

Abstract
Whole exome sequencing of metastatic castration-resistant prostate cancer (mCRPC) reveal that 5~7% of tumors harbor promyelocytic zinc finger protein (PLZF) homozygous deletions. PLZF is a canonical androgen-regulated putative tumor suppressor gene whose expression is inhibited by androgen deprivation therapy (ADT). Here, we demonstrate that knockdown of PLZF expression promotes a CRPC and enzalutamide resistant phenotype in prostate cancer cells. Reintroduction of PLZF expression is sufficient to reverse androgen-independent growth mediated by PLZF depletion. PLZF loss enhances CRPC tumor growth in a xenograft model. Bioinformatic analysis of the PLZF cistrome shows that PLZF negatively regulates multiple pathways including the MAPK pathway. Accordingly, our data support an oncogenic program activated by ADT and this acquired mechanism together with the finding of genetic loss in CRPC implicate PLZF inactivation as a mechanism promoting ADT resistance and the CRPC phenotype.
Introduction
A long-standing challenge in the management of prostate cancer is the development of resistance to androgen deprivation therapy (ADT), a standard treatment to disrupt the androgen receptor (AR) signaling pathway, since AR has a profound effect on prostate carcinogenesis through the regulation of transcriptional networks, genomic stability, and gene fusions (1). While ADT is initially effective and presumably extends the survival of most prostate cancer patients, prostate cancer inevitably becomes resistant to ADT and castration resistant prostate cancer (CRPC) emerges (2). Newer agents targeting the androgen signaling axis (AR-targeted therapies), such as abiraterone and enzalutamide, have yielded improved outcomes for patients with CRPC. Unfortunately, not all patients with CRPC respond to these AR targeted therapies, and moreover, these agents are not curative in this setting (3). The main subset of mechanisms of resistance to these antagonists involve the AR signaling pathway, including AR gene overexpression, gain-of-function mutations, constitutively active AR splice variants, dysregulation of its coregulators, and de novo androgen synthesis (4). Additional categories of resistance mechanisms consist of de-repression of pro-growth pathways in response to ADT (5) or transformation to a distinct, androgen and AR-indifferent cell state (4).
The recent surge of genomic and transcriptomic information may permit molecular classification of CRPC and future clinical development of precision medicine based on predictive biomarkers (5). Intriguingly, whole exome sequencing of metastatic CRPC (mCRPC) revealed that 5~7% of tumors harbor promyelocytic leukemia zinc finger (PLZF) focal homozygous deletions. PLZF, also known as BTB-containing protein 16 (ZBTB16), was originally identified as a gene fused to RARα in acute promyelocytic leukemia (APL) patients (6). PLZF has been shown to play an important role in the regulation of major developmental and biological processes and carcinogenesis as a tumor suppressor gene, since it regulates the cell cycle and apoptosis in various cell types (7). Overexpression of PLZF was shown to inhibit cellular proliferation in AR positive LNCaP and AR-negative DU-145 prostate cancer cell lines (8,9). Herein, our data show that PLZF emerged as the top gene from an AR cistrome analysis, credentialing PLZF as an androgen-regulated putative tumor suppressor gene in prostate cancer. Accordingly, we report a resistance mechanism to ADT mediated by PLZF, which appears to result from the activation of pro-growth pathways in response to ADT. Furthermore, the findings of PLZF genetic loss in mCRPC tumors supports that PLZF may be an important mediator in a subset of CRPC tumors.
Materials and Methods
Cell Culture, Lentiviral Infection and Xenografts
LNCaP/22Rv1 and VCaP cells were cultured in RPMI1640 and DMEM medium with 10% FBS. 22Rv1 xenografts were established in the flanks of male nude mice by injecting ~2 million stable 22Rv1 cells with shCtrl or shPLZF knockdown in 50% matrigel 3 days after castration. Tumors were measured 3 times every week and harvested after 3 weeks. All animal experiments were approved by the Beth Israel Deaconess Institutional Animal Care and Use Committee and were performed in accordance with institutional and national guidelines
Cell Proliferation (Crystal violet staining/WST1)
Cell growth was examined using the crystal violet (CV) staining and WST1 assays (Roche) following the manufacturer’s protocol. CV was dissolved in 10% acetic acid and cell proliferation calculated relative to the negative control cells, by measuring the absorbance at 595 nm.
