

Abstract
Age-related structural changes and gradual loss of key enzymes significantly affect the ability of the Endoplasmic Reticulum (ER) to facilitate proper protein folding and maintain homeostasis. In this work we present several lines of evidence supporting the hypothesis that the age-related decline in expression of the ER chaperone glucose regulated protein GRP78 (GRP78) could be related to the development of Parkinson’s disease (PD). We first determined that old (24 month) rats exhibit significantly lower levels of GRP78 protein in the nigrastriatal system as compared to young (2 month) animals. Then using recombinant adeno-associate virus (rAAV) mediated gene transfer, we found that GRP78 down-regulation by specific small interfering RNAs (siRNAs) aggravates alpha-synuclein (α-syn) neurotoxicity in nigral dopamine (DA) neurons. Moreover, the degree of chaperone decline corresponds with the severity of neurodegeneration. Additionally, comparative analysis of nigral tissues obtained from old and young rats revealed that aging affects the capacity of nigral DA cells to upregulate endogenous GRP78 protein in response to human α–syn neurotoxicity. Finally, we demonstrated that a sustained increase of GRP78 protein over the course of nine months protected aging nigral DA neurons in the α–syn-induced rat model of Parkinson’s-like neurodegeneration. Our data indicate that the ER chaperone GRP78 may have therapeutic potential for preventing and/or slowing age-related neurodegeneration.
Introduction
Activation of the ER stress response, also known as the Unfolded Protein Response (UPR), and accumulation of intracellular or extracellular protein aggregates are common to many age-related neurodegenerative disorders, including Parkinson’s disease (PD) (Muchowski and Wacker, 2005, Hoozemans et al., 2007, Hetz, 2012, Hoozemans et al., 2012, Naidoo and Brown, 2012). Glucose regulated protein 78 (GRP78), also known as BiP, is a key mediator of the UPR. It is a detector and regulator of the ER stress response that dissociates from three main “stress mediator” proteins when confronted with unfolded/misfolded proteins, thus activating them and the UPR. These stress sensors include Activating Transcription Factor 6 (ATF6), Inositol Requiring protein 1 (IRE1) and PKR-like Endoplasmic Reticulum Kinase (PERK). Early, pro-survival UPR signaling attempts to restore ER homeostasis. If this fails, the subsequent proapoptotic (late) UPR signaling tries to kill the malfunctioning cell, and involves crosstalk between the ER and mitochondria (Rutkowski and Kaufman, 2004, Szegezdi et al., 2009).
A number of age-related changes in cellular structure and function can predispose individuals to Parkinson’s disease. Among these is a sustained increase in α-syn protein, which when combined with the less efficient proteasome system in aging individuals, leads to accumulation of misfolded protein (Auluck et al., 2010). This age-related accumulation of α-syn, as well as an accompanying loss of TH-positive (TH+) neurons, were both previously documented in monkeys and humans (Kahle et al., 2000, Chu and Kordower, 2007). Overexpression of α-syn by an rAAV vector in the SNc has also been shown to trigger a progressive nigro-striatal degeneration, which featured α-syn-positive cytoplasmic and axonal inclusions. This degeneration resulted in dystrophic and fragmented neuritis and ultimately lead to cell death in both rat and mouse models of PD (Kahle et al., 2000, Masliah et al., 2000, Kirik et al., 2002). While it appears that α-syn protein is less efficiently cleared from the cytosol, leading to its accumulation and Lewy body formation, the exact mechanism by which α-syn triggers neurodegeneration remains unclear.
Immunoreactivity for markers of UPR activation, such as phosphorylated PERK (pPERK) and eIF2α (peIF2α), were previously found in post-mortem tissues of PD patients but not control cases (Hoozemans et al., 2007), indicating UPR involvement that was unique to the diseased state. The accumulation of pPERK also colocalized with accumulations of α-syn (Hoozemans et al., 2007), which was not surprising given that ER-accumulation of α-syn was shown to trigger the UPR and subsequent pro-apoptotic changes (Colla et al., 2012a, Colla et al., 2012b). In addition to describing a close association between UPR signaling and α-syn accumulation and aggregation there have also been reports demonstrating the potential for attenuating protein misfolding through pharmacological manipulation of ER stress in animal models of PD (Matus et al., 2008, Colla et al., 2012b). Despite these findings, little remains known about the age-related changes in individual UPR pathways and stress markers, which are associated with PD.
The age-related loss of critical UPR components, as well as structural changes in the ER, significantly affect its ability to control proper protein folding and maintain homeostasis (Naidoo and Brown, 2012). This breakdown in ER function is believed to occur, in part, from the oxidative wear of key ER resident proteins such as GRP78, PDI, calnexin and GRP94 during aging (Hinds and McNelly, 1978, Nuss et al., 2008).
Old mice (20–24 months) have demonstrated a 20% decrease in GRP78 ATPase activity, as compared to young (3–5 month) mice, which is consistent with a 2-fold increase in GRP78 carbonylation. Such findings support the hypothesis that loss of ER or other cellular functions, often seen in age-related diseases, is caused by the life-long accumulation of oxidative damage to key proteins (Nuss et al., 2008). Another study by Erickson, Dunning et al. (2006), describes a decline of ≤ 73% in GRP78 mRNA in old (900 day-old) vs young (21 day-old) rats, suggesting the loss of GRP78 activity and the associated physiological declines occur at both the protein and transcript levels (Erickson et al., 2006). This suggests that the loss of GRP78 function could be a predisposing factor for neurodegenerative disorders associated with age (Naidoo and Brown, 2012).
We have previously demonstrated (Gorbatyuk et al., 2012) the involvement of UPR signaling in the degeneration of nigral DA cells caused by α-syn-induced pathology in rats. Parkinson-like neurodegeneration was induced by rAAV-mediated expression of human α-syn in the rat substantia nigra pars compacta (SNc). We found that the mechanism of α-syn neurotoxicity in nigral DA neurons was associated with an activation of the UPR. This increase in α-syn correlated with an increase in markers for two of the three pathways in the UPR at 4 weeks post-injection, when α-syn expression reached maximum levels but significant neurodegeneration had not yet occurred. We observed a 23% elevation in peIf2α protein levels, a nine-fold induction in ATF4 (PERK pathway), and a greater than two-fold elevation of the cleaved form of pATF6 protein (ATF6 pathway). These increases were also accompanied by a three-fold elevation of pro-apoptotic CHOP protein that is known to be a downstream marker of the PERK and ATF6 pathways. Elevation of this protein is known to directly promote apoptosis and is thus perhaps responsible for DA cell death. Thus, we have concluded that overexpression of human α-syn activates the UPR signaling in the SNc.
In the current study we attempted to answer the question: does a decline in GRP78 during aging make nigral neurons more susceptible to human α-syn cytotoxicity? To this end, we used rAAV-mediated transduction to simultaneously express human α-syn and anti-GRP78 siRNAs in the rat SNc. Since aging sees the decline of numerous genes that could contribute to nigral degeneration, we conducted our experiments in young (2 month) animals so as to isolate the effects of GRP78 reduction from that of other genes. We also tested the hypothesis that a long-term increase in GRP78 expression could protect nigral DA neurons against α-syn cytotoxicity in older animals.
Results
Aging affects the nigral DA cell survival in response to human α–syn neurotoxicity
To examine the possibility that overexpression of human α–syn might have a different effect on DA cell survival in old (24 month) versus (vs) young (2 month) rats, we injected rAAV expressing human α-syn into the rat SNc. Control animals were injected with rAAV containing a blank expression cassette (BV), which was an α-syn vector with an early stop codon. All of the viral vectors in this study utilized the rAAV5 serotype capsid. Injections were done on the right side, while the left was kept as an uninjected control. In this study we used the same human α-syn rAAV stock as in our previous publication (Gorbatyuk et al., 2012), where we established that α-syn expression in the SNc at 4 weeks post vector administration was approximately 3.8 fold higher when compared with the noninjected control. Animals were sacrificed at 1.5 and 3 months (m) after injection. These time points were selected based on our previous findings (Burger et al., 2004, Gorbatyuk et al., 2010a, Gorbatyuk et al., 2010b, Gorbatyuk et al., 2012), which demonstrated that expression of α-syn reached a peak at 3–4 weeks after rAAV delivery and that neuronal degeneration in the SNc became significant after 2 months. We assumed that 3 months after injection provided sufficient time to reach a significant level of nigral degeneration in both experimental groups. The intermediate 1.5-m time point was selected to help determine if the rate of DA neuronal loss differed between young and old animals.
