

Abstract
Clinical reports have demonstrated that higher rates of non-diabetic glomerulosclerosis in African Americans can be attributed to two coding sequence variants (G1 and G2) in the APOL1 gene; however, the underlying mechanism is still unknown. Kidney biopsy data suggest enhanced expression of APOL1/APOL1 variants (Vs) in smooth muscle cells (SMCs) of renal vasculature. Since APOL1 is a secretory protein of relatively low molecular weight (41 kDa), SMCs may be a contributory endocrine/paracrine source of APOL1 wild type (WT)/APOL1Vs in the glomerular capillary perfusate percolating podocytes. In the present study, we tested the hypothesis that an HIV milieu stimulated secretion of APOL1 and its risk variants by arterial SMCs contributes to podocyte injury. Human umbilical artery smooth muscle cells (HSMCs)-treated with conditioned media (CM) of HIV-infected peripheral mononuclear cells (PBMC/HIV-CM), CM of HIV-infected U939 cells, or recombinant IFN-γ displayed enhanced expression of APOL1. Podocytes co-cultured in trans-wells with HSMCs-over expressing APOL1WT showed induction of injury; however, podocytes co-cultured with HSMC-over expressing either APOL1G1 or APOL1G2 showed several fold greater injury when compared to HSMC- over expressing APOL1WT. Conditioned media collected from HSMC-over-expressing APOL1G1/APOL1G2 (HSMC/APOL1G1-CM or HSMC/APOL1G2-CM) also displayed higher percentages of injured podocytes in the form of swollen cells, leaky lysosomes, loss of viability, and enhanced sensitivity to adverse host factors when compared to HSMC/APOL1WT-CM. Notably, HSMC/APOL1WT-CM promoted podocyte injury only at a significantly higher concentrations compared to HSMC/APOL1G1/G2-CM. We conclude that HSMCs could serve as an endocrine/paracrine source of APOL1Vs, which mediate accelerated podocyte injury in HIV milieu.
Introduction
Compared with European Americans (EAs), African Americans (AAs) develop 4–5 fold higher rates of progressive nephropathy including focal segmental glomerulosclerosis (FSGS), and hypertension-attributed chronic kidney disease (CKD) [Tzur et al, 2010; Kopp et al, 2011; Quaggin and George, 2011], and this disparity reaches a greater than 10-fold difference in the case of HIV-associated nephropathy (HIVAN) [Genovese et al, 2010]. Recent clinical reports have shown that this major health disparity is strongly associated with two coding sequence variants (G1 and G2) in APOL1 [Friedman et al, 2011;; Genovese et al, 2010, 2013; Foster et al, 2013], but the underlying mechanisms are only beginning to be elucidated [Lan et al, 2014; Nichols et al, 2014; Thomson et al, 2014]. It is controversial whether APOL1 nephropathy risk alleles associate with atherosclerosis or protection from calcified atherosclerotic plaque and improved survival (Freedman et al 2015; Ito et al, 2014; Langefeld, 2015). Ito et al reported heightened risk for myocardial infarction with APOL1 risk variants, despite simultaneous association with lower coronary artery calcified plaque (Ito et al, 2014); however, these findings are not supported by two follow-up analyses (Freedman et al, 2015; Langefeld et al, 2015).
Podocytes, the highly differentiated cells play a cardinal role in the maintenance of the glomerular filtration barrier. Podocytopathy (altered podocyte phenotype, reduction in number and effacement of foot processes) is usually associated with proteinuric diseases including HIVAN [Mundel and Shankland, 2002; Medapalli et al, 2011]. In previous studies, we demonstrated that APOL1 risk variants (Vs) G1 and G2 induce necrosis in podocytes and make them vulnerable to adverse host factors (AHFs) such as HIV infection [Lan et al, 2014]. Those studies revealed a causal relationship between podocyte injury and expression of APOL1Vs, thus providing a potential mechanistic basis for the observed disparity in chronic kidney disease (CKD) in relation to APOL1 genotype. An observation of interest in these studies was that inhibition of uptake of APOL1 reduced podocyte injury. These findings suggested a possible role for extracelluar APOL1 in podocyte injury, which is of potential relevance in view of the fact that APOL1 is the only member of the APOL1-6 family cluster with a signal peptide [Page et al, 2001]; however, in vivo, secretion of APOL1 may be from podocytes (autocrine), adjacent cells (paracrine), or from distant cells (endocrine).
Although APOL1Vs enhance podocyte injury in HIV milieu, it is not clear whether HIV contributes to accelerated podocyte injury through enhanced APOL1 expression in podocytes themselves or whether other sources also contribute. Of note, kidney biopsy specimens from HIVAN patients did not reveal upregulation of APOL1 in podocytes [Madhavan et al, 2011]. On the other hand, podocyte APOL1 expression has been reported to be attenuated both in FSGS and HIVAN [Madhavan et al, 2011]. Thus, it appears that either the source of APOL1 contributing to kidney cell injury was not from these cells or those mechanisms wherein loss of APOL1 may be compromising the viability of podocytes is responsible for injury. The latter formulation seems less likely given the finding that absence of APOL1 does not alter the podocyte phenotype, and that APOL1 is dispensable for kidney health [Johnstone et al, 2002; Lan et al, 2014; Thomson et al, 2014]. Nonetheless, the source of APOL1 contributing to kidney cell injury in HIVAN remains elusive. This leads to the alternative formulation that the mere presence of HIV infection either renders APOL1Vs expressing podocytes vulnerable to injury and/or compromises their rescue strategy in the presence of adverse factors. This concept is also supported by recent reports in which circulating levels of APOL1 did not correlate with CKD parameters in a cohort of patients with HIVAN (Bruggeman et al, 2014).
Human podocytes have been reported to endocytose exogenous APOL1 [Ma et al, 2015]. We also demonstrated that uptake of secretory APOL1Vs was associated with podocyte injury [Lan et al, 2014]. Madhavan et al reported that APOL1 expression in small arterial and arteriolar smooth muscle cells (SMCs) is increased both in FSGS and HIVAN [Madhavan et al, 2011]. Based on these studies, we hypothesize that podocyte uptake of the exogenous APOL1 Vs could originate from smooth muscles of arteries and arterioles and contribute to accelerated podocyte injury. Thus, it is likely that smooth muscle cells may be serving either an endocrine or paracrine source of APOL1/APOL1Vs.
In the current study, we report the expression profile of APOL1 in arterial SMCs in an experimental HIV milieu, and have also examined the effects of smooth muscle cell APOL1Vs on human podocytes.
Materials and Methods
Cell Culture
Human podocytes were cultured as previously reported [Saleem et al, 2002; Hussain et al, 2009]. Briefly, immortalized human podocytes proliferated in the growth medium containing RPMI 1640 supplemented with 10% fetal bovine serum, 1 X penicillin-streptomycin, 1 mM L-glutamine, and 1 X insulin, transferrin, and selenium (ITS) (Invitrogen, Grand Island, NY) at permissive temperature (33°C). Podocytes at 80% confluence were transferred to 37°C for differentiation in a medium without ITS for 5–7 days.