RT-qPCR, Immunoblotting, and Immunohistochemistry
RNAs were extracted using Trizol according to the manufacturer’s protocol. Primers are listed in Supplemental Information. qPCR data are represented as mean ±STD for more than 3 replicates. Blots were incubated with anti-PLZF (MAB2944, R&D Systems), anti-actin (A5316, Sigma), total p44/42 MAK (Erk1/2) (4695, Cell Signaling) or phospho- p44/42 MAK (Erk1/2) (4370, Cell Signaling). Paraffin sections underwent antigen retrieval and were subjected to the staining protocol using Dako EnVision+System-HRP DAB. Anti-PLZF (MAB2944, R & D systems), anti-Ki67 (Dako), or non-specific IgG was then added overnight at 4°C. Sections compared in each figure were stained at the same time and photographed under identical conditions.
Chromatin Immunoprecipitation (ChIP) assay and ChIP-Seq Data Analysis
ChIP experiments were performed as previously described (10). The PLZF antibody (MAB2944, R&D Systems), or nonspecific IgG were used for ChIP. ChIP-Seq raw data was mapped by Bowtie 2 with default parameters. The identification of ChIP-seq peaks (bound regions and summit) was performed using MACS (PMID: 18798982). Regions of enrichment comparing to input control exceeding a given threshold (p <1e-5) were called as peaks. The primers for qPCR are provided in the Supplemental Information.
Gene expression experiments and analysis
LNCaP cells were transfected with either control shRNA (shCtrl) or shRNAs targeting PLZF (shPLZF). Forty-eight hours after shRNA transfection, total RNA was isolated and hybridized to Affymetrix human U133 plus 2.0 expression array (Affymetrix, Santa Clara, CA). Raw data is preprocessed using RMA (PMID: 12538238) and the cutoff of 1.5 fold change and P value <0.05 is applied for differential expressed gene analysis.
Results and Discussion
The hope of precision medicine is to tailor treatment based on each patient’s genomic and transcriptomic characteristics. This approach has proven to be challenging for the management of prostate cancer because of the paucity of actionable mutations found thus far. The recent finding (11) that 7% (4/61) of mCRPC tumors harbored PLZF homozygous deletions captured our interest (Fig. 1A). Indeed, homozygous deletion of PLZF was further seen in two independent cohorts; 6% (4/63) and 5% (8/152) from the University of Washington and the Stand Up to Cancer/Prostate Cancer Foundation (SU2C/PCF) which will be part of a larger SU2C genomic landscape paper (personal communications). This prompted us to explore the role of PLZF in prostate cancer. Here, we postulated that since PLZF was androgen regulated, AR might activate PLZF, an intermediate tumor suppressor gene which might derepress an oncogenic program with androgen depletion. This was based on the observation that ADT induces the expression of androgen-repressed genes that normally regulate androgen synthesis, DNA replication and cell cycle progression in CRPC models (12).
Figure 1. PLZF is an androgen-regulated gene involved in growth suppression.

(A) Venn diagram showing the frequency (%) of PLZF homozygous deletions (n=homozygous deletions/total mCRPC tumors) (11) and PLZF as a putative tumor suppressor gene with strongest AR binding merged from two AR cistrome datasets. RT-qPCR and Western blotting were used to measure PLZF mRNA and protein expression of LNCaP cells which were cultured in charcoal-stripped serum (CSS), followed by (B) 10nM of DHT and/or 10μM of bicalutamide (Bic.) treatment. The colonies were stained by crystal violet (CV) and photographed. The efficiency and efficacy of (C) PLZF shRNA knockdown and (D) ectopic re-expression of PLZF was measured by Western blot. Each column was relative to the corresponding the first column and shown as mean ± SD (n ≥ 3), and *p < 0.05.
Taking an agnostic approach, we sought androgen regulated candidate tumor suppressor genes. We compiled two AR cistrome datasets and showed that PLZF was the canonical tumor suppressor gene with strongest androgen-induced AR recruitment to its putative enhancer regions in two androgen-dependent prostate cancer cell lines, LNCaP and VCaP (Supplementary Fig. S1 and S2A), implying that the tumor suppression function of PLZF may be diminished upon ADT treatment.
