPI (blue). (f) Immunostaining to detect the p-TrkB receptor (green) in the hippocampus of mice 24 hours after moderate TBI and treated with DHF and ANA12. The section was counterstained with DAPI (blue).
Immunostaining with antibody to evaluate the distribution and protein level of p-TrkB in the HDG showed that TrkB is highly phosphorylated in the sham-operated hippocampus, mainly in the neurons located in the inner one third of the GCL, where the newborn neurons reside (Fig. 4b). TBI markedly reduced the intensity of p-TrkB immunoreactivity in the hippocampus (Fig. 4c). The immunostaining of p-TrkB in the inner one third of the GCL of TBI-injured mice was significantly reduced compared to the mice with sham operation. DHF treatment significantly increased p-TrkB in the hippocampus, particularly those neurons in the inner one third of the GCL (Fig. 4d). Treatment with ANA12 attenuated DHF-induced p-TrkB in the inner one third of the GCL following TBI (Fig. 4e, f). These results provide further evidence that DHF restored BDNF signaling through activating its receptor TrkB in the hippocampus following TBI.
DHF Pretreatment Restored TrkB Phosphorylation in Immature Neurons in the HDG Following TBI
Because the distribution of p-TrkB is highly correlated with the location of immature neurons in the hippocampus, we next determined the effect of p-TrkB in newborn neurons following TBI by double immunostaining with antibodies against Dcx and p-TrkB. As shown in Figure 4b, p-TrkB is highly phosphorylated in the neurons at the inner GCL (Fig. 5a). The p-TrkB is highly expressed in Dcx-positive immature neurons (Fig. 5b–d), indicating that BDNF is relatively more active in the immature neurons than other mature neurons in the hippocampal GCL. After TBI, the strong fluorescence of p-TrkB in the inner GCL was diminished. The Dcx-positive cells were significantly reduced in the HDG following TBI (Fig. 5e). Expression of p-TrkB in the spared immature neurons became difficult to detect (Fig. 5f–h). These data indicate that TBI not only induces immature neuron death but also significantly reduces the activity of BDNF signaling in the newborn neurons. In contrast, in the CCI-injured mice treated with DHF, p-TrkB was extensively expressed in the HDG, i.e. p-TrkB was not only observed in the inner GCL, it was strongly expressed in both inner and outer granular cell layers (Fig. 5i). These results indicate that DHF treatment activated the BDNF signaling pathway in both immature and mature granular neurons in the hippocampus following TBI. When BDNF signaling is activated by DHF, the numbers of immature neurons were also increased (Fig. 3e, 5j–l). Treatment with ANA12 attenuated the protective effect of DHF on immature neuronal survival and on DHF-induced p-TrkB in immature neurons following TBI (Fig. 5m–p). These data suggest that DHF treatment activates the BDNF signaling pathway and protects immature neurons from death triggered by TBI.
Figure 5.

7, 8-Dihydroxyflavone (DHF) pre-treatment restored tyrosine-related kinase B (TrkB) phosphorylation in immature neurons in the hippocampal dentate gyrus after traumatic brain injury (TBI). (a–p) Double immunostaining to assess the expression of phosphorylated TrkB (p-TrkB) receptor in the newborn neurons with antibodies against p-TrkB (Green) and doublecortin (Dcx, red) in the hippocampal dentate gyrus at 24 hours after sham treatment (a), TBI (e), TBI and treated with DHF (i), and TBI and treated with DHF and ANA12 (m). Sections were counterstained with DAPI. Amplified images from the boxes in panels (a, e, i, m) show the expression of p-TrkB in the dentate gyrus (b, f j, n). Enlarged images from the boxes in panels (a, e, I, m) show immature neurons expressing Dcx (c, g, k, o). Merged images (d, h, i, p) of (b) and (c), (f) and (g), (j) and (k), and (n) and (o), respectively.