Human umbilical artery smooth muscle cells (HSMC) were purchased from ScienCell Research Laboratories (Carlsbad, CA), and were cultured with Smooth Muscle Cell Medium (SMC/CM, ScienCell) at 37°C.
Lentivirus preparation
APOL1 lentivirus was produced by transfection of 293T cells by using the Effectene Transfection Reagent (Qiagen), and the virus titer was determined by using QuickTiter Lentivirus Titer Kit (Cell Biolabs, San Diego, CA) as described [Lan et al, 2014].
Preparation of Conditioned Media from APOL1 and APOL1Vs-transduced HSMCs and from HIV-infected immune cells
To collect conditioned media (CM) from smooth muscle cells, confluent HSMCs were transduced with lentivirus LG12-APOL1 (0.4 pg p24 protein/cell)[Lan et al, 2014 ]. To collect conditioned media (CM) from immune cells, PBMCs were isolated from blood as previously described [Mikulak et al, 2010], and infected with HIV strains NL4-3 (AIDS Reagent, NIH). After 3 h, the virus was removed, and the cells were rinsed with PBS, and were cultured in fresh RPMI medium with 10% FBS. Three days later, CM were harvested, cleared of free-floating cells by centrifugation for 5 min at 12000 rpm, and stored at −80ºC. Similarly, conditioned medium of HIV-infected U939 cells (U1 cells, monocytoid cell line, AIDS Reagent, NIH) was collected and stored until further use.
HIV infection of HSMCs
HSMCs at 80% confluence were infected with HIV-1 (NL4-3 strain, NIH AIDS Research and Reference Reagent Program, Germantown, MD) for 4 h, and then were cultured in RPMI medium containing 10% FBS at 37°C.
Doxycycline inducible APOL1-expressing HSMCs
A lentiviral expressing vector LW12-TetOn3G-Apol1 was constructed (Figure 1). Promoter PTRE3G was from plasmid pTRE3G (Clontech Laboratories, Mountain View, CA), and promoter PCMVIE and cDNA Tet-On 3G were from plasmid pCMV-Tet3G (Clontech Laboratories). Two primers APOL1-FW (GGAATTCATGGAGGGAGCTGCTTTGCTGAGAG, EcoRI cleavage site underlined) and APOL1-RV (GGGGTACCTCACAGTTCTTGGTCCGCCTGCAG, KpnI cleavage site underlined) were used to amplify the full length ORF of the APOL1 gene. Lentivirus was prepared following our previous protocol [Lan et al, 2014]. Human podocytes were transfected with these LW12-TetOn3G-APOL1/WT/Vs harboring virus for 5 days, and GFP positive cells were sorted by Fluorescence-Activated Cell Sorting (FACS). The resulting cells were cultured in SMC-CM for amplification and in RPMI medium containing 10% FBS for CM production.
Figure 1.
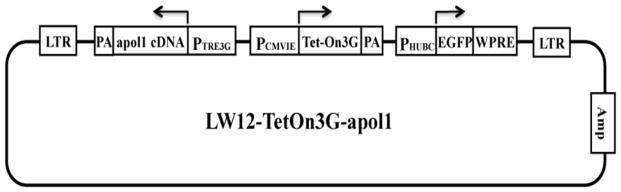
Schematic diagram of lentiviral expressing vector LW12-TetOn3G-apol1
Determination of APOL1 concentration
APOL1 concentrations in media were determined by ELISA with the Human APOL1 Kit (Proteintech Group Inc, Chicago, IL), according to the manufacturer’s instructions.
Trypan blue staining
Cells were rinsed with PBS (to remove the floating cells and cell debris), detached with Accutase (Innovative Cell Technologies, San Diego, CA), and then labeled with 0.2% Trypan blue and counted under a light microscope. The unstained cells (white cells) were counted as live cells, while the stained (blue) cells were counted as dead cells.
Propidium iodide (PI) and Hoechst staining
PI and Hoechst staining were performed as previously described [Lan et al, 2014]. Briefly, after each treatment condition of interest, the culture medium was removed from the cells and fresh medium containing Hoechst 33342 (10 μg/ml) was added. Cells were subsequently incubated for 10 min at 37°C. Then, PI solution was added and culture dishes were kept on ice for 7 min. Cell images were recorded with a ZEISS microscope (Carl Zeiss Micro Imaging GmbH, Jena, Germany) equipped with a digital imaging system.
Assessment of lysosomal integrity
Control and experimental cells were stained with Lucifer Yellow (Invitrogen) and LysoTracker Red (Invitrogen) as described previously [Castro et al, 2004; Chandel et al, 2013]. Briefly, cells were cultured overnight in medium containing 1 mg/ml Lucifer Yellow, and then 500 nM LysoTracker was added followed by another 30 min culture. Then the medium was removed, and the cells were rinsed 3 times with PBS. The cells were then fixed with 4% paraformaldehyde (PFA) for 10 min. Lysosomal imaging was carried under a fluorescence microscope (ZEISS)
Real time (RT) PCR
RT-PCR was performed as previously described [Lan et al, 2014], with addition of HIV-1 Gag primers SK38/SK39: 5′-ATAATCCACCTATCCCAGTAGGAGA-3′ (Gag-FW), and 5′-TTTGGTCCTTGTCTTATGTCCAGAATGC-3′ (Gag-RV).
Western blotting
Western blotting was performed as described previously [Lan et al, 2014]. Briefly, cells incubated under each treatment or corresponding control condition were washed with PBS and lysed in RIPA buffer (1 X PBS, pH7.4, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 1.0 mM sodium orthovanadate, 10 μl of protease inhibitor cocktail (100 x, Calbiochem) per 1 ml of buffer, and 100 μg/ml PMSF). Proteins (20–30 μg) were separated by 12% SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred on an Immuno-Blot polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA). After blocking in PBS/Tween (0.1%) with 5% nonfat milk, the membrane was incubated with primary antibodies overnight at 4°C followed by horseradish peroxidase-conjugated secondary antibodies (1:3000, Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and then developed using Enhanced Chemiluminescent (ECL) solution (Pierce). Primary antibodies used were goat anti-APOL1 (1:1000, Acris Antibody, Inc., San Diego, CA, USA), and goat anti-actin (1:3000, Santa Cruz). For protein expression quantification, the films were scanned with a CanonScan 9950F scanner and the acquired images were then analyzed using the public domain NIH image program (http://rsb.info.nih.gov/nih-image/).
Depletion of APOL1 from conditioned media
Soluble APOL1 in CM was removed by using anti-APOL1 antibody. Briefly, 96-well microtiter plates (Costar) were coated overnight at 4°C with goat anti-APOL1 (0.5 μg/ml, Acris Antibody), in NaHCO3 solution (0.1 M, pH9.5). As a control, goat serum was used to coat the plates. Excess antibody was removed through washing with 0.1% Tween in PBS, three times and non-specific binding was blocked through exposure with 3% BSA in PBS for 6 hours. CM were incubated in these wells overnight at 4°C. Residual APOL1 in the CM was determined by ELISA. APOL1-free CM were used to treat human podocytes.