The androgen-stimulated effect on PLZF expression was demonstrated in LNCaP, VCaP and 22Rv1 cells (Fig. 1B, Supplementary S2B and S2C). More importantly, PLZF expression was repressed by an antiandrogen (bicalutamide) in LNCaP cells (Fig. 1B, bottom panel).
To explore the tumor suppressive function of PLZF on prostate cancer, we examined the biological effect of altered PLZF expression on cell growth in androgen-depleted condition. Knockdown of PLZF using 4 different shRNA constructs (shPLZF#1~#4) promoted androgen-independent growth in LNCaP cells (Fig. 1C and Supplementary S3A). Conversely, re-expression of PLZF reversed the androgen-independent growth mediated by PLZF depletion (Fig. 1D).
To determine whether PLZF perturbation might promote androgen-independent growth in vivo, we also analyzed the growth of 22Rv1 prostate cancer xenograft expressing PLZF shRNAs, in castrated nude mice. Consistent with the in vitro observations, PLZF knockdown enhanced tumor formation in castrate levels of androgen (Fig. 2A). Altogether, our results show that PLZF is a putative AR-regulated tumor suppressor gene.
Figure 2. PLZF functions as a tumor suppressor in vivo.

(A) Tumor formation assays of castrated male nude mice injected with shCtrl and PLZF stable silencing 22Rv1 cells. Bottom right: averaged xenograft tumors (mean±SEM); left: PLZF and Ki-67 immunohistochemistry (IHC) were used to monitor the efficacy of PLZF knockdown and cell proliferation in 22Rv1 xenografts. (B) PLZF gene expression from 27 hormone-sensitive prostate cancer (HSPC) and 29 bone metastatic castration-resistant prostate cancer (mCRPC).
Next, we interrogated the extent to which PLZF expression might be altered in patient-derived CRPC tumor samples and found that mean PLZF expression was significantly lower in CRPC bone metastases compared to primary tumors (Fig. 2B) consistent with the notion that ADT suppresses serum and tissue androgens and results in diminished AR pathway activity (9).
The subcellular localization of PLZF is mainly in the nucleus where it achieves its transcriptional repression by binding to the regulatory elements in the promoter region of the target genes (7). In order to uncover PLZF-regulated transcriptome, we defined the PLZF cistrome using PLZF ChIP-seq and gene profiling data sets (Fig. 3A, Supplementary Table 1 and Table 2). Next, we investigated the potential biological consequence of PLZF suppression. Genes whose expression was up-regulated in PLZF-depleted LNCaP cells were subjected to bioinformatic pathway analysis. KEGG analysis revealed that PLZF-repressed genes were significantly enriched in the MAPK signaling pathway, including 5 genes with PLZF binding sites, RRAS, MKNK2, DDIT3, JUND and JUN (Supplementary Table 3). ChIP- and RT-qPCR confirmed that these genes are part of the PLZF-repressed cistrome (Fig. 3B). More importantly, PLZF knockdown substantially induced phospho-ERK (1/2) expression upon epidermal growth factor (EGF) stimulation in LNCaP (Fig. 3C, left). We also observed elevated levels of phospho-ERK activity in PLZF-depleted 22Rv1 cells (Fig. 3C, right). The inhibitory effect of MAPK inhibitors (UO126 and AZD6244) on PLZF depleted LNCaP was assessed. Cells with PLZF depletion responded better to MAPK inhibitors as compared to shCtrl, implying that the MAPK pathway may be activated due to loss of PLZF expression (Supplementary Fig. S4).
Figure 3. Bioinformatic analysis of the PLZF transcriptional program.