Blocking BDNF Signaling Attenuated DHF-mediated Neuroprotection In Vitro and In Vivo
To determine whether DHF plays a neuroprotective role through activating TrkB receptors, we blocked the activity of BDNF signaling with ANA12. We first applied ANA12 before the administration of DHF to inhibit the TrkB activating effect of DHF on hippocampal immature neurons in vitro. Consistent with the experiments above (Fig. 1n), DHF protected neurons from death induced by glutamate/glycine-mediated excitotoxicity in vitro (Fig. 6a, F = 80.30, p < 0.001). In contrast, ANA12 reduced the protective effect of DHF, suggesting that DHF mediated neuroprotective functions through activating the BDNF signaling pathway in vitro (Fig. 6a, F = 6.215, p < 0.05). ANA12 is stable in body fluids and can cross through the blood-brain barrier (BBB). Inhibition of TrkB phosphorylation by ANA12 reduced the protective effect of DHF on newborn immature neurons following CCI (Fig. 6b, F = 20.401, p < 0.001). It also attenuated the neuroprotective effect of DHF on cell death (Fig. 6c–f). The density of dead cells in the GCL was 19505 ± 600/mm3 in the TBI-injured mice treated with DHF; in mice treated with both DHF and ANA12, the density of dead cells in the GCL was increased to 24776 ± 1237/mm3 (p < 0.01). This number of dead cells was similar to that in TBI-injured mice without treatment (p > 0.05) (Fig. 6f). These data indicate that DHF exerts the neuroprotection mainly through activating the TrkB signaling-mediated BDNF signaling pathway following TBI.
Figure 6.

Blocking brain derived neurotrophic factor (BDNF) signaling attenuates 7, 8-dihydroxyflavone (DHF)-mediated neuroprotection in vitro and in vivo. (a) Quantification show that treatment with ANA12 attenuated 7, 8-dihydroxyflavone (DHF)-mediated neuroprotection in vitro, assessed with PI staining (n = 3/group, p < 0.05). (b) Quantifications show that ANA12 treatment attenuated DHF-mediated neuroprotection in vivo using immunostaining with antibody against doublecortin (Dcx) (n = 5/group, p < 0.05). (c) Fluoro-Jade B (FJB) staining (green) demonstrates dead cells in the hippocampus 24 hours after moderate traumatic brain injury (TBI). (d) FJB staining shows dead cells in the hippocampus 24 hours after moderate TBI pre-treated with DHF. (e) FJB staining shows dead cells in the hippocampus 24 hours after moderate TBI and pre-treated with DHF and ANA12. (f) Quantifications show that ANA12 treatment attenuated DHF-mediated neuroprotection in vivo (n = 5/group, *p < 0.05; **p < 0.01; ***p < 0.001). Sections b, c, d were counterstained with DAPI (blue).
DISCUSSION
Despite advances in research and improved neurological intensive care in recent years there is still a lack of effective pharmacological approaches that aim to protect neurons from death following TBI. The discovery of NSCs in the adult brain raises a potentially promising strategy for repairing CNS injury. However, we recently found that newborn neurons are particularly vulnerable to TBI. Thus, although TBI transiently enhances NSC proliferation, it does not result in an increase in neurogenesis. DHF is a small molecule (MW = 254.25 g/mol) with the ability to penetrate through the BBB and reach the brain areas that are affected by TBI (57). Administration of DHF can upregulate the phosphorylation of TrkB and protect newborn neurons from death in vitro and in vivo.
A large body of work has demonstrated the pro-survival role of BDNF in CNS neurons (58), including those in the cortex (28) and the hippocampus (59). In particular, BDNF is reported to show a pro-survival effect on immature neurons in the developing neurons (29). BDNF is the most abundant neurotrophin in the hippocampal formation of both the adult rodent cortex and human cortex (2, 32). BDNF has been shown to relate to functional recovery following ischemia (60).
TBI has been shown both to increase BDNF expression in the hippocampus (61–63), and to decrease BDNF expression in the cortex (64), suggesting that BDNF might be involved in modulating neurological function following TBI. Prior work has shown that there is a correlation between neurotrophic factor expression and outcomes in children with severe TBI (65). Recently, studies with TBI animal models demonstrated that increased BDNF might increase neurogenesis in the adult hippocampus (66), and contribute to functional recovery in brain injured rats with treatment of erythropoietin (67) or statins (66). D-amphetamine treatment or voluntary exercise also improved cognitive functions and increased levels of BDNF in the hippocampus after TBI (68). Dietary omega-3 fatty acids or curcumin (69), voluntary exercise (70), intravenous administration or intracerebral transplantation of marrow stromal cells (71), simvastatin (66), and erythropoietin (72) improved recovery from TBI with upregulation of BDNF or even seemed to require BDNF. We found that conditional knockout of BDNF in the hippocampus further increased the cell death of immature and mature neurons in the hippocampus following TBI (30), indicating that BDNF is involved in regulating neuronal survival following TBI. This strongly suggests that BDNF signaling is a promising target for promoting the survival of the immature neurons after TBI. The above reports support the speculation that BDNF-TrkB signaling is involved in the process and later outcomes of TBI.