Statistical analyses
Data are presented as means ± standard deviation (SD) unless otherwise noted. All experiments were conducted and repeated at least three times, either in duplicate or triplicate for each assay. All data were evaluated statistically by analysis of variance (ANOVA), followed by Newman-Keuls multiple comparison tests using Prism 4.0 GraphPad software. In the case of single mean comparison, data were analyzed by student t-test. P values < 0.05 were regarded as statistically significant.
Results
APOL1 expression in HSMCs is increased in HIV milieu
Since arterial SMCs have been reported to be infected by HIV [Duchateau et al, 1997], we asked whether HIV infection of SMCs might increase their APOL1 expression. HSMCs were infected with HIV-1 strain NL4-3 for 1, 3, 6 and 9 days, and the expression of HIV Gag and APOL1 was quantified by RT-PCR. HIV Gag expression increased with the passage of time, consistent with HIV replication in HSMCs (Fig. 2A). HIV infection enhanced SMC expression of APOL1 up to five-fold at 72 hours, but the levels subsequently declined at later time points (Figure 2B). ELISA results also displayed a 2.5 fold increased secretory APOL1 in the medium at 72 hours but no further rise at the later time periods despite evidence of continued viral replication as indicated by continued rise of Gag expression (Figure 2C). Similar results were obtained by infecting SMCs with another primary HIV strain (92HT599) (data not shown). These results suggest that HIV infection enhances APOL1 expression, but does so only transiently and not in a manner which rises continuously in conjunction with the continued rise in HIV replication.
Figure 2. Effects of HIV infection on APOL1 expression in SMCs.
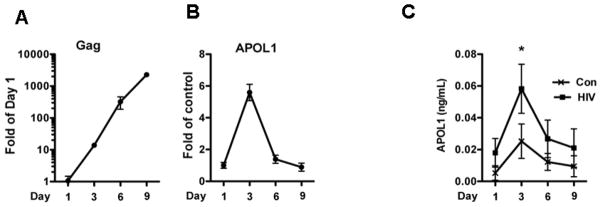
SMCs were transduced with HIV NL4-3 for 3 h, washed and re-incubated in fresh medium.
Cells and media were harvested at days 1, 3, 6 and 9.
A–B. RNA was extracted and the expression of HIV Gag and APOL1 was detected by RT-PCR.
C. APOL1 concentrations in incubation media were determined with ELISA kit.
The results (mean ± SD) are from three independent sets of experiments carried out in triplicate.
* p < 0.05 vs. control.
We then asked whether HIV-infected immune cells will affect APOL1 expression in vascular SMCs. Conditioned media (CM) of PBMC- infected with HIV NL4-3 or U939 cells-infected with HIV (U1, monocytoid cell line) were collected after 3 days of infection. The CM were then used to treat HSMCs to examine the expression of APOL1. Western blot analysis revealed that both CM increased APOL1 expression in HSMCs (Figure 3A–B). PBMC/HIV-CM also significantly stimulated APOL1 secretion by HSMCs, and APOL1 concentrations reached 1.0 ng/ml at 48 hours (Figure 3C).
Figure 3. Effects of immune cell/HIV-conditioned media on APOL1 expression in SMCs.
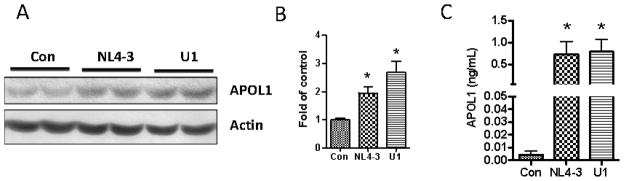
SMCs were treated with conditioned medium collected from HIV (NL4-3 or U1)-infected PBMCs (n=3). After 48 h, cells and incubation media were harvested for Western blot assay and ELISA respectively.
A. Protein blots were probed for APOL1, stripped and re-probed for actin.
B. Cumulative densitometric data from of three independent experiments shown as bar graphs.
* p < 0.05 compared to control
C. APOL1 concentrations in incubation media were determined with an ELISA kit. The results (mean ± SD) represent three sets of experiments carried out in triplicate.
* p < 0.05 compared with control.
To further confirm the induction of APOL1 expression in SMCs by exogenous factors, we treated HSMCs with IFN-γ. Western blot analysis showed that IFN-γ stimulated APOL1 expression in both a dose- and time-dependent manner (Figure 4A–D). IFN-γ also increased APOL1 secretion by HSMCs in a time dependent manner (Figure 4E). At the end of 72 hours, the concentration of APOL1 in media reached up to 5 ng/ml. Taken together, the foregoing results suggest that HSMCs in the inflammatory milieu (interaction with PBMC/HIV-CM) could potentially have a role in vascular and glomerular injury.
Figure 4. Effects of IFN-γ on APOL1 expression in HSMCs.
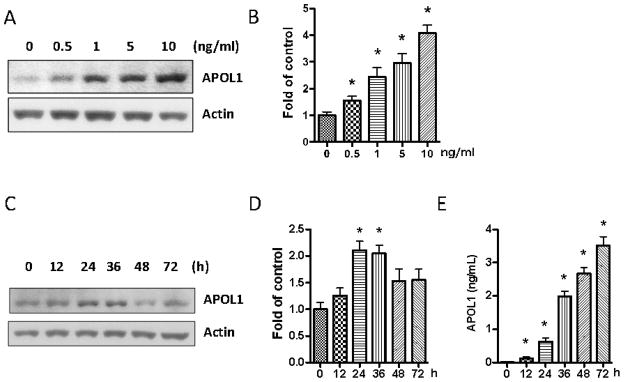
A. SMCs were treated with recombinant human IFN-γ for 36 hours at the concentrations indicated (n=3). Protein blots were probed for APOL1 and the same blots were stripped and reprobed for actin.
B. Cumulative densitometric data shown in bar graphs. * p < 0.05 compared with control.
SMCs were treated with 10 ng/ml of recombinant human IFN-γ for the indicated time periods (n=3). Subsequently, cells and incubation media were harvested.
C. Protein blots were probed for APOL1, stripped and reprobed for actin.
D. Cumulative densitometric analysis shown in a bar diagram. * p < 0.05 compared with 0, 12, 48, and 72 h.
E. APOL1 concentrations in incubation media were determined by ELISA. * p < 0.05 compared with control.
HSMC/APOL1Vs induce human podocyte injury
First, we assessed baseline APOL1 expression in HSMCs. After culturing HSMCs for 24, 48, and 72 h, we collected both cell lysates and medium for Western blot and for ELISA, respectively. We found that in the cell lysates, APOL1 expression did not increase with the passage of time (Figure 5A–B). APOL1 in the medium accumulated slowly, but only reached concentrations approaching 0.01 ng/ml at 72 h (Figure 5C), indicating that the baseline APOL1 expression in HSMCs was very low when compared to the concentrations in the inflammatory milieu.