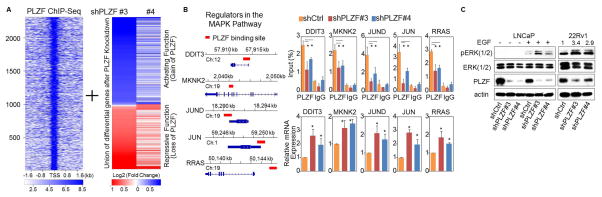
(A) Heat maps of PLZF ChIP-seq signal ±2.0 kb around the PLZF peak summit in LNCaP. The color scale indicates average signal. The numbered index of PLZF peaks is shown to the left. A cluster of differentially expressed genes in the LNCaP with stable knockdown of shPLZF# 3 or 4. (B) KEGG pathway analysis of PLZF-repressed genes. PLZF direct targets were highlighted in bold red. (C) Left panel: Schematic graph shows the PLZF binding sites (red bars) within the PLZF target gene loci as defined by PLZF. Right panel: ChIP-seq in LNCaP cells. ChIP- and RT-qPCR validation of PLZF binding and gene expression to selected PLZF targets. Values were the mean ± SD (n≥3) and *p < 0.05. (D) WB of LNCaP and 22Rv1 was used to measure of PLZF expression and ERK (1/2) activity with or without EGF (10ng/ml) stimulation.
Although our results suggest that PLZF regulates ERK (1/2) activity, it is unlikely that the mechanism underlying ERK activation only depends on PLZF transcriptional modulation. Moreover, PLZF has been shown to regulate a variety of downstream targets at the post-translational level (13). PLZF-regulated intracellular signaling molecules may also cross-talk with other regulatory pathways. Nonetheless, taken together, our data suggest that suppression of PLZF may permit sustained prostate cancer cell growth under conditions of androgen deprivation in part by de-repressing key tumorigenic mechanisms such as ERK (1/2) signaling. This finding may partly explain and is in agreement with previous findings that MAPK signaling is up-regulated in some CRPC murine models and patient-derived tumor samples (14–16). Thus, in the subset of patients with low PLZF expression including genetic loss, MAPK pathway inhibition may be of particular importance.
To begin to explore the potential effect of PLZF loss on AR-targeted therapy, we evaluated the impact of enzalutamide on the growth of prostate cancer cells in the absence or presence of PLZF knockdown. As expected, enzalutamide completely killed the shLuc-silenced cells when cultured in the regular conditioned medium. PLZF-depleted cells showed the ability to grow, although to a lesser extent, even in presence of enzalutamide. (Fig. 4A). When we conducted the same experiment culturing the cells in androgen-deprived medium (CSS), we observed a similar growth pattern. While shLuc cells remained quiescent in absence of androgens, shPLZF cells showed slight sensitivity to enzalutamide at early time point, becoming progressively resistant to the drug at late time points (Fig. 4B). To determine whether the presence of AR is required for PLZF-dependent growth, we introduced an IPTG-inducible shAR in the shPLZF stable LNCaP cells. Strikingly, our data showed that PLZF loss enables LNCaP cells to proliferate even in the absence of androgens or AR expression (Fig. 4C). These results imply that PLZF inactivation may be a key factor in the development of resistance to AR-directed therapeutics, such as enzalutamide. Collectively, our data suggest that PLZF suppression or genetic loss may underlie a novel mode of resistance to ADT, wherein an AR-repressed oncogenic program facilitates residual prostate tumor cells to adjust to castrate levels of androgens to survive or grow.
Figure 4. PLZF depletion alters the growth inhibitory effect of enzalutamide.

LNCaP cells with or without PLZF knockdown were cultured in (A) 5% FBS or (B) CSS medium and treated with or without 2.5 μM of enzalutamide or (C) IPTG-inducible shAR knockdown, followed by CV staining at the time as indicated. Each column was relative to the corresponding the first column and shown as mean ± SD (n ≥ 3), and *p < 0.05.
In view of the AR-dependent mechanisms for CRPC development, ADT may directly or indirectly activate an AR-repressed network, although we cannot completely exclude the involvement of oncogenic activation mediated by persistent AR expression in residual prostate tumors. Accordingly, we report that the up-regulation of the PLZF-repressed oncogenic program is an acquired mechanism in response to ADT and that genetic loss of PLZF in the course of disease are important molecular events in the emergence of CRPC and development of resistance to ADT and perhaps enzalutamide. Presumably, prostate cancer genomic and transcriptomic information may permit better molecular classification and provide new insights into the mechanisms of resistance to androgen/AR signaling blockade, thus aiding the design of future therapeutic combinations to overcome drug resistance.