Some groups have shown that with continuous infusion of BDNF to injured areas of cortical or hippocampal areas, AAV-assisted transfer of TrkB gene or their combination neither saved neuron death in the cortex, CA3, and hilus, nor helped cognitive recovery after TBI (73, 74). Neither study showed a protective effect on the mature neurons against cell death in cortex and the hippocampal CA3 and hilus area. Therefore, the effects of BDNF on recovery in learning memory and cognitive functions following TBI are still controversial.
When we investigated the activity of BDNF signaling, instead of BDNF expression, by assessing both TrKB expression and TrkB phosphorylation in the hippocampus following TBI, we found that TBI significantly reduced TrkB phosphorylation in the hippocampus by Western blot. Further immunostaining analyses confirmed that the level of TrkB phosphorylation was significantly reduced (Fig. 4), particularly in the inner GCL (Fig. 4c) and in newborn immature neurons (Fig. 5). This result indicates that TBI attenuates BDNF signaling in the hippocampus. It may partially explain, in the previous studies, moderate level of BDNF increase following TBI was not enough to affectively protect neurons from death following TBI, i.e. suggesting that stronger activation of the TrK receptor is required.
DHF is 8 times more potent in activating the BDNF pathway than BDNF itself (33). Although by Western blot DHF treatment significantly increased TrkB phosphorylation. Western blot cannot distinguish whether DHF treatment increases TrkB phosphorylation in the immature neurons, mature neurons, or both in the hippocampus. Based on the location and cell-type specific marker, we found that DHF treatment increased TrkB phosphorylation in both immature neurons and mature neurons in the hippocampus. It also reduced glutamate/glycine-induced cell excitotoxicity in newborn neuron-enriched cultures in vitro and systemic administration attenuated newborn neuron death and preserved newborn neurons in the adult hippocampus following TBI. DHF can penetrate the BBB, bind the extracellular domain of TrkB, and provoke its dimerization and autophosphorylation and has shown neuroprotective effects in several neurodegenerative disease models (33, 75).
No specific pharmacological therapy is currently available that prevents the adult born immature neuron death following TBI. In the present study, pre-treatment of DHF was proven to have the protective effect against the newborn neuronal death after TBI via activating the TrkB in the hippocampus. Since the pretreatment paradigm is not relevant to clinical application, it is important to assess whether post-injury treatment of DHF is also neuroprotective against newborn neuron death.
Supplementary Material
Acknowledgments
This work was supported through funding from the Indiana Spinal Cord & Brain Injury Research Grants, the Ralph W. and Grace M. Showalter Research Award, Indiana University Biological Research Grant (BRG), and 1R21NS072631-01A.