We first used unbiased estimation of nigral TH+ neurons to compare the rAAV-injected SNc with the uninjected SNc for each rat (Figure 1A and B). We did not observe any significant differences between rats injected with the control BV vector at the 1.5 and 3-m time points. Animals injected with human α-syn showed a progressive loss of TH+ neurons in both young and old animals. However, this loss reached significance at the 1.5-m time point only in old animals (69% reduction compared with BV control, P < 0.0001; N=7 for both control and experimental groups). Young rats demonstrated a 17% loss of TH+ cells, which did not reach significance vs BV (both N=8). This however was dramatically less than the loss seen from human α-syn in old rats (P < 0.001, N=6 and 8 for BV and α-syn injected rats respectively). At the 3-m time point we observed a significant decrease in the number of nigral TH+ cells in both young (59% reduction) and old (85% reduction) groups of human α-syn treated vs BV control animals. However, the human α-syn-induced effect on nigral TH+ cells in young rats was significantly less severe as compared with old animals (P < 0.01, N=7 for both control and experimental groups).
Figure 1.
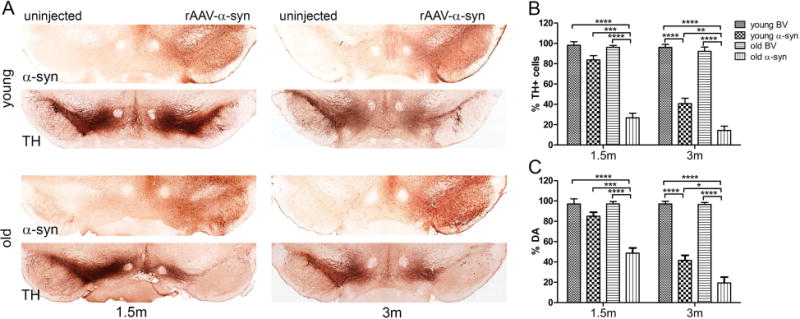
Aging increases vulnerability of nigral DA cells to human α–syn neurotoxicity. This figure compares the effect of human α-syn expression on SNc neuron survival in young and old rats. (A) Bright-field photomicrographs showing nigral degeneration in representative young and old animals at 1.5 and 3 months after virus administration. Images illustrate cells immunostained for human α-syn and a reduction of TH immunoreactivity on the injected side of parallel sections. (B) Graphs showing unbiased estimation of TH+ cells remaining in the SNc and (C) quantification of striatal DA level (bottom) at both time points. The percentage of TH+ cells and DA level were calculated by comparison with the uninjected side in the same animal. Two-way ANOVA was used. Tukey’ multiple comparisons test is presented as ± SE (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; N=6–8).
Next, dopamine (DA) levels were evaluated in striatal tissues of the injected vs uninjected sides. Extracted DA was fractionated by HPLC, and measured by using electrochemical detection (see Material and Methods for details). Our results revealed that striatal DA levels tracked with the number of surviving nigral neurons (Figure 1C).
Old animals displayed a more severe reduction in DA pools in response to human α-syn neurotoxicity. In response to α-syn overexpression, old animals at 1.5-m postinjection, displayed a 48% decrease in striatal DA (P < 0.0001 compared with BV, both N=7), which was accompanied by a ~69% loss of TH+ cells (compare Figures 1B and 1C). As with the stereology count results, young rats overexpressing human α-syn demonstrated a depletion of DA that was not significant at 1.5 m as compared with the BV control (both N=8) and was markedly less severe than in old animals (P < 0.0001; N=6 for both control and experimental groups).
At 3-m post treatment, DA depletion in both young and old animals corresponded well with the number of surviving TH+ neurons. However, the degree of significance between DA depletion in young (59% reduction, N=8) and old (81% reduction, N=6) rats injected with human α-syn was lower (P < 0.05) then at the 1.5-m time point.
These results allow us to conclude that old animals demonstrate a more rapid rate of progression and a greater degree of nigral degeneration in response to human α-syn cytotoxicity.
Age-associated decline of GRP78 in the rat SNc
While an age-related reduction of GRP78 has been documented in a number of tissues, including some parts of the brain, it has not been previously demonstrated in the SNc. To determine if this phenomenon does occur in the SNc, we performed RT-PCR and quantitative western blot analysis on dissected nigral tissues from 2- and 24-month-old F344 rats (Figure 2, Supplementary Figure 1A). We did not observe significant age-related changes in mRNA expression of GRP78, PERK or CHOP when normalized to β-actin or GAPDH (data not shown). Western blot analysis did not demonstrate any significant changes in the levels of tyrosine hydroxylase (TH) but α-syn protein levels were increased by 25% only in old versus (vs.) young males (P<0.006; both N=5). GRP78 protein levels were significantly lower in old vs young animals for both male (by 36.2%; P<0.02′ N=5) and female (by 27%; P<0.02; N=4 old/5 young) groups. These comparisons were made by normalizing to levels of GAPDH (Figure 2, Supplementary Figure 1B). Using β-actin as an additional house-keeping control, we confirmed the reduction in GRP78 (49%; P<0.001; N = 4 old, 3 young) that was initially observed when comparing to GAPDH. We also observed a dramatic increase of ATF4 protein levels (101.7%; P<0.001) in old vs young animals (Suplementary Figure 1C).
Figure 2.
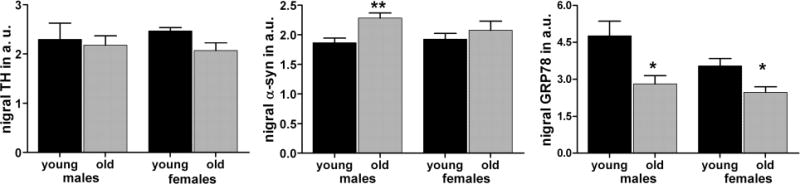
Downregulation of GRP78 expression accompanies aging in the rat SNc. Nigral tissue extracts from young (2 month) and old (24 month) rats were western blotted with GRP78, TH, α-syn and GAPDH antibodies (see Supplementary Figure 1A). The amount of analyzed proteins was normalized to GAPDH and shown on graphs in arbitrary units (a.u.). One tailed unpaired t test. Data are presented as mean ± SE (*P<0.05, **P<0.01; N=4 young females; 5 all other groups).
Efficient expression of multiple rAAV vectors in rat SNc
To determine the efficiency of rAAV-mediated transduction in the nigrostriatal tract, we performed a preliminary experiment using three different single siRNAs specific to rat GRP78. These were cloned as small hairpin RNA (shRNA) vectors expressing both the shRNA and green fluorescent protein (GFP) under different promoters. Following standard procedure, we used three different shRNA constructs against GRP78 as well as a scrambled (si-C) control to avoid off-target effects. For this preliminary study, titers for all three rAAV-shRNA constructs and the scrambled control (si-C) (Supplementary Table 1) were equalized to 6 ×1012 vg/ml. Rats were injected unilaterally with a total of 1.5 μl of each virus and quantitative western blot analysis of GRP78 expression was performed with an anti-GRP78 antibody in pooled (N=3) nigral tissues obtained at 4 weeks post injection. All three GRP78 shRNAs, but not si-C, induced a distinct reduction in target protein levels on the injected right (R) vs non-injected left (L) side in the SNc (Figure 3A). As expected, GFP was detected only on the injected side. It is worthy to note that none of the rAAV vectors used in this experiment affected nigral TH expression (Figure 3B).
Figure 3.