Figure 5. Overexpression of APOL1 in human vascular smooth muscle cells.

A–C: HSMCs were cultured in SMCM until reaching 80–90% confluence, and were transferred to RPMI medium. After another 24, 48, and 72 h, cell lysates and medium were collected for detection of APOL1 with Western blots (A-B) and ELISA (C), respectively.
D–F: HSMCs were infected with lentivirus (titrated as 0.4 pg HIV p24 protein per cell) for 3 h, and then re-incubated in fresh medium for 48 h (n=3). Cells and media were collected.
D. Protein blots were probed for APOL1, stripped and re-probed for actin.
E. Densitometric analysis shown as bar graphs. * p < 0.05 compared with control (vector).
F. APOL1 concentrations were determined in media with an ELISA kit.
Results (mean ± SD) are from three sets of experiments carried out in triplicate. * p < 0.05 compared with control (vector). Vec, vector alone; WT, APOL1 wild type; G1, APOL1 G1 variant; G2, APOL1 G2 variant.
We then overexpressed APOL1Vs (G1 and G2) as well as APOL1WT (G0) in HSMCs employing a lentivirus expression system, as previously reported [Lan et al, 2014]. HSMCs were transduced with equal quantities of APOL1WT or APOL1Vs lentivirus (titrated as 0.4 pg p24 protein/cell). After 48 h, only very few cells (less than 0.1%) showed swelling or necrosis in APOL1G1/G2, and no cell swelled in APOL1WT or vector control. Cells were harvested for protein blots and media were collected for determination of APOL1 concentrations by ELISA. HSMCs displayed overexpression of APOL1WT and APOL1Vs (Figures 5D–E). ELISA concentrations of APOL1 secreted into the medium by SMCS/Vec were minimal (< 0.01 ng/ml at 48 hpi), whereas HSMC/APOL1WT and HSMC/APOL1G1/G2 displayed increased APOL1 secretion (3–5 ng/ml) (Fig. 5F), representing more than 300-fold elevated APOL1 secretion.
To test the hypothesis whether APOL1 secreted by HSMCs exerts an endocrine/paracrine effect on podocyte phenotypes, we co-cultured HSMCs and human podocytes in a double chamber (Figure 6A). HSMCs were cultured in the bottom chamber and then transduced with lentivirus LG12-APOL1. After 48 h, differentiated human podocytes were cultured in the upper chamber. In this experimental system, HSMC- secreted proteins have access to the co-cultured podocytes through the filter. After 3 days of co-culture, podocytes were assessed for their viability by Trypan Blue staining. Podocyte co-cultured with HSMC/APOL1 WT displayed augmented cell death and this effect was markedly more prominent in podocytes co-cultured with either HSMC/APOL1G1 or HSMC/APOLG2 (Figure 6B).
Figure 6. Co-culture of podocytes with APOL1 risk variants-producing vascular smooth muscle cells.
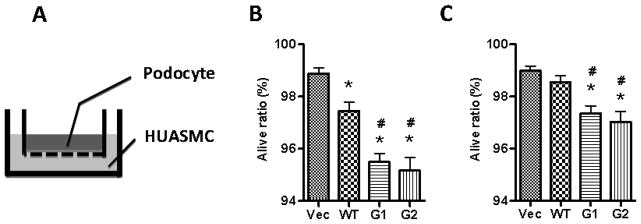
A. Schematic diagram of co-culture of podocytes and HSMCs.
B. HSMCs at the bottom of 6-well plates were transduced with lentivirus (titrated as 0.4 pg HIV p24 protein per cell). After for 3 h, the cells were washed with PBS, and reincubated in fresh medium. At 24 hpi, inserts containing human podocytes on the filter were inserted. 72 hours later, the inserts were taken out, and the cells were subjected to Trypan Blue staining to count the alive cells. * p < 0.05 compared with vector, while # p < 0.05 compared to APOL1WT.
C. SMCs were transduced with half the dose of APOL1-gene harboring virus (0.2- 0.4 pg HIV p24 protein per cell), and alive podocytes were counted after co-cultured as mentioned above. * p < 0.05 compared with vector, while # p < 0.05 compared to APOL1WT.
In order to account for possible concentration differences and disentangle this from to the effect of the different molecular structures of the WT vs. Vs moieties of APOL1, we conducted dose dependency studies. HSMCs were transduced with half the dose of lentivirus (titrated as 0.2 pg p24 protein/cell) for 3 hours and then incubated in fresh media for 48 hours. Co-culture with human podocytes was performed, and living cells were counted. As shown in Figure 6C, at these reduced transfection doses, APOL1WT-expressing HSMCs did not display overt podocyte injury; on the other hand, HSMCs-expressing APOLG1 or G2 displayed a decreased percentage of viable cells even at this reduced dosage. These findings indicate that APOL1-induced podocyte injury was dependent on concentration as well as conformation of APOL1, such that the risk variants have a lower dose threshold for inducing injury.
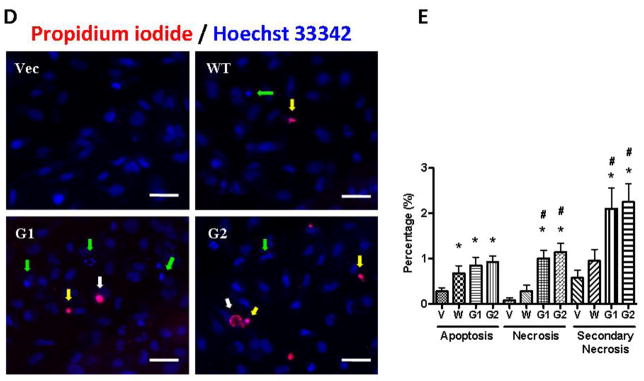
To confirm the role of HSMC-CM in podocyte injury, we collected the conditioned media from over expressing APOL1WT and APOL1Vs HSMCs and evaluated the effect of these CM on podocytes. CM-G1 and CM-G2, but not CM-WT, induced podocyte swelling and death (Figure 7A–C). G1-CM and G2-CM induced apoptosis, necrosis, and secondary necrosis in a significantly greater percentage of podocytes when compared to podocytes treated with WT-CM. (Figure 7D–E).
Figure 7. Conditioned medium of SMCs promote podocyte injuries.


HSMCs were transduced with lentivirus (titrated as 0.4 pg HIV p24 protein per cell) for 3 h, and reincubated in fresh medium (n=3). At 72 hpi, the media were collected. After centrifugation, supernatants were collected (conditioned media, CM), and added to human podocytes in 6-well plates.
A. Podocytes were treated with either HSMC/Vector-CM, HSMC/APOLWT-CM, HSMC/APOL1G1-CM, or HSMC/APOL1G2-CM for 48 hours (n=3). Cells were stained with Trypan blue and dead cells were counted by two investigators, unaware of the experimental conditions. * p < 0.05 compared to HSMC/Vector-CM, while # p< 0.05 compared to HSMC/-APOL1WT-CM.