References
- 1.Hall ED, Sullivan PG, Gibson TR, et al. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma. 2005;22:252–65. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- 2.Maisonpierre PC, Belluscio L, Friedman B, et al. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–9. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 3.Faden AI, Demediuk P, Panter SS, et al. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 4.Isoniemi H, Kurki T, Tenovuo O, et al. Hippocampal volume, brain atrophy, and APOE genotype after traumatic brain injury. Neurology. 2006;67:756–60. doi: 10.1212/01.wnl.0000234140.64954.12. [DOI] [PubMed] [Google Scholar]
- 5.Ariza M, Serra-Grabulosa JM, Junqué C, et al. Hippocampal head atrophy after traumatic brain injury. Neuropsychologia. 2006;44:1956–61. doi: 10.1016/j.neuropsychologia.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Wilde EA, Bigler ED, Hunter JV, et al. Hippocampus, amygdala, and basal ganglia morphometrics in children after moderate to severe traumatic brain injury. Dev Med Child Neurol. 2007;49:294–9. doi: 10.1111/j.1469-8749.2007.00294.x. [DOI] [PubMed] [Google Scholar]
- 7.Tate DF, Bigler ED. Fornix and hippocampal atrophy in traumatic brain injury. Learning & Memory. 2000;7:442. doi: 10.1101/lm.33000. [DOI] [PubMed] [Google Scholar]
- 8.Bigler ED, Blatter DD, Anderson CV, et al. Hippocampal volume in normal aging and traumatic brain injury. American J Neuroradiol. 1997;18:11. [PMC free article] [PubMed] [Google Scholar]
- 9.Umile EM, Sandel ME, Alavi A, et al. Dynamic imaging in mild traumatic brain injury: support for the theory of medial temporal vulnerability. Arch Phys Med Rehab. 2002;83:1506–13. doi: 10.1053/apmr.2002.35092. [DOI] [PubMed] [Google Scholar]
- 10.Saatman KE, Feeko KJ, Pape RL, et al. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23:1241–53. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- 11.Bonislawski DP, Schwarzbach EP, Cohen AS. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis. 2007;25:163–9. doi: 10.1016/j.nbd.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mount Sinai J Med. 2009;76:97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- 13.Leuner B, Kozorovitskiy Y, Gross CG, et al. Diminished adult neurogenesis in the marmoset brain precedes old age. Proc Nat Acad Sci USA. 2007;104:17169–73. doi: 10.1073/pnas.0708228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–33. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cameron HA, McKay RD. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol. 2001;435:406–17. doi: 10.1002/cne.1040. [DOI] [PubMed] [Google Scholar]
- 16.Eriksson PS, Perfilieva E, Bjork-Eriksson T, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–7. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 17.Kornack DR, Rakic P. Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc Nat Acad Sci USA. 1999;96:5768–73. doi: 10.1073/pnas.96.10.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–50. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- 19.Kempermann G, Gage FH. Neurogenesis in the adult hippocampus. Novartis Found Symp. 2000;231:220–35. discussion 35–41, 302–6. [PubMed] [Google Scholar]
- 20.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–50. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao X, Deng-Bryant Y, Cho W, et al. Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J Neurosci Res. 2008;86:2258–70. doi: 10.1002/jnr.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rola R, Mizumatsu S, Otsuka S, et al. Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp Neurol. 2006;202:189–99. doi: 10.1016/j.expneurol.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, Chen L, Gao X, et al. Moderate traumatic brain injury triggers rapid necrotic death of immature neurons in the hippocampus. J Neuropathol Exp Neurol. 2012;71:348–59. doi: 10.1097/NEN.0b013e31824ea078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pullela R, Raber J, Pfankuch T, et al. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev Neurosci. 2006;28:396–409. doi: 10.1159/000094166. [DOI] [PubMed] [Google Scholar]
- 25.Yavuz BB, Ariogul S, Cankurtaran M, et al. Hippocampal atrophy correlates with the severity of cognitive decline. Int Psychogeriatr. 2006:1–11. doi: 10.1017/S1041610206004303. [DOI] [PubMed] [Google Scholar]
- 26.Pitkanen A, McIntosh TK. Animal models of post-traumatic epilepsy. J Neurotrauma. 2006;23:241–61. doi: 10.1089/neu.2006.23.241. [DOI] [PubMed] [Google Scholar]