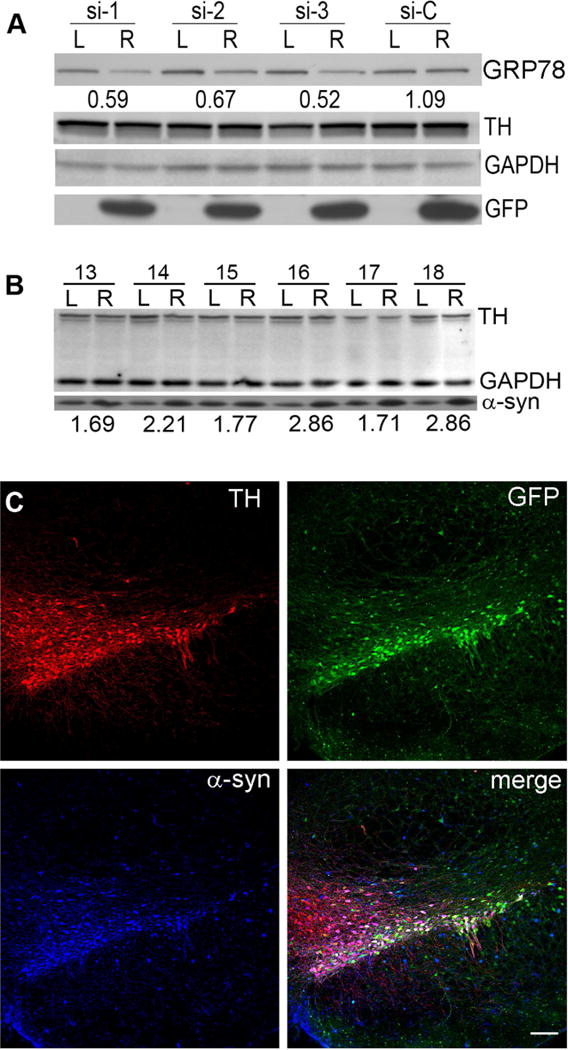
GRP78 expression can be augmented with specific shRNAs. Expression of GRP78 shRNA (A) and human wt α-syn alone (B) and together (C) in the SNc at one month after rAAV injection. (A) Comparison of three different shRNAs (si-1, si-2, si-3) against rat GRP78 and a scrambled control shRNA (si-C) in the SNc of rats at one month after vector injection. All four vectors also express GFP. Pooled (N=3 for each group) nigral tissue extracts were western blotted with GRP78, TH and GAPDH antibodies. The amount of GRP78 was normalized to GAPDH and the ratio of the injected (R) vs uninjected (L) sides was calculated (I/U). Note there was no loss of TH-positive staining in any of the groups at this time point. (B) α-Syn protein level in the SNc at one month after vector injection. Nigral tissue was excised at one month post injection from uninjected (L) and injected (R) sides of individual animals (13–18) that had been treated with rAAV-human α-syn. Nigral tissue extract was western blotted with anti-α-syn antibody that equally recognizes rat and human α-syn (Gorbatyuk et al., 2012). The ratio of α-syn on the injected and uninjected sides (I/U) was calculated for each animal. Note there was no loss of TH-positive staining in any of the animal groups at this time point. (C) Confocal images demonstrate co-expression of both GFP (green) (expressed by the GRP78 shRNA vectors) and human wt α-syn (blue) in TH-positive (red) neurons of the SNc injected with both viruses simultaneously (bar = 150 μm).
We next wanted to establish the appropriate level of human α-syn overexpression needed to reveal the difference in nigral degeneration between the animals transduced with α-syn alone or in combination with GRP78 shRNAs. Expecting that shRNAs would induce downregulation of GRP78 and thereby increase the vulnerability of nigral neurons, we decreased the α-syn vector titers to 6 × 1012 (the same as rAAV-shRNAs) thus allowing us to obtain a 2.2-fold increase in α-syn protein levels (Figure 3B) and to increase the timescale over which we observed neurodegeneration.
We then co-injected a mixture of the rAAV-shRNA and rAAV-α-syn viruses (1.5 μl of each), into the rat SNc. The delivery of rAAV-shRNAs to the SNc was verified by detection of native GFP fluorescence (Figure 3C). Using antibodies specific to TH, as a marker for DA neurons, and to human α-syn we analyzed neuronal co-localization of GFP fluorescence and human α-syn at 4 weeks post injection. We found that both viruses mediated expression in the same pattern of nigral TH-positive cells (Figure 3C). However, the transduction area of GFP/ α-syn neurons was not limited to the SNc since there were some GFP/α-syn-positive neurons in the pars reticulata as well as in the ventral tegmental area. These findings are in agreement with our previous observation (Burger et al., 2004, Gorbatyuk et al., 2008, Gorbatyuk et al., 2012). These observations lead us to conclude that we had expressed significant amounts of human α–syn and shRNA in most of the TH-positive neurons in the SNc.
siRNA-mediated downregulation of GRP78 aggravates α-syn-induced neurotoxicity in the nigrastriatal system
Two-month-old female Sprague-Dawley rats were injected in the SNc with 3 μl of the appropriate mixture of two vectors. In control groups (N=6 for each) we mixed si-1, si-2, si-3 or si-C with an rAAV vector containing a blank expression cassette (BV). This was used as a control to maintain the same volume and particle numbers for comparison with experimental groups injected with two viruses simultaneously. In experimental groups (N=10 for each) we mixed si-1, si-2, si-3 or si-C with an rAAV vector producing human α–syn. Injections were done on one side of the brain while keeping the other side as a non-transduced internal control. We used an amphetamine induced rotation test to monitor the same animals every 30 days for onset of behavioral deficit. At 3 months after treatment we observed a significant number of rotations toward the injected side in si-3 + α–syn experimental group of animals, as compared to control rats (see below). We did not observe statistically significant changes in the behavior test of the other experimental groups, including rats injected with si-C + α–syn. At this point the experiment was stopped and the animals were sacrificed for further analysis.
Confocal microscopy revealed visually discernible differences between experimental and control animals (Supplementary Figure 2). To give these results context, the degree of reduction in GRP78 protein was estimated in control rats injected with si-1, si-2, si-3 or si-C, combined in each case with BV. For this, the nigral sections were immunostained for GRP78 and TH (Figure 4A), and the level of GRP78 staining was estimated in GFP-positive nigral neurons that co-stained for TH on the injected side. These measurements were repeated and compared for both the injected and uninjected sides of the brain. There was a substantial reduction of GRP78 in injected vs. non-injected nigras. Using confocal image analysis software (Leica Applications Suite, Leica Microsystems CMS GmbH, Mannheim, Germany), it was determined that the level of GRP78 staining in TH positive neurons was 62 % of the control side in animals injected with si-1 (P < 0.001), 65% for those injected with si-2 (P < 0.05) and 51% in animals injected with si-3 (P < 0.0001; N=4 for all groups, 300 cells/group). No significant changes in GRP78 were seen in rats injected with si-C.
Figure 4.