B. Podocytes were treated with either HSMC/Vector-CM, HSMC/APOLWT-CM, HSMC/APOL1G1-CM, or HSMC/APOL1G2-CM for 48 hours (n=3). Subsequently, cells were examined under a phase microscope. Representative microphotographs of swollen cells are shown. Scale bar: 100 μm.
C. Cumulative data on percentage of swollen cells in each subgroup. Results (mean ± SD) are from three sets of experiments. * p < 0.05 compared to HSMC/Vector-CM, while # p< 0.05 compared to HSMC/APOL1WT-CM.
D. Podocytes were treated with either HSMC/Vector-CM, HSMC/APOLWT-CM, HSMC/APOL1G1-CM, or HSMC/APOL1G2-CM for 48 hours (n=3). Subsequently, cells were subjected to PI/Hoechst staining and then examined under an immunofluorescence microscope. Representative microphotographs are displayed showing apoptotic (green arrow), necrotic (white arrow), and secondary necrotic (yellow arrow) cells. Scale bar: 50 μm.
E. Cumulative data on percentage of apoptotic and necrotic cells in each subgroup are shown. Results (mean ± SD) are from three sets of experiments. * p < 0.05 compared with HSMC/Vector-CM, # p < 0.05 compared to HSMC/APOL1WT-CM
F. Podocytes were treated with either HSMC/Vector-CM, HSMC/APOLWT-CM, HSMC/APOL1G1-CM, or HSMC/APOL1G2-CM for 48 hours (n=3). Podocytes were subjected to LysoTracker Red and Lucifer Yellow and examined under an immunofluorescence microscope. Representative microphotographs are displayed. Scale bar: 10 μm.
G. Cumulative data for each variable of the protocol F (Lyso Tracker Red labeling) are displayed in bar graphs. Results (mean ± SD) are from three sets of experiments. * p < 0.05 compared to HSMC/Vector-CM, # p < 0.05 compared to HSMC/APOL1WT-CM
H. Cumulative data for each variable of the protocol F (Lucifer Yellow labeling) are displayed as bar graphs. Results (mean ± SD) are from three sets of experiments. * p < 0.05 compared to HSMC/Vector-CM, # p < 0.05 compared to HSMC/APOL1WT-CM
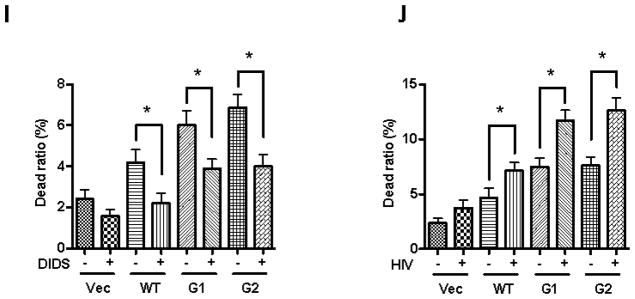
I. Podocytes were treated with either HSMC/Vector-CM, HSMC/APOLWT-CM, HSMC/APOL1G1-CM, or HSMC/APOL1G2-CM in the presence or absence of DIDS (100 μM, a chloride channel blocker) for 48 hours (n=3). Subsequently, cells were stained with Trypan blue and dead cells were counted. * p < 0.05 compared to respective variables.
J. Empty vector or NL4-3(HIV)-transduced podocytes were incubated in media containing either HSMC/Vector-CM, HSMC/APOLWT-CM, HSMC/APOL1G1-CM, or HSMC/APOL1G2-CM for 48 hours (n=3). Subsequently, cells were stained with Trypan blue and dead cells were counted. * p < 0.05 compared to respective variables.
To determine the effects of CM on lysosomal integrity, Lucifer Yellow and LysoTracker Red staining was performed. APOL1Vs enhanced lysosomal leakage and decreased the LysoTracker Red staining, indicating that CM-G1 and CM-G2 increased lysosomal membrane permeability (LMP) (Figure 7F–H). On the other hand, a chloride channel blocker significantly decreased CM-Vs-induced podocyte injuries (Figure 7I); while HIV transduction significantly increased CM-Vs-mediated podocyte death (Figure 7J).
To validate the role of APOL1 Vs in the CM-induced podocyte injuries, we used microtiter plates to remove the APOL1 proteins from SMC-CMs. ELISA assay results showed that the anti-APOL1 antibody could successfully remove almost all the APOL1 protein from the CM, while the goat serum control did not (data not shown). Human podocytes were treated with CM and cell injury was determined by counting live cell ratios. APOL1 depletion from the CM, significantly decreased podocyte injury (Figure 8). These findings indicate that podocyte injury was partially mediated through the cytotoxicity of APOL1Vs secreted into the media.
Figure 8. APOL1 depletion attenuates CM-induced podocyte injury.

HSMC-CM were incubated in micro titer plates coated with anti-APOL1 antibody (+) or goat serum control (-). After overnight, media were collected. Podocytes were treated with these media for 48 hours (n=3). Subsequently, podocytes were assayed for viability. * p < 0.05 compared with respective variables.
To further validate these results, we established stabile cell lines over-expressing APOL1 under the control of Tet-on system (Fig. 1). After sorting, more than 95% of the cells were GFP positive even after 3 passages, indicating that the ectopic inducible transgenes could be stably expressed in these cells. Doxycycline (100 ng/ml) was added to the medium, and the cells were cultured at 37°C for 4 days. ELISA results showed that doxycycline successfully induced APOL1 production and secretion (Figure 9A). The medium was collected as CM and was used to treat human podocytes. After another 48 h, cell injury was measured by counting alive cell ratio. CM-Vs (Doxy) also showed podocyte cytotoxicity, while CM-WT (Doxy) didn’t induce podocyte cytotoxicity (Figure 9B). These results confirmed the podocyte cytotoxicity of APOL1Vs derived from HSMCs.
Figure 9. Doxycycline induced APOL1Vs from HSMCC cause podocyte injury.
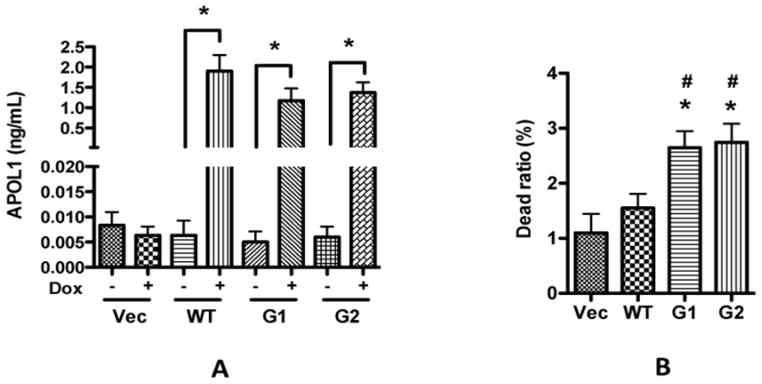
After FACS sorting, LW12-TetOn3G-APOL1/WT/Vs harboring HMSCs were cultured in SMCM to around 80–90% confluence, and then were changed to RPMI medium containing 10% FBS and 100 ng/ml doxycycline. After another 4 days, the medium was collected, and free-floating cells were cleared by centrifugation. APOL1 concentrations in the collected CMs were determined by ELISA (A). The CMs were used to treat differentiated human podocytes in cell culture dishes, and the cells were subjected to Trypan Blue staining to count the alive cells (B). * p < 0.05 compared with vector, while # p < 0.05 compared to APOL1WT.