Supplementary Material
Acknowledgments
We extend our gratitude to Drs. Shaoyong Chen and Gwo-Shu Mary Lee for their contributions in experimental and technical assistance, suggestions, and critical reading of the manuscript. The work from the laboratory of P.W.K. was supported by DF/HCC Prostate Cancer SPORE (P50 CA090381-12) and C-L.H. was supported by DoD Postdoctoral Training Award (W81XWH-13-1-0383). The rapid autopsy program and tissue/tumor collection and analyses were supported by PNW SPORE (P50 CA097186), NIH (P01CA163227) and NIH (P01CA085859). S.P.B was supported by NIH (P01 CA163227). M.B. was supported by NIH (P01CA163227). C.C. was supported by NIH K99CA166507. E.M.V was supported by NIH 1 K08 CA188615-01, SU2C/PCF Young Investigator Award. H.H.H. was supported by the Princess Margaret Cancer Foundation.
Footnotes
The authors declare no conflict of interest.
References
- 1.Mills IG. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nature reviews Cancer. 2014;14(3):187–98. doi: 10.1038/nrc3678. [DOI] [PubMed] [Google Scholar]
- 2.Egan A, Dong Y, Zhang H, Qi Y, Balk SP, Sartor O. Castration-resistant prostate cancer: adaptive responses in the androgen axis. Cancer treatment reviews. 2014;40(3):426–33. doi: 10.1016/j.ctrv.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene. 2014 doi: 10.1038/onc.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claessens F, Helsen C, Prekovic S, den Broeck TV, Spans L, Poppel HV, et al. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nature reviews Urology. 2014 doi: 10.1038/nrurol.2014.243. [DOI] [PubMed] [Google Scholar]
- 5.Roychowdhury S, Chinnaiyan AM. Advancing precision medicine for prostate cancer through genomics. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(15):1866–73. doi: 10.1200/JCO.2012.45.3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConnell MJ, Chevallier N, Berkofsky-Fessler W, Giltnane JM, Malani RB, Staudt LM, et al. Growth Suppression by Acute Promyelocytic Leukemia-Associated Protein PLZF Is Mediated by Repression of c-myc Expression. Molecular and Cellular Bology. 2003;23(24):9375–88. doi: 10.1128/MCB.23.24.9375-9388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suliman BA, Xu D, Williams BR. The promyelocytic leukemia zinc finger protein: two decades of molecular oncology. Front Oncol. 2012;2:74. doi: 10.3389/fonc.2012.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang F, Wang Z. Identification and characterization of PLZF as a prostatic androgen-responsive gene. Prostate. 2004;59(4):426–35. doi: 10.1002/pros.20000. [DOI] [PubMed] [Google Scholar]
- 9.Kikugawa T, Kinugasa Y, Shiraishi K, Nanba D, Nakashiro K, Tanji N, et al. PLZF regulates Pbx1 transcription and Pbx1-HoxC8 complex leads to androgen-independent prostate cancer proliferation. Prostate. 2006;66(10):1092–9. doi: 10.1002/pros.20443. [DOI] [PubMed] [Google Scholar]
- 10.Hsieh CL, Fei T, Chen Y, Li T, Gao Y, Wang X, et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(20):7319–24. doi: 10.1073/pnas.1324151111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33(22):2815–25. doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi J, Vogt PK. Posttranslational regulation of Myc by promyelocytic leukemia zinc finger protein. International journal of cancer Journal international du cancer. 2009;125(7):1558–65. doi: 10.1002/ijc.24449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(49):E4762–9. doi: 10.1073/pnas.1319948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia S, Gao X, Lee SH, Maira SM, Wu X, Stack EC, et al. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013;3(1):44–51. doi: 10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Velasco MA, Tanaka M, Yamamoto Y, Hatanaka Y, Koike H, Nishio K, et al. Androgen deprivation induces phenotypic plasticity and promotes resistance to molecular targeted therapy in a PTEN-deficient mouse model of prostate cancer. Carcinogenesis. 2014;35(9):2142–53. doi: 10.1093/carcin/bgu143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.