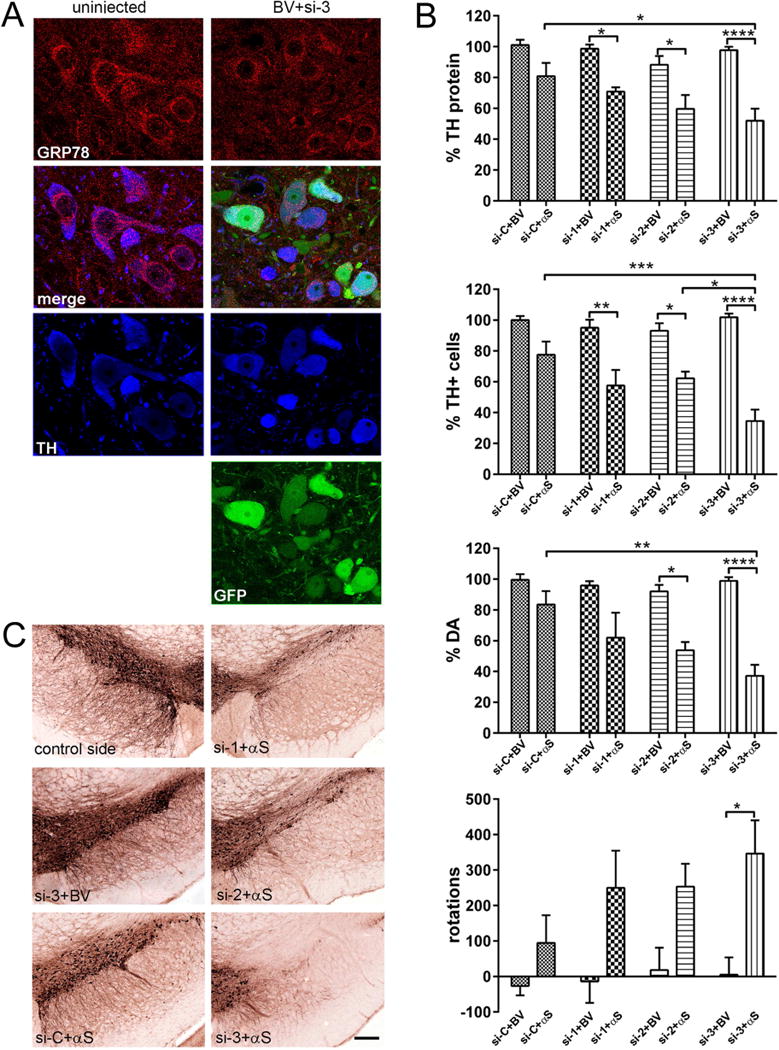
Degree of decline in GRP78 expression correlates with striatal TH protein levels, nigral DA cell survival and amplitude of behavioral asymmetry in response to human α-syn (αS) overexpression. (A) GRP78 protein expression level in DA neurons of the SNc at 3 months after rAAV-GRP78 shRNA injection. Confocal images show GRP78 (red) and TH (blue) as well as native GFP (green) in nigral neurons at 3 month after combined injection of viruses expressing si-3 and BV. TH is used as a marker for DA cells. Images illustrate protein expression levels of GRP78 (red) in nigral TH+ neurons (blue) on non-injected (left panel) and injected (right panel) sides. (B) The effect of GRP78 decline and human wt α-syn expression on striatal TH protein levels (upper graph), unbiased estimation of nigral TH+ neuron survival as well as striatal DA level and rotational behavior (bottom graph) at 3 months after rAAV injection. Striatal TH-protein level and the number of nigral TH+ cells remaining on the injected side shown as a percentage of the uninjected side ± SE. Amount of striatal TH protein, DA and number of nigral TH+ neurons were most dramatically reduced in si-3 + αS injected rats compared to control si-3 + BV as well as to si-C + αS (and si-2 + αS for TH cells count) injected animals. The amphetamine induced rotation test revealed a significant number of ipsilateral rotations only in rats injected with si-3 + αS versus control si-3 + BV animals. Two-way ANOVA analysis. Tukey’ multiple comparisons test are indicated as *, **, ***, **** = P < 0.05, 0.01, 0,001 and 0.0001, respectively vs. appropriate control rats; N = 6 (for control groups) and 10 (for experimental groups). (C) Bright-field photomicrographs showing remaining TH-positive cells in the SNc of representative animals from different experimental groups. Bar = 250 μm.
Next, striatal tissues from all animals were western blotted to determine the levels of TH on the injected right (R) vs. uninjected left (L) side (Supplementary Figure 3). We found that all experimental groups containing animals injected with α–syn and shRNAs, but not α–syn and si-C, demonstrated significant reduction in striatal TH protein levels, which was most prominent in rats injected with si-3 + α–syn (Figure 4B). Rats, which were injected with a mixture of α–syn and si-1, si-2 or si-3, retained 72.1 ± 3.0 %, 62.9 ± 8.7 % or 52 ± 7.8 % of the TH protein seen on the uninjected side, respectively. This was in contrast to the more modest, non-significant reduction in TH (80.7 ± 8.7 %) seen for the α–syn + si-C injection. In contrast, the level of TH in the striatum of control rats injected with BV and si-1, si-2 or si-3 was not significantly reduced (N=10 for experimental and N=6 for control groups).
Unbiased stereology counts of the number of TH+ cells in the SNc (Figure 4C) were done to quantitate the extent of neurodegeneration. We found that lowering GRP78 with si-1, si-2 and si-3 significantly exacerbated nigral degeneration in response to human α–syn expression (Figure 4B). Among the rAAV-shRHAs, si-3 was more potent at aggravating α–syn neurotoxicity using the same input genome dose as si-1 and si-2. This was consistent with its higher knockdown activity against GRP78 as determined by WB in nigral tissues at 4 week post injection (Figure 3A) and by confocal image analysis of control groups at three months post treatment (Figure. 4A). There was no significant TH+ cell loss identified in the α–syn + si-C injected SNc (Figure 4B).
The measurement of striatal DA levels in all experimental groups (N=6) agreed with TH protein and stereology count results. The α–syn + si-3 injected rats demonstrated dramatic DA depletion in the striatum compared with both control BV+si-3 as well as α–syn + si-C injected animals (Figure 4C).
The loss of TH+ neurons in the SNc and striatal DA was accompanied by a significant amphetamine-induced rotational asymmetry in rats injected with α–syn + si-3 (Figure 4C). All other experimental groups injected with α–syn displayed ipsilateral rotation (toward the injected side) however those results did not reach statistical significance (data not shown). This is consistent with previous observations that significant impairment in drug-induced and spontaneous motor behavior would appear only in animals which experienced a >50–60% loss of nigral DA neurons or striatal DA levels (Kirik et al., 1998, Kirik et al., 2002).
Aging affects the nigral cell capacity to upregulate endogenous GRP78 protein in response to α–syn neurotoxicity
To evaluate if ER stress induced by human α–syn in the SNc leads to equal upregulation of GRP78 in young (2 months) and old (24 months) rats, we injected rAAV expressing human α-syn on the right side of the brain in both groups. rAAV with blank expression cassette (BV) was administrated into the SNc on the contralateral side as control virus injection. Quantitative western blot analysis of GRP78, TH and α-syn expression was performed on dissected nigral tissues of individual animals at 4 weeks post injection (Figure 5, Supplementary Figure 4). As expected, no human α-syn expression was detected on the control left side. The level of total rat and human α-syn expression in the nigral tissue 4 weeks after injection with wt α-syn vector was approximately 4.1 (young) and 3.8 (old) fold higher than the control side of the brain. In addition, there was no significant difference between the levels of TH protein when normalized to total protein. However, comparative analysis of nigral tissues obtained from old and young rats revealed that aging affected the capacity of nigral cells to upregulate endogenous GRP78 protein in response to human α–syn expression. We identified a significant (up to 152 %; N=4, P<0.01) upregulation of GRP78 protein in the injected SNcs compared to control sides of young rats. At the same time, overexpression of human α–syn in the SNc of old rats did not induce a significant upregulation of GRP78 (~121 %, N=4, P>0.05).
Figure 5.
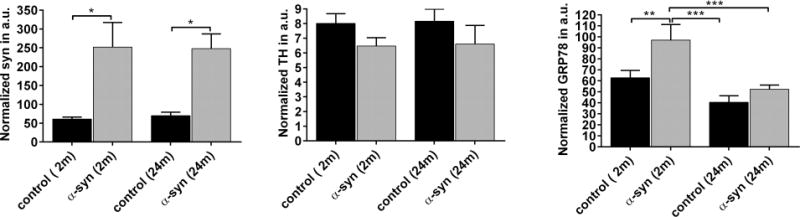
Aged nigral cells have a reduced capacity to upregulate endogenous GRP78 in response to human α-syn overexpression. Graphs show the level of TH, GRP78 or total α-syn expression in rAAV-human α-syn injected side compared to control rAAV-BV administered in young (2 month) and old (24 month) animals (see immunoblot images in Supplementary Figure 4). Each graph shows the change induced in each of the three proteins in response to human α-syn neurotoxicity. The amount of detected proteins was normalized to GAPDH. Two-way ANOVA analysis. Tukey’ multiple comparisons test are indicated as *, **, *** = P < 0.05, 0.01 and 0.001; N = 4 (for TH and GRP78) and N = 3 (for α-syn) per group.
Sustained overexpression of GRP78 protects aging nigral neurons against α-syn neurotoxicity
To test the hypothesis that a prolonged increase in GRP78 could be therapeutically beneficial for aging nigral DA cells, we injected rAAV vectors in the SNc of 12-month old female Sprague-Dawley rats to overexpress GRP78 and human α-syn simultaneously. We also performed injections with each virus separately. BV was once again used to maintain a consistent volume and particle number between single and double vector injections and was used as a virus injection control. In this study we used the same virus titers for human α-syn and GRP78 as we used in our previous publication (Gorbatyuk et al., 2012) in which the level of total α-syn expression in the SNc at 4 weeks post vector administration was approximately 3.8 fold higher compared with the control non-injected side. At the same time, GRP78 protein in the injected SNc was upregulated 1.4 fold compared to the non-injected side (Gorbatyuk et al., 2012). We had previously shown that the co-transduction with human α-syn and tagged GRP78 constructs did not alter protein expression, as compared with individual injections. The exogenous (tagged) GRP78 was also shown to co-immunoprecipitate with human α-syn, suggesting that it is indeed active (Gorbatyuk et al., 2012).