Discussion
Vascular smooth muscle cells are important constituents of arterial walls and among other functions regulate the caliber of the blood vessels. Contractility of these cells serves to regulate perfusion to body organs. Additionally, vascular SMCs are an important source of interleukins, chemokines and growth factors in several organ systems [Gerthoffer and Singer, 2002]. Since vascular SMCs were shown to express of APOL1 in renal biopsy specimens of patients with HIVAN and FSGS [Madhavan et al, 2011], and the APOL1 gene product is a secreted protein, it is likely that arterial smooth muscle cells may be contributing to the APOL1 content of blood perfusing glomerular capillaries. In the present study, we demonstrated that both HIV-CM and IFN-γ enhanced APOL1 production by vascular SMCs. APOL1Vs displayed an overt toxicity in response to podocytes, whereas, APOL1WT was toxic to podocytes only at higher concentration. Depletion of APOL1 from the CM mitigated podocyte toxicity, confirming that it is APOL1 in the CM which is directly responsible for the induction of podocyte injury under these experimental conditions. These results indicate that vascular SMCs carry the potential to contribute to podocyte injury through generation of APOL1/APOL1Vs and that there is a disparity in the toxicity of the WT compared to the risk APOL1Vs. This is the first report to validate a causal relationship between the upregulation of HSMC/APOL1 and podocyte injury.
Although HIV infection displayed the potential to enhance smooth muscle cell expression of APOL1 during the time course examined, this response was not sustained over time, nor did it correlate with the continuous increase in viral replication. Moreover, data on HIV infection of smooth muscle cells are limited (Eugenin et al, 2008). Therefore, more studies are needed to confirm productive HIV infection of smooth muscle cells. Furthermore, secretion of APOL1 by HIV/HSMCs was relatively low (only 5%) when compared to the secretory potential of conditioned media of HIV-infected PBMCs and U939 cells. Thus, it appears that APOL1 expression maybe more likely attributed to the HIV milieu (secretory products of HIV-infected immune cells) rather than direct HIV infection. The HIV milieu contains interferons and other antiviral innate immune molecules [Benlahrech and Patterson, 2011; Segal et al, 2012]. It has been reported that IFN-γ can dramatically increase APOL1 expression in podocytes, renal endothelial cells, and other cells [Nichols et al, 2014; Zhaorigetu et al, 2008]. Consistent with these reports, we also found that both PBMC/HIV-CM and recombinant IFN-γ could enhance APOL1 expression in HSMCs. These findings provide a mechanistic insight into enhanced occurrence of FSGS in patients with HIV infection.
Notably, in the present study, APOL1 depletion from the CM provided only partial protection against podocyte injury. Therefore, it is likely that under these phlogogenic conditions, SMCs may also be secreting other inflammatory mediators which are also injurious to podocytes. Since SMCs are known to secrete interleukins [Gerthoffer and Singer, 2002], it will be interesting to investigate the effect of APOL1Vs on the production of inflammatory cytokines such as IL-1β and IL6 in future studies.
Although we have confirmed that HIV-CMs and IFN-γ have the potential to enhance the production of APOL1 by SMCs, the detailed mechanism is far from clear. P53 has been reported to regulate APOL1 expression in endometrial cancer cell [Wan et al, 2008]. It will be interesting to investigate whether P53 also plays a role in the regulation of HIV-induced APOL1 expression in SMCs in future studies. APOL1 has also been shown to be involved in autophagy, and podocytes are critically dependent on a careful balance of autophagic flux pathways, and also on intact endososomal trafficking, so that disruption of one or more key molecules in these pathways may also have a cardinal role [Wan et al, 2008; Weide and Huber, 2011; Bechtel et al, 2013].
In a recent report, no correlation was observed between circulating APOL1 levels with HIV infection status or inflammatory mediators in an HIV positive cohort with kidney disease (Bruggeman et al, 2014). This makes it all the more relevant to consider local effects, whether they be APOL1 that is produced and acting by an endogenous intracellular mechanism, or via paracrine exposure from adjacent cell. In the present study, HIV transiently increased APOL1 production by vascular smooth muscle cells and direct HIV infection did not enhance smooth muscle cell APOL1 expression significantly. Since there was no relationship between severity of HIV infection and development of kidney disease, we speculate that APOL1- induced vulnerability to HIV effects are more critical for the development of HIVAN rather than the level of viral load; on that account, it seems of paramount importance to delineate the mechanism for APOL1-induced podocyte injury in an HIV milieu. Another example of a sharp disparity between circulating levels and local intra-renal mechanisms has been well studied, and that is the renin angiotensin system (RAS), wherein local activation may predominate in physiologic, pathophysiologic and pharmacologic responses [Gonzalez-Villalobos et al, 2013]. By analogy, it is important to consider and examine the effects of local production of APOL1 by either smooth muscle cells or kidney cells to the manifestation of kidney disease. Therefore, the theme of the present study was to test whether smooth cells have the potential to serve as a source of secretory APOL1 in the presence or absence of HIV infection. Interestingly, levels of viral load in HIV patients correlate better with low CD4 T cell count (Segal et al., 2012). On that account, a higher level of viral load would be associated with a decreased pool of T cells. Since local generation of T cell cytokines may be modulating smooth muscle cell APOL1 expression, this may not necessarily correlate with viral load. However, the present study clearly demonstrates that it is not the HIV-infected smooth muscle cells but rather interaction between HIV infected T cells with smooth muscle cells which serve as a potentially relevant source of APOL1 production for kidney injury. Interestingly, this may also provide an explanation for the lack of correlation between the circulating viral load/ cytokine mediators and APOL1 levels in HIVAN patients (Bruggeman et al, 2014). Additionally, human biopsy data support our notion that smooth muscle cells express APOL1 and that its expression is increased in patients with HIVAN (Madhavan et al, 2011). Whether, this increase in APOL1 expression in HIVAN patients can be attributed to smooth muscle cell HIV infection is a conjecture. While the results of the study are consistent, they do not definitively refute nor confirm this conjuncture, but provide the lead needed to rationalize in vivo or clinical investigation.