- 27.Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–69. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 29.Liu L, Cavanaugh JE, Wang Y, et al. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc Nat Acad Sci USA. 2003;100:8532. doi: 10.1073/pnas.1332804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao X, Chen J. Conditional knockout of brain-derived neurotrophic factor in the hippocampus increases death of adult-born immature neurons following traumatic brain injury. J Neurotrauma. 2009;26:1325–35. doi: 10.1089/neu.2008.0744. [DOI] [PubMed] [Google Scholar]
- 31.Kirschenbaum B, Goldman SA. Brain-derived neurotrophic factor promotes the survival of neurons arising from the adult rat forebrain subependymal zone. Proc Nat Acad Sci USA. 1995;92:210. doi: 10.1073/pnas.92.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timmusk T, Belluardo N, Persson H, et al. Developmental regulation of brain-derived neurotrophic factor messenger RNAs transcribed from different promoters in the rat brain. Neuroscience. 1994;60:287–91. doi: 10.1016/0306-4522(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 33.Jang SW, Liu X, Yepes M, et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Nat Acad Sci USA. 2010;107:2687–92. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andero R, Daviu N, Escorihuela RM, et al. 7,8-dihydroxyflavone, a TrkB receptor agonist, blocks long-term spatial memory impairment caused by immobilization stress in rats. Hippocampus. 2012;22:399–408. doi: 10.1002/hipo.20906. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Chan CB, Jang SW, et al. A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J Med Chem. 2010;53:8274–86. doi: 10.1021/jm101206p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng Y, Liu Y, Wu M, et al. Activation of TrkB by 7,8-dihydroxyflavone prevents fear memory defects and facilitates amygdalar synaptic plasticity in aging. J Alzheimers Dis. 2012;31:765–78. doi: 10.3233/JAD-2012-120886. [DOI] [PubMed] [Google Scholar]
- 37.Devi L, Ohno M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2012;37:434–44. doi: 10.1038/npp.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brewer G, Torricelli J, Evege E, et al. Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosc Res. 1993;35:567–76. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 39.Lau AC, Cui H, Tymianski M. The use of propidium iodide to assess excitotoxic neuronal death in primary mixed cortical cultures. Methods Mol Biol. 2007;399:15–29. doi: 10.1007/978-1-59745-504-6_2. [DOI] [PubMed] [Google Scholar]
- 40.Gao X, Deng P, Xu ZC, et al. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PloS One. 2011;6:e24566. doi: 10.1371/journal.pone.0024566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romine J, Gao X, Chen J. Controlled cortical impact model for traumatic brain injury. J Vis Exp. 2014;90:e51781. doi: 10.3791/51781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brody DL, Mac Donald C, Kessens CC, et al. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma. 2007;24:657–73. doi: 10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montalbano A, Baj G, Papadia D, et al. Blockade of BDNF signaling turns chemically-induced long-term potentiation into long-term depression. Hippocampus. 2013;23:879–89. doi: 10.1002/hipo.22144. [DOI] [PubMed] [Google Scholar]
- 44.Cazorla M, Premont J, Mann A, et al. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–57. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao X, Chen J. Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral Cortex. J Neuropathol Exp Neurol. 2011;70:183–91. doi: 10.1097/NEN.0b013e31820c6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmued L, Hopkins K. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–30. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 47.Amaral D, Witter M. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–91. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- 48.Palmer AM, Marion DW, Botscheller ML, et al. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem. 1993;61:2015–24. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- 49.Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–5. doi: 10.1038/35016083. [DOI] [PubMed] [Google Scholar]
- 50.Barnes AP, Polleux F. Establishment of axon-dendrite polarity in developing neurons. Annual Rev Neurosci. 2009;32:347–81. doi: 10.1146/annurev.neuro.31.060407.125536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 52.Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 53.Schmued LC, Hopkins KJ. Fluoro-Jade: novel fluorochromes for detecting toxicant-induced neuronal degeneration. Toxicol Pathol. 2000;28:91–9. doi: 10.1177/019262330002800111. [DOI] [PubMed] [Google Scholar]
- 54.Yan Q, Radeke MJ, Matheson CR, et al. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–57. [PubMed] [Google Scholar]