Amphetamine rotation tests were performed at 1, 3, 6 and 9 month after vector administration (Figure 6A) and revealed a notable number of rotations toward the injected side only in human α-syn overexpressing rats. It reached significance at 3 month post injection compared to BV, α-syn+GRP78 or GRP78-injected animals (accordingly, 423.7 ± 144.1 versus −1.8 ± 9.7, P < 0.01; −91.7, P < 0.001 or 17.6 ± 48.4, P < 0.001; N = 9 for all groups) and continued to demonstrate a significant number of ipsilateral rotations at 6 and 9 month time points compared to all control and experimental animal groups.
Figure 6.
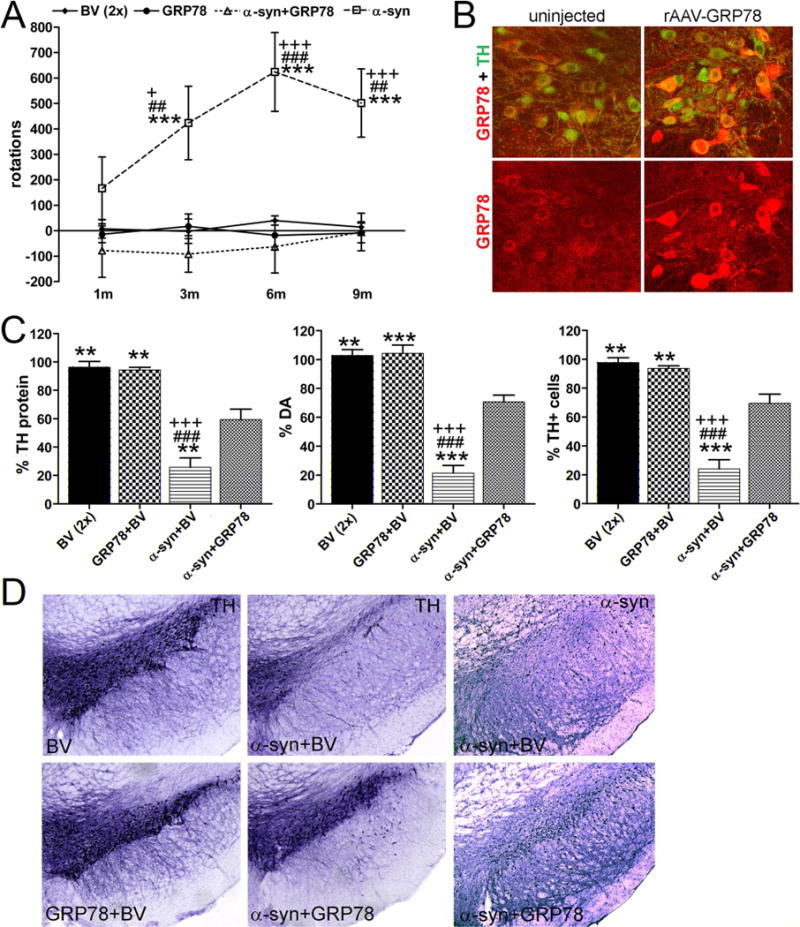
Overexpression of GRP78 in aging nigral DA neurons diminishes α-syn induced neurodegeneration. (A) Amphetamine-induced rotation test vs time post rAAV injections. Starting at 3 month a significant number of rotations toward the injected side (positive, ipsilateral rotation) was revealed only in α-syn injected rats compared to all other animal groups. Amphetamine induced rotation was measured for 90 min and the average rotation + SE is shown. Two-way ANOVA analysis. Tukey’s multiple comparison test is indicated as +, ## and +++ (### or ***) = P < 0.05, 0.01 and 0.001 vs. BV (#), GRP78 (+) and α-syn plus GRP78 (*); N= 9 per group. (B) GRP78 protein expression in DA neurons of the SNc at 9 months after AAV-GRP78 shRNA injection. Confocal Images illustrate protein expression level of GRP78 (red) in nigral TH-positive (green) neurons on non-injected (left panel) and injected (right panel) sides. (C) Quantitate analysis of striatal TH protein revealed by western blot (left graph, see Supplementary Figure 5). The graph shows the level of TH protein remaining in the striatum on the injected side as a percentage of the uninjected side ± SE at 9 months post injection. Two-way ANOVA analysis. Tukey’s multiple comparison test is indicated as ** and *** (### or +++) = P < 0.01 and 0.001, respectively vs. BV (#), GRP78 (+) and α-syn plus GRP78 (*); N = 8–9 per group. Middle graph shows loss of DA in the striatum injected with a vector expressing α-syn. Overexpression of GRP78 protein along with α-syn significantly (by 49%) preserved DA levels as compared with α-syn injected alone. This was also significantly different from the BV control. Overexpression of GRP78 protein alone did not have an effect on DA in injected SNcs compared to control BV injection. Two-way ANOVA analysis. Tukey’s multiple comparison test is indicated as ** and *** (### or +++) = P < 0.01 and 0.001, respectively vs. BV (#), GRP78 (+) and α-syn plus GRP78 (*); N = 5 (GRP78 control) – 7 (all others) per group. Right graph shows unbiased estimation of TH positive cells remaining in SNc on the injected side as a percentage of the uninjected side ± SE at 9 months post injection. The number of TH-positive neurons counted in the α-syn injected SNc was dramatically reduced compared to control BV rats. Co-expression of α-syn and GRP78 proteins led to significant rescue of TH-positive cells preventing cell death compared to a single human α-syn injection. However, these animals demonstrate a 28% and 24% loss of TH-positive cells compared to BV and GRP78 animals respectively, which is significant in both cases. GRP78 overexpression alone did not reduce the number of TH-positive cells. Two-way ANOVA analysis. Tukey’s multiple comparison test is indicated as ** and *** (### or +++) = P < 0.01 and 0.001, respectively vs. BV (#), GRP78 (+) and α-syn plus GRP78 (*); N = 8–9 per group. (D) Bright-field photomicrographs showing remaining TH-positive cells and human α-syn immunostaining in the SNc of representative animals from different experimental groups.
Rats were sacrificed 4 days after the last rotation test, when they were 21 months old. Rats injected with GRP78 showed higher expression of the chaperone in the SNc on the right side (Figure 6B). Using the same confocal image analysis software approach as we used in the above shRNAs study, we determined that the level of GRP78 staining in TH+ neurons (Figure 6B) in the injected SNc was 2.1 fold higher than the control side (P < 0.001). No significant change was seen in GRP78 levels in animals injected with control BV (N=4 for both groups, 300 cells/group).
As expected immunoblotting of striatal tissue extracts with human α-syn antibody revealed expression of human α-syn only on the injected side. Examination with an anti-TH antibody demonstrated severe loss of TH protein (25.8±6.6 %, N=9) in the striatum of animals overexpressing human α-syn only (Figure 6C, Supplementary Figure 5). The level of TH protein was significantly lower compared to control BV and GRP78 injected groups (for both N=8, P < 0.001). Co-expression of human α-syn with exogenous GRP78 significantly increased striatal TH protein (59.3±7.3 %, N=9; P < 0.001) compared to single human α-syn overexpression. However, this level of expression was still significantly lower compared to both BV and GRP78 groups of animals (P<0.01).
In parallel with TH protein measurements, we also assayed striatal DA in rAAV injected animals (Figure 6C). Striatal DA was diminished in rats injected with human α-syn compared with animals injected with control BV (21.3 ± 5.2, n = 7 vs. 102.7±4.2%, N = 5; P < 0.001) and with GRP78 (21.3 ± 5.2% versus 104.2 ± 5.7%, N = 6; P < 0.001). Co-injection of GRP78 with human α-syn significantly ameliorated the decline of striatal DA compared with animals injected with human α-syn alone (70.5 ± 4.7%, N = 7 vs. 21.3 ± 5.2%; P < 0.001). Overexpression of GRP78 alone did not affect DA levels in the striatum compared to control BV injections (104.2.5 ± 5.7% vs. 102.7 ± 4.2%, P > 0.05).