APOL1-associated kidney disease may lead to secondary hypertension and this phenomenon is often mistaken for essential hypertension as the initiator of chronic kidney disease [Kopp, 2013; Skorecki and Wasser, 2013; Ito et al, 2014]. We previously suggested that prognosis of hypertension-attributed kidney disease between AAs and EAs is not dependent on their blood pressure control but a reflection of differences in their genetic vulnerability of kidney cell injury [Skorecki and Wasser, 2013]. We further speculate that smooth muscle cells in AAs may be acting as a source of circulating APOL1Vs. In such a scenario, uptake of circulating APOL1Vs by podocytes may be contributing to the initiation and progression of FSGS in this population. In this regard, it is important to note that APOL1 has a signal peptide [SP] and moreover that multiple splice variants some of which may not bear the SP have been described, and moreover it is known that splicing variation can be influenced by viral, inflammatory and oncogenic signals [Tzur et al, 2010; Skorecki and wasser, 2013]. Therefore, depletion or inhibition of APOL1Vs may be a logical approach, in addition to meticulous blood pressure control to prevent or slow the progression of chronic kidney disease.
Patients with HIV infection have been reported to have greater incidence of coronary artery disease and atherosclerosis [Falusi and Aberg, 2001]. Partly, this may be related to dysplipidemia associated with anti-retroviral therapy [Duro et al, 2013]. It has also been suggested that HIV-Nef protein has a potential to block the efflux of cholesterol from their intracellular stores and therefore cells such as macrophages may convert into foam cells [Mujawar et al, 2010]. Moreover, ongoing lipid peroxidation may also contribute to enhanced atherosclerosis in patients with HIV infection [Cui et al, 2014]. Diamond and Karnovsky proposed that both atherosclerosis and focal segmental glomerulosclerosis have similar mechanisms involved for their development and progression [Diamond and Karnovsky, 1988]. In both cases, there is influx of macrophages, subintimal space in case of atherosclerosis and mesangial region in case of FSGS, contribute to the respective phenotype. In both instances, HIV milieu may be serving as a source of interferon-gamma and may thus enhance APOL1 expression by SMCs. Whether modulation of APOL1Vs expression in the altered cytokine milieu also contributes to deleterious or beneficial effects in the development of cardiovascular events remains a controversial issue at present [Ito et al, 2014; Freedman et al, 2015; Langefeld et al, 2015].
In summary, APOL1 expression in vascular SMCs can be dramatically increased in HIV milieu, and has the potential to serve as an endocrine/paracrine source. Extracellualr APOL1 can be taken up by podocytes, leading to cytotoxicity. In case of podocyte uptake of APOL1Vs, the magnitude and or rate of podocyte loss may be several folds higher, which may overwhelm the repair capability of the vulnerable population [Wiggins et al, 2014]. The present study provides additional mechanistic insight into occurrence of accelerated loss of podocytes and possibly vascular involvement in HIV milieu in patients carrying the kidney disease risk variants of APOL1.
Figure 10.

Highlights.
Human artery smooth muscle cells (HSMC) displayed enhanced expression of APOL1 in HIV milieu
Podocytes co-cultured with HSMC-expressing APOL1G1 or APOL1G2 showed several fold greater injury
Conditioned media collected from HSMC-expressing APOL1G1/APOL1G2 also inflicted injury in higher percentages of podocytes.
HSMCs could serve as an endocrine/paracrine source of APOL1Vs, which mediate accelerated podocyte injury in HIV milieu.
Acknowledgments
This work was supported by grants RO1DK 098074, RO1DK084910, RO1 DK083931 (PCS) from National Institutes of Health, Bethesda, MD. KS was supported by grants from the Ernest and Bonnie Beutler Grant Program at Rambam Medical Center, the Binational Science Foundation, and the Israel Science Foundation. We are grateful to AIDS Reagent Program, NIH, for providing primary viral strains.
Footnotes
Conflict of Interest
There is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bechtel W, Helmstädter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, Schell C, Kretz O, Liu S, Geist F, Kerjaschki D, Walz G, Huber TB. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol. 2013;24:727–43. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benlahrech A, Patterson S. HIV-1 infection and induction of interferon alpha in plasmacytoid dendritic cells. Curr Opin HIV AIDS. 2011;6:373–8. doi: 10.1097/COH.0b013e328349592a. [DOI] [PubMed] [Google Scholar]
- 3.Bruggeman LA, O'Toole JF, Ross MD, Madhavan SM, Smurzynski M, Wu K, Bosch RJ, Gupta S, Pollak MR, Sedor JR, Kalayjian RC. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol. 2014;25:634–44. doi: 10.1681/ASN.2013070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castro J, Bittner CX, Humeres A, Montecinos VP, Vera JC, Barros LF. A cytosolic source of calcium unveiled by hydrogen peroxide with relevance for epithelial cell death. Cell Death Differ. 2004;11:468–478. doi: 10.1038/sj.cdd.4401372. [DOI] [PubMed] [Google Scholar]
- 5.Chandel N, Sharma B, Husain M, Salhan D, Singh T, Rai P, Mathieson PW, Saleem MA, Malhotra A, Singhal PC. HIV compromises integrity of the podocyte actin cytoskeleton through downregulation of the vitamin D receptor. Am J Physiol Renal Physiol. 2013;304:F1347–1357. doi: 10.1152/ajprenal.00717.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui HL, Ditiatkovski M, Kesani R, Bobryshev YV, Liu Y, Geyer M, Mukhamedova N, Bukrinsky M, Sviridov D. HIV protein Nef causes dyslipidemia and formation of foam cells in mouse models of atherosclerosis. FASEB J. 2014;287:2828–39. doi: 10.1096/fj.13-246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diamond JR1, Karnovsky MJ. Focal and segmental glomerulosclerosis: analogies to atherosclerosis. Kidney Int. 1988;33:917–24. doi: 10.1038/ki.1988.87. [DOI] [PubMed] [Google Scholar]
- 8.Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O’Connor PM, Malloy MJ, Kane JP. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem. 1997;272:25576–25582. doi: 10.1074/jbc.272.41.25576. [DOI] [PubMed] [Google Scholar]
- 9.Duro M, Sarmento-Castro R, Almeida C, Medeiros R, Rebelo I. Lipid profile changes by high activity anti-retroviral therapy. Clin Biochem. 2013;46:740–4. doi: 10.1016/j.clinbiochem.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 10.Eugenin EA, Morgello S, Klotman ME, Mosoian A, Lento PA, Berman JW, Schecter AD. Human immunodeficiency virus (HIV) infects human arterial smooth muscle cells in vivo and in vitro: implications for the pathogenesis of HIV-mediated vascular disease. Am J Pathol. 2008;172:1100–1111. doi: 10.2353/ajpath.2008.070457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falusi OM, Aberg JA. HIV and cardiovascular risk factors. AIDS Read. 2001;11:263–8. [PubMed] [Google Scholar]