- 55.Leal G, Afonso PM, Salazar IL, et al. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2014 Oct 23; doi: 10.1016/j.brainres.2014.10.019. pii: S0006–8993(14)01421–8 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 56.Cazorla M, Prémont J, Mann A, et al. Identification of a low–molecular weight TrkB antagonist wih anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–67. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andero R, Heldt SA, Ye K, et al. Effect of 7, 8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psych. 2011;168:163–72. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barde YA. Neurotrophins: a family of proteins supporting the survival of neurons. Prog Clin Biol Res. 1994;390:45. [PubMed] [Google Scholar]
- 59.Lindholm D, Carroll P, Tzimagiorgis G, et al. Autocrine-paracrine regulation of hippocampal neuron survival by IGF-1 and the neurotrophins BDNF, NT-3 and NT-4. Eur J Neurosci. 1996;8:1452–60. doi: 10.1111/j.1460-9568.1996.tb01607.x. [DOI] [PubMed] [Google Scholar]
- 60.Almli CR, Levy TJ, Han BH, et al. BDNF protects against spatial memory deficits following neonatal hypoxia-ischemia. Exp Neurol. 2000;166:99–114. doi: 10.1006/exnr.2000.7492. [DOI] [PubMed] [Google Scholar]
- 61.Hicks RR, Martin VB, Zhang L, et al. Mild experimental brain injury differentially alters the expression of neurotrophin and neurotrophin receptor mRNAs in the hippocampus. Exp Neurol. 1999;160:469–78. doi: 10.1006/exnr.1999.7216. [DOI] [PubMed] [Google Scholar]
- 62.Yang K, Perez-Polo JR, Mu XS, et al. Increased expression of brain-derived neurotrophic factor but not neurotrophin-3 mRNA in rat brain after cortical impact injury. J Neurosci Res. 1996;44:157–64. doi: 10.1002/(SICI)1097-4547(19960415)44:2<157::AID-JNR8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 63.Mattson MP, Scheff SW. Endogenous neuroprotection factors and traumatic brain injury: mechanisms of action and implications for therapy. J Neurotrauma. 1994;11:3–33. doi: 10.1089/neu.1994.11.3. [DOI] [PubMed] [Google Scholar]
- 64.Wu CH, Hung TH, Chen CC, et al. Post-injury treatment with 7,8-dihydroxyflavone, a TrkB receptor agonist, protects against experimental traumatic brain injury via PI3K/Akt signaling. PLoS One. 2014;9:e113397. doi: 10.1371/journal.pone.0113397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chiaretti A, Piastra M, Polidori G, et al. Correlation between neurotrophic factor expression and outcome of children with severe traumatic brain injury. Intensive Care Med. 2003;29:1329–38. doi: 10.1007/s00134-003-1852-6. [DOI] [PubMed] [Google Scholar]
- 66.Wu H, Lu D, Jiang H, et al. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25:130–9. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- 67.Mahmood A, Lu D, Qu C, et al. Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J Neurosurg. 2007;107:392–7. doi: 10.3171/JNS-07/08/0392. [DOI] [PubMed] [Google Scholar]
- 68.Griesbach GS, Hovda DA, Gomez-Pinilla F, et al. Voluntary exercise or amphetamine treatment, but not the combination, increases hippocampal brain-derived neurotrophic factor and synapsin I following cortical contusion injury in rats. Neuroscience. 2008;154:530–40. doi: 10.1016/j.neuroscience.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu A, Ying Z, Gomez-Pinilla F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp Neurol. 2006;197:309–17. doi: 10.1016/j.expneurol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 70.Griesbach GS, Hovda DA, Gomez-Pinilla F. Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res. 2009;1288:105–15. doi: 10.1016/j.brainres.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mahmood A, Lu D, Chopp M. Intravenous administration of marrow stromal cells (MSCs) increases the expression of growth factors in rat brain after traumatic brain injury. J Neurotrauma. 2004;21:33–9. doi: 10.1089/089771504772695922. [DOI] [PubMed] [Google Scholar]
- 72.Lu D, Mahmood A, Qu C, et al. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma. 2005;22:1011–7. doi: 10.1089/neu.2005.22.1011. [DOI] [PubMed] [Google Scholar]
- 73.Blaha G, Raghupathi R, Saatman K, et al. Brain-derived neurotrophic factor administration after traumatic brain injury in the rat does not protect against behavioral or histological deficits. Neuroscience. 2000;99:483–93. doi: 10.1016/s0306-4522(00)00214-1. [DOI] [PubMed] [Google Scholar]
- 74.Conte V, Raghupathi R, Watson DJ, et al. TrkB gene transfer does not alter hippocampal neuronal loss and cognitive deficits following traumatic brain injury in mice. Rest Neurol Neurosci. 2008;26:45–56. [PMC free article] [PubMed] [Google Scholar]
- 75.Andero R, Daviu N, Escorihuela RM, et al. 7, 8 dihydroxyflavone, a TrkB receptor agonist, blocks long term spatial memory impairment caused by immobilization stress in rats. Hippocampus. 2010;22:399–408. doi: 10.1002/hipo.20906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.