Unbiased estimation of nigral TH+ cells revealed that overexpression of human α-syn caused a severe loss of neurons in the SNc at 9 month post injection (Figure 6C and D). We determined that the number of TH+ cells was reduced dramatically (76%) in animals overexpressing human α-syn compared to BV control (24 ± 6.3%, N = 9 vs. 97.7 ± 3.4%, N = 8; P < 0.001). Co-expression of GRP78 with human α-syn prevented much of the loss of TH+ neurons caused by human α-syn (24 ± 6.3%, n = 9 vs. 69.4 ± 6.3%, N = 9; P < 0.001). However, these animals exhibited a 28% loss of TH+ cells compared to BV, which was significant (P > 0.01). GRP78 overexpression alone did not affect the number of nigral TH+ neurons. Likewise, in our previous study (Gorbatyuk et al., 2010b), we co-expressed a-syn with phospholipase D2 (PLD2) or GFP and found that neither protein had a protective effect unlike GRP78. Moreover, PLD2 possessed a more pronounced toxicity compared with a-syn.
Discussion
The results outlined in this study suggest that a decline in GRP78 expression during aging increases vulnerability of nigral DA neurons to human α-syn cytotoxicity and might be a predisposing factor for the onset and progression of synucleinopathies in humans. Our results demonstrate a correlation between the level of GRP78 knockdown provided by each siRNA and the level of neurodegeneration seen in our samples, making off-target effects highly unlikely as the cause of our observed results. Our findings of a significant loss of GRP78 protein in the SNc of old animals echo previous findings that have described a similar phenomenon in other tissues (Erickson et al., 2006, Nuss et al., 2008). This loss of GRP78 does not extend to the mRNA level, suggesting that GRP78 protein levels are controlled either on a translation level or via increased protein turnover.
GRP78 is a multifunctional chaperone that facilitates the refolding or proteasomal degradation of misfolded proteins and serves as a regulator of UPR signaling (Lee, 2005, Naidoo and Brown, 2012). It is through these roles that GRP78 imparts some of its anti-apoptotic activity as it serves to resolve conditions that lead to chronic ER stress, such as accumulation of misfolded proteins, as well as the associated late-stage, proapoptotic UPR signaling (Pfaffenbach and Lee, 2011, Naidoo and Brown, 2012). Conversely, loss of GRP78 activity reduces the cellular capacity to maintain healthy protein folding and to deal with stress, thus shifting the intracellular environment towards a proapoptotic balance (Li et al., 2014). Indeed our shRNA results indicate that loss of GRP78 activity sensitizes the cells to stress, which in our case was provided by the overexpression of human α-syn. The fact that the three tested siRNAs caused differing degrees of GRP78 knockdown proved to be instructional, since it revealed that a greater reduction of GRP78 corresponded with greater α-syn toxicity. This relationship was observed at the molecular, cellular and behavioral levels (Figure 4B).
An age-associated rise in the level of α-syn was previously reported (Kahle et al., 2000) and, taken together with our findings, could explain why pathology onset is often later in life, when neurons are in a more vulnerable state by virtue of worn-down cellular machinery. With this said, it needs to be confirm by future clinical studies whether the increase in α-syn is an independent phenomenon, or if it stems from the age-related decrease in GRP78 activity, which would leave the cell progressively less able to degrade misfolded α-syn. Indeed a major component of Lewy Bodies, a hallmark of PD, is phosphorylated α-syn (Cookson, 2005, Cookson, 2009). This could simply be protein that was initially marked for proteasomal degradation, but instead due to aging cellular machinery became sequestered in aggregates in an attempt to minimize harm to the cell.
Our own data demonstrated that older animals were less able to respond to challenge with α-syn (Figure 5). However it is not clear if the subsequent cell loss was due to acute α-syn toxicity, or to chronic activation of the UPR. Perpetual UPR activation caused by ever-increasing amounts of aggregated protein could lead to cell death through apoptosis; a well characterized consequence of unresolved ER stress (Muchowski and Wacker, 2005).
Age-associated deficits in a number of other chaperones and the proteasome have been previously described (Nuss et al., 2008), revealing a template for a broad range of age-related degenerative diseases. These diseases could vary in their localization and/or the main protein constituent comprising the aggregates (α-syn, β amyloid, etc.) but may fundamentally be the result of the same age-related breakdown of the cell’s capacity to deal with misfolded proteins. A gradual, age-related loss of ER function would thus condemn cells to create a backlog of misfolded proteins, which they could no longer clear. In failing to resolve the ER stress, chronic UPR signaling would lead to apoptosis. In this light, it is conceivable that the particular degenerative disease an individual would develop would be heavily influenced by the stresses endured in a particular tissue. Indeed, a lifetime of oxidative damage may very well result in ER stress, cell death and physiological deficits by the mechanism outlined above.
Fortunately, the emerging picture of the molecular events that may underlie the development and onset of PD also points to a potential therapeutic approach. We have previously shown that rAAV-mediated upregulation of GRP78 could be neuro-protective when challenging 2-month-old rats with overexpression of human α-syn in the SNc. We found that GRP78 significantly attenuated the ER stress response by diminishing upregulation of mediators of two UPR signaling pathways: ATF6 and PERK (Gorbatyuk et al., 2012). A downregulation of the latter was very recently shown to prevent neurodegeneration, as well as accumulation and aggregation of misfolded proteins, in prion-infected mice (Moreno et al., 2013). While the findings of those studies in young rats were interesting, GRP78 would need to be effective in older animals to show real therapeutic promise. Taking into account our current data and results of others (Kim et al., 2011) showing that aged nigral neurons are more vulnerable to human α-syn cytotoxicity, our current study has provided the preliminary evidence that long-term upregulation of GRP78 appears to be safe in our animal model. Comparing obtained results in animal model with human pathology this is important since PD symptoms do not manifest until at least 60–80% of the DA neurons in the SNc are already lost. Baring the development of a reliable test for the early stages of this process, prophylactic treatment could prove to be an effective course of action in preventing pathology vs. simply treating disease symptoms.
While rAAV mediated gene transfer has proven to be an invaluable research tool, it is not the only approach to elevating GRP78 expression. Alternative approaches include the use of 2-deoxy-D-glucose (2-DG; a non-metabolizable analogue of glucose; tissue culture only) (Duan and Mattson, 1999), a selective inducer of GRP78/BiP, BiP Inducer X (BIX) (Oida et al., 2010), methoxyflavones (Takano et al., 2007) and histone deacetylase (HDAC) inhibitors (Hoozemans et al., 2012). Treatment with HDAC inhibitors has been shown as an effective way to boost GFP78 expression as well as activity of existing GRP78 protein (Chen et al., 2012, Moreno et al., 2013).
Future studies will need to focus on these or other pharmacological means of altering GRP78 expression and activity as well as the potential effects of GRP78 on α-syn induced neuropathology, such as inclusions and axonal swelling. We believe, these results show promise for the next generation of therapeutics aimed at the treatment and prevention of synucleinopathies, including PD.
Material and Methods
rAAV vectors
All vectors have been packaged in AAV5 capsid and purified as described previously (Zolotukhin et al., 2002). DNA sequences (Supplementary Table 1) encoding shRNA targeting GRP78 under the control of human U6 promoter were excised with EcoRI/SalI from the respective plasmid DNAs pGFP-V-RS supplied by the provider (OriGene). The EcoRI site was converted into SalI by adding the respective adapter sequence. The fragment was then subcloned into SalI site of the pTR-UF11 in the clockwise orientation, downstream of the GFP transgene cassette and substituting the neoR cassette. The control vector (si-C) incorporated scrambled DNA sequence (Supplementary Table 1) of the same length driven by the hU6 promoter. Virus titers (vector genomes per ml) were determined by dot blot assay (Zolotukhin et al., 1999, Zolotukhin et al., 2002) and were as follows: human α-syn = 9.7 × 1012; GRP78 = 9.1 × 1012.