- 12.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, Kao WL. APOL1 Variants Associate with Increased Risk of CKD among African Americans. J Am Soc Nephrol. 2013;24:1484–1491. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freedman BI, Langefeld CD, Lu L, Palmer ND, Carrie Smith S, Bagwell BM, Hicks PJ, Xu J, Wagenknecht LE, Raffield LM, Register TC, Jeffrey Carr J, Bowden DW, Divers J. APOL1 associations with nephropathy, atherosclerosis, and all-cause mortality in African Americans with type 2 diabetes. Kidney Int. 2015;87:176–81. doi: 10.1038/ki.2014.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol. 2011;22:2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genovese G, Friedman DJ, Pollak MR. APOL1 variants and kidney disease in people of recent African ancestry. Nat Rev Nephrol. 2013;9:240–244. doi: 10.1038/nrneph.2013.34. [DOI] [PubMed] [Google Scholar]
- 16.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of Trypanolytic ApoL1 Variants with Kidney Disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerthoffer WT, Singer CA. Secretory functions of smooth muscle: cytokines and growth factors. Mol Interv. 2002;2:447–56. doi: 10.1124/mi.2.7.447. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest. 2013;123:2011–2023. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Husain M, Meggs LG, Vashistha H, Simoes S, Griffiths KO, Kumar D, Mikulak J, Mathieson PW, Saleem MA, Del Valle L, Pina-Oviedo S, Wang JY, Seshan SV, Malhotra A, Reiss K, Singhal PC. Inhibition of p66ShcA longevity gene rescues podocytes from HIV-1-induced oxidative stress and apoptosis. J Biol Chem. 2009;284:16648–16658. doi: 10.1074/jbc.M109.008482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito K, Bick AG, Flannick J, Friedman DJ, Genovese G, Parfenov MG, Depalma SR, Gupta N, Gabriel SB, Taylor HA, Jr, Fox ER, Newton-Cheh C, Kathiresan S, Hirschhorn JN, Altshuler DM, Pollak MR, Wilson JG, Seidman JG, Seidman C. Increased burden of cardiovascular disease in carriers of APOL1 genetic variants. Circ Res. 2014;114:845–50. doi: 10.1161/CIRCRESAHA.114.302347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnstone DB, Shegokar V, Nihalani D, Rathore YS, Mallik L, Ashish, Zare V, Ikizler HO, Powar R, Holzman LB. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS One. 2012;7:e51546. doi: 10.1371/journal.pone.0051546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopp JB. Rethinking hypertensive kidney disease: arterionephrosclerosis as a genetic, metabolic, and inflammatory disorder. Curr Opin Nephrol Hypertens. 2013;22:266–7. doi: 10.1097/MNH.0b013e3283600f8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, Mikulak J, Aviram S, Malhotra A, Skorecki K, Singhal PC. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307:F326–36. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langefeld CD, Divers J, Pajewski NM, Hawfield AT, Reboussin DM, Bild DE, Kaysen GA, Kimmel PL, Raj DS, Ricardo AC, Wright JT, Jr, Sedor JR, Rocco MV, Freedman BI. Apolipoprotein L1 gene variants associate with prevalent kidney but not prevalent cardiovascular disease in the Systolic Blood Pressure Intervention TrialKidney Int. 2015;87(1):169–75. doi: 10.1038/ki.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D, Saleem MA, Satchell SC, Banas B, Mathieson PW, Kretzler M, Hemal AK, Rudel LL, Petrovic S, Weckerle A, Pollak MR, Ross MD, Parks JS, Freedman BI. Localization of APOL1 Protein and mRNA in the Human Kidney: Nondiseased Tissue, Primary Cells, and Immortalized Cell Lines. J Am Soc Nephrol. 2015 Jul 10;26(2):339–348. doi: 10.1681/ASN.2013091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madhavan SM, O'Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol. 2011;22:2119–28. doi: 10.1681/ASN.2011010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medapalli RK, He JC, Klotman PE. HIV-associated nephropathy: pathogenesis. Curr Opin Nephrol Hypertens. 2011;20:306–311. doi: 10.1097/MNH.0b013e328345359a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikulak J, Teichberg S, Arora S, Kumar D, Yadav A, Salhan D, Pullagura S, Mathieson PM, Saleem MA, Singhal PC. DC-specific ICAM-3-grabbing nonintegrin mediates internalization of HIV-1 into human podocytes. Am J Physiol Renal Physiol. 2010;299(3):F664–F673. doi: 10.1152/ajprenal.00629.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13:3005–3015. doi: 10.1097/01.asn.0000039661.06947.fd. [DOI] [PubMed] [Google Scholar]
- 31.Mujawar Z, Tamehiro N, Grant A, Sviridov D, Bukrinsky M, Fitzgerald ML. Mutation of the ATP cassette binding transporter A1 (ABCA1) C-terminus disrupts HIV-1 Nef binding but does not block the Nef enhancement of ABCA1 protein degradation. Biochemistry. 2010;49:8338–49. doi: 10.1021/bi100466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D'Agati V, Markowitz G, Kopp JB, Alper SL, Pollak MR, Friedman DJ. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2014 Aug 6; doi: 10.1038/ki.2014.270. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Page NM, Butlin DJ, Lomthaisong K, Lowry PJ. The human apolipoprotein L gene cluster: identification, classification, and sites of distribution. Genomics. 2001;74:71–8. doi: 10.1006/geno.2001.6534. [DOI] [PubMed] [Google Scholar]
- 34.Quaggin SE, George AL. Apolipoprotein l1 and the genetic basis for racial disparity in chronic kidney disease. J Am Soc Nephrol. 2011;22:1955–1958. doi: 10.1681/ASN.2011090932. [DOI] [PubMed] [Google Scholar]
- 35.Saleem MA, O'Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 36.Segal JL, Thompson JF, Charter RA. A novel immunogen to modulate cytokine production and promote immune system reconstitution in HIV-AIDS. Am J Ther. 2012;19:317–23. doi: 10.1097/MJT.0b013e3182204fd9. [DOI] [PubMed] [Google Scholar]
- 37.Skorecki KL, Wasser WG. Hypertension-misattributed kidney disease in African Americans. Kidney Int. 2013;83:6–9. doi: 10.1038/ki.2012.369. [DOI] [PubMed] [Google Scholar]
- 38.Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, Winkler CA, Kopp J, Rotimi C, Adeyemo A, Doumatey A, Ayodo G, Alper SL, Pollak MR, Friedman DJ, Raper J. Evolution of the primate trypanolytic factor APOL1. PNAS. 2014;111 :E2130–E2139. doi: 10.1073/pnas.1400699111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Human genetics. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283:21540–9. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weide T, Huber T. Implications of autophagy for glomerular aging and disease. Cell and Tissue Research. 2011;343:467–473. doi: 10.1007/s00441-010-1115-0. [DOI] [PubMed] [Google Scholar]
- 42.Wiggins RC, Alpers CE, Holzman LB, He JC, Salant DJ, Chugh SS, Natarajan R, Trachtman H, Brasile L, Star RA, Rys-Sikora KE, Moxey-Mims MM, Flessner MF. Kidney Research National Dialogue. Glomerular Disease: Looking beyond Pathology. Clin J Am Soc Nephrol. 2014;9:1138–1140. doi: 10.2215/CJN.01450214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Chien-an AH. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]