Animal experiments
Ten young (2 month; 5 male and 5 female) and 10 old (24 month; 5 male and 5 female) F344 rats (National Institute of Aging) were used to study the age-associated decline of GRP78 in the SNc. Additionally, sixty-four F344 female rats were used to determine if aging affects nigral DA cell survival in response to human α–syn neurotoxicity. Sixty-four young (2 month) female Sprague-Dawley rats (Charles River) were used to evaluate GRP78 siRNA constructs. An addition 40 Sprague-Dawley female rats (12 month) were used for the long-term GRP78 expression study. Finally, 10 young (2 month) and 10 old (24 month) female F344 rats were used to evaluate GRP78 upregulation in response to challenge with α-syn. The animals were housed at the University of Florida animal facility in a 12:12 light-dark cycle with free access to food and water. All procedures followed the National Institutes of Health guidelines and were approved by the University of Florida Institutional Animal Care and Use Committee. All surgical procedures were performed using aseptic techniques and isofluorene gas anesthesia as previously described (Gorbatyuk et al., 2008, Gorbatyuk et al., 2010a, Gorbatyuk et al., 2012). The rats were placed in the stereotaxic frame and injected into the SNc (anterior posterior −5.6 mm, lateral −2.4 mm from bregma and dorsoventral −7.2 mm from dura), through a glass micropipette at a rate of 0.5 μl per minute. Animals were injected with a total of 1.5 μl for single gene or 3 μl for gene combination.
Isolation and Processing of Tissues
Animals were deeply anesthetized by pentobarbital injection. Brains were removed and divided into two parts by a coronal blade cut at approximately −3.5 mm behind the bregma. The caudal part containing the SNc was fixed in ice-cold 4% paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4. The fixed portions of the brains were stored overnight at 4°C and then transferred into 30% sucrose in 0.1 M PBS for cryoprotection. Forty μm thick coronal sections were cut on a freezing stage sliding microtome and processed for immunohistochemistry. The rostral piece of brain tissue was used immediately to dissect the right and left striatum. Tissue samples were weighed, frozen separately on dry ice and kept at −80°C until assayed. Some brains were used to obtain SNc tissue samples for western blot analysis. Frozen brains were sectioned on a cryostat (Leica Microsystems) into 200 μm slices with SNc tissue subsequently dissected out under a microscope. Each sample was dissected individually and refrozen on dry ice for future western blot or quantitative RT-PCR analysis. Total tissue weight of nigral tissue obtained from one animal did not exceed 0.2–0.4 mg.
Immunohistochemistry
For bright-field microscopy analysis sections were pre-incubated first with 1% H2O2–10% methanol for 15 min and then with 5% normal goat serum for 1 h. Sections were incubated overnight at room temperature with anti-TH (1:2000; MAB318, mouse; Millipore) or anti-α-syn (1:1000; 61–787, mouse; BD laboratories) antibodies. Incubation with biotinylated secondary anti-mouse antibody was followed by incubation with avidin–biotin–peroxidase complex (ABC; Vector Laboratories, Burlingame, CA, USA). Reactions were visualized using 3,3-diaminobenzidine (DAB) as a chromagen.
For confocal microscopy, sections were incubated with primary antibodies against human α-syn (1:1000; 32–8100, mouse; Invitrogen), TH (1:1000; MAB318, mouse; Millipore) and GRP78 (1:400; ab-32618, rabbit; Abcam) and secondary fluorescent antibodies labeled with Alexa Fluor 488, 555 and 647 (1:500 for all; Invitrogen). The sections were examined using a Leica TCS SP5 confocal laser scanning microscope. Alexa Fluor 488 fluorescence was visualized by 488-nm excitation with a Kr/Ar laser, and emissions were examined at 500 to 530 nm. For imaging of Alexa Fluor 555 fluorescence, excitation at 543 nm (He/Ne laser) was used, whereas emission was observed at 560 to 580 nm. Alexa Fluor 647 fluorescence was induced at 633 nm (He/Ne laser) and observed at 660 to 700 nm. Sequential scanning was used to suppress optical cross talk between the fluorophores in stationary-structure colocalization assays. All manipulations of contrast and illumination on color images as well as color replacement were made using Adobe Photoshop CS software.
Unbiased Stereology
The unbiased stereological estimation of the total number of TH+ neurons in the SNc was performed using the optical fractionator method as previously described (Kirik et al., 2002, Burger et al., 2004). This sampling technique is not affected by tissue volume changes and does not require reference volume determinations. Sampling of cells to be counted was performed using the MicroBrightfield StereoInvestigator System. The software was used to delineate the transduction area at 5× on 40 μm sections and generate counting areas of 100 × 100 μm. A counting frame was placed randomly on the first counting area and systematically moved through all counting areas until the entire delineated area was sampled. Actual counting was performed using a 100× oil objective (NA 1.4). The estimate of the total number of neurons and coefficient of error were calculated according to the optical fractionator formula as previously described (Burger et al., 2004).
Immunoblotting
Tissues were suspended in 300 μl of lysis buffer (50mM Tris, pH 7.5, 0.15M NaCl) containing a protease inhibitor cocktail (0.1 mM PMSF, 0.5 μg/ml Leupeptin, 0.7 μg/ml pepstatin A) (Roche) and homogenized for 10 seconds. One hundred μl aliquots were added immediately to 250 μl of 0.1M HClO4 and stored at −80° for subsequent measurements. The remainder of each aliquot was adjusted to a final concentration of 1% NP-40, 0.1%SDS, incubated on ice for 30 min and centrifuged for 15 min at 4°. Lysates from each group were pooled and protein concentrations were determined by a Bradford assay. Fifty μg of each protein pool were adjusted to 2% SDS, 0.1% 2-mercaptoethanol, heated for 5 min at 95°, separated on Biorad precast 4–20% SDS-PAGE gradient gels and transferred to PVDF-LFP (Amersham) membranes for western blotting. Mouse anti-α-syn (1:2000; BD Transduction Laboratories, Zymed Laboratories), mouse or rabbit anti-GAPDH, and rabbit anti-TH (both 1:2000; Millipore) were used as recommended by the supplier. Cy5 and Cy3-conjugated goat anti-mouse and rabbit anti-goat secondary antibodies were purchased from GE Bioscience. Immunoblotted α-syn was detected and quantified with a Typhoon scanner (Amersham) by using purified α-syn-TAP protein as a standard. Westerns used for TH quantitation included GAPDH for normalization.
Striatal DA measurements
DA samples were thawed and the equivalent of 3 mg starting tissue was diluted into 1 ml of 0.1N HClO4 containing dihydrobenzylamine as an internal control, and thoroughly homogenized. The sample was centrifuged and the supernatant was filtered through a 0.2μ filter. DA and DOPAC levels were analyzed on a Beckman Gold System using a C18 Waters Symmetry column with an ESA and a Coulochem electrochemical detector equipped with a 5011A analytical cell. The mobile phase was composed as follows: 8.2 mM citric acid, 8.5 mM sodium phosphate monobasic, 0.25 mM EDTA, 0.30 mM sodium octyl sulfate, and 7.0% acetonitrile, pH adjusted to 3.5 then filtered through a 0.2 μm filter membrane. The flow rate was set at 1.5ml/min with a 30 μl injection volume.
Rotational behavior
Drug-induced rotational behavior was measured following an injection of d-amphetamine sulfate (2.5 mg/kg i.p.; Sigma, St Louis, MO). Rotations were measured during a 90-minute period, and only full 360° turns were counted.
Statistical Analysis
Data were analyzed using one tailed unpaired t test and two-way ANOVA with Tukey’s multiple comparisons post tests using Prism 6 (GraphPad Software, Inc.). Data are presented as mean ± SE.
Supplementary Material
Supplementary Figure 1. Western blot images of nigral tissues dissected from 150μm cryostat sections (A) in young (2 months) and old (24 months) rats with antibodies specific to GRP78, TH, rat α-syn and GAPDH (B) as well as to GRP78, ATF4 and β-actin (C). Quantitative results presented in the Figure 2 and in the Results section.
Supplementary Figure 2. Confocal microscopy of representative sections illustrating nigral neurons at 3 month after injection of shRNAs and human α-syn expressing vectors. Fluorescent images show co-expression of both GFP (expressed by the GRP78 shRNA vectors) and human α-syn (blue) in TH-positive (red) neurons of the SNc of different experimental and control groups. Scale bar: 150 μm.
Supplementary Figure 3. Western blot analysis of striatal TH protein levels on injected right (R) vs. uninjected left (L) side in all animals used in shRNA experiments. GAPDH was used as loading control.
Supplementary Figure 4. Immunoblot images show GRP78, TH and α-syn expression in young (2 months) and old (24 months) rats at 4 weeks after rAAV mediated gene transfer of human α-syn. Samples of nigral extracts taken from the left control side (L) injected with BV and from the right side injected with rAAV human α-syn (R), were electrophoresed on acrylamide gels. Fifty micrograms of tissue extract was analyzed by immunoblots using antibodies that recognize both human and rat α-syn, as well as GRP78, TH and GAPDH antibodies. The amount of detected protein was normalized to GAPDH (see Figure 5).
Supplementary Figure 5. Western blots of striatal TH protein levels on injected right (R) vs. uninjected left (L) side of individual animals at 9 months after rAAV mediated transduction with human α-syn alone, GRP78 alone or human α-syn and GRP78 in combination as well as BV as a vector injection control. GAPDH serves as loading control.
Highlights.
Ageing affects the nigral DA cell survival in response to human α–synuclein toxicity.
GRP78 protein is declined in the SNc of old rats.
GRP78 decline in the SNc of young rats aggravates α-synuclein neurotoxicity.
Ageing affects the nigral cell capacity to upregulate endogenous GRP78.
Sustained overexpression of GRP78 protects ageing nigral neurons against α-syn toxicity.
Acknowledgments
This work was supported by M.J.Fox Foundation (Target Validation Program) grant to O.S.G. and National Institutes of Health grants R01EY020846 to J.H.L., R01EY020905 to M.S.G, P30 EY003039 core grant to VSUAB.
Abbreviations
- GRP78
glucose regulated protein 78
- DA
dopamine
- PD
Parkinson Disease
- TH
tyrosine hydroxylase
- rAAV
recombinant Adeno-associated virus
- SNc
substantia nigra pars compacta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Auluck PK, Caraveo G, Lindquist S. a-Synuclein: Membrane Interactions and Toxicity in Parkinson’s Disease. Annual Review of Cell and Developmental Biology. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N. Recombinant AAV Viral Vectors Pseudotyped with Viral Capsids from Serotypes 1, 2, and 5 Display Differential Efficiency and Cell Tropism after Delivery to Different Regions of the Central Nervous System. Mol Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Chen SH, Wu HM, Ossola B, Schendzielorz N, Wilson BC, Chu CH, Chen SL, Wang Q, Zhang D, Qian L, Li X, Hong JS, Lu RB. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. British Journal of Pharmacology. 2012;165:494–505. doi: 10.1111/j.1476-5381.2011.01575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease. Neurobiol Dis. 2007;25:134–149. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK. Endoplasmic Reticulum Stress Is Important for the Manifestations of α-Synucleinopathy In Vivo. The Journal of Neuroscience. 2012a;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of Toxic α-Synuclein Oligomer within Endoplasmic Reticulum Occurs in α-Synucleinopathy In Vivo. The Journal of Neuroscience. 2012b;32:3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson M. alpha-Synuclein and neuronal cell death. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson’s disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Duan W, Mattson MP. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J Neurosci Res. 1999;57:195–206. doi: 10.1002/(SICI)1097-4547(19990715)57:2<195::AID-JNR5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Erickson RR, Dunning LM, Holtzman JL. The effect of aging on the chaperone concentrations in the hepatic, endoplasmic reticulum of male rats: the possible role of protein misfolding due to the loss of chaperones in the decline in physiological function seen with age. J Gerontol A Biol Sci Med Sci. 2006;61:435–443. doi: 10.1093/gerona/61.5.435. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk MS, Shabashvili A, Chen W, Meyers C, Sullivan LF, Salganik M, Lin JH, Lewin AS, Muzyczka N, Gorbatyuk OS. Glucose Regulated Protein 78 Diminishes [alpha]-Synuclein Neurotoxicity in a Rat Model of Parkinson Disease. Mol Ther. 2012 doi: 10.1038/mt.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, Mandel RJ, Chen W, Meyers C, Manfredsson FP, Muzyczka N. In Vivo RNAi-Mediated [alpha]-Synuclein Silencing Induces Nigrostriatal Degeneration. Mol Ther. 2010a;18:1450–1457. doi: 10.1038/mt.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Nguyen FN, Manfredsson FP, Kondrikova G, Sullivan LF, Meyers C, Chen W, Mandel RJ, Muzyczka N. [alpha]-Synuclein Expression in Rat Substantia Nigra Suppresses Phospholipase D2 Toxicity and Nigral Neurodegeneration. Mol Ther. 2010b;18:1758–1768. doi: 10.1038/mt.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human α-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proceedings of the National Academy of Sciences. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Hinds JW, McNelly NA. Dispersion of cisternae of rough endoplasmic reticulum in aging CNS neurons: a strictly linear trend. Am J Anat. 1978;152:433–439. doi: 10.1002/aja.1001520311. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJM, van Haastert ES, Nijholt DAT, Rozemuller AJM, Scheper W. Activation of the Unfolded Protein Response Is an Early Event in Alzheimer’s and Parkinson’s Disease. Neurodegenerative Diseases. 2012;10:212–215. doi: 10.1159/000334536. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Ries V, Cheng H-C, Kareva T, Oo TF, Yu WH, Duff K, Kholodilov N, Burke RE. Age and α-synuclein expression interact to reveal a dependence of dopaminergic axons on endogenous Akt/PKB signaling. Neurobiol Dis. 2011;44:215–222. doi: 10.1016/j.nbd.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Bjorklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. 1998;152:259–277. doi: 10.1006/exnr.1998.6848. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS. The ER chaperone and signaling regulator GRP78//BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu X, Fang F, Jiang D, Tang L. Down-regulation of GRP78 enhances apoptosis via CHOP pathway in retinal ischemia-reperfusion injury. Neuroscience Letters. 2014;575:68–73. doi: 10.1016/j.neulet.2014.05.042. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr Mol Med. 2008;8:157–172. doi: 10.2174/156652408784221324. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Science Translational Medicine. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Brown M. The endoplasmic reticulum stress response in aging and age-related diseases. Frontiers in Physiology. 2012;3 doi: 10.3389/fphys.2012.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss JE, Choksi KB, DeFord JH, Papaconstantinou J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem Biophys Res Commun. 2008;365:355–361. doi: 10.1016/j.bbrc.2007.10.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oida Y, Hamanaka J, Hyakkoku K, Shimazawa M, Kudo T, Imaizumi K, Yasuda T, Hara H. Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neurosci Lett. 2010;484:43–46. doi: 10.1016/j.neulet.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Current Opinion in Cell Biology. 2011;23:150–156. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941–953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- Takano K, Tabata Y, Kitao Y, Murakami R, Suzuki H, Yamada M, Iinuma M, Yoneda Y, Ogawa S, Hori O. Methoxyflavones protect cells against endoplasmic reticulum stress and neurotoxin. Am J Physiol Cell Physiol. 2007;292:C353–361. doi: 10.1152/ajpcell.00388.2006. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Western blot images of nigral tissues dissected from 150μm cryostat sections (A) in young (2 months) and old (24 months) rats with antibodies specific to GRP78, TH, rat α-syn and GAPDH (B) as well as to GRP78, ATF4 and β-actin (C). Quantitative results presented in the Figure 2 and in the Results section.
Supplementary Figure 2. Confocal microscopy of representative sections illustrating nigral neurons at 3 month after injection of shRNAs and human α-syn expressing vectors. Fluorescent images show co-expression of both GFP (expressed by the GRP78 shRNA vectors) and human α-syn (blue) in TH-positive (red) neurons of the SNc of different experimental and control groups. Scale bar: 150 μm.
Supplementary Figure 3. Western blot analysis of striatal TH protein levels on injected right (R) vs. uninjected left (L) side in all animals used in shRNA experiments. GAPDH was used as loading control.
Supplementary Figure 4. Immunoblot images show GRP78, TH and α-syn expression in young (2 months) and old (24 months) rats at 4 weeks after rAAV mediated gene transfer of human α-syn. Samples of nigral extracts taken from the left control side (L) injected with BV and from the right side injected with rAAV human α-syn (R), were electrophoresed on acrylamide gels. Fifty micrograms of tissue extract was analyzed by immunoblots using antibodies that recognize both human and rat α-syn, as well as GRP78, TH and GAPDH antibodies. The amount of detected protein was normalized to GAPDH (see Figure 5).
Supplementary Figure 5. Western blots of striatal TH protein levels on injected right (R) vs. uninjected left (L) side of individual animals at 9 months after rAAV mediated transduction with human α-syn alone, GRP78 alone or human α-syn and GRP78 in combination as well as BV as a vector injection control. GAPDH serves as loading control.