

Abstract
During development a network of transcription factors functions to differentiate foregut cells into pancreatic endocrine cells. Differentiation of appropriate numbers of each hormone-expressing endocrine cell type is essential for the normal development of the pancreas and ultimately for effective maintenance of blood glucose levels. A fuller understanding of the details of endocrine cell differentiation may contribute to development of cell replacement therapies to treat diabetes. In this study, by using morpholino and gRNA/Cas9 mediated knockdown we establish that differential levels of the basic-helix loop helix (bHLH) transcription factor Neurod are required for the differentiation of distinct endocrine cell types in developing zebrafish. While Neurod plays a role in the differentiation of all endocrine cells, we find that differentiation of glucagon-expressing alpha cells is disrupted by a minor reduction in Neurod levels, whereas differentiation of insulin-expressing beta cells is less sensitive to Neurod depletion. The endocrine cells that arise during embryonic stages to produce the primary islet, and those that arise subsequently during larval stages from the intra-pancreatic duct (IPD) to ultimately contribute to the secondary islets, show similar dependence on differential Neurod levels. Intriguingly, Neurod-deficiency triggers premature formation of endocrine precursors from the IPD during early larval stages. However, the Neurod-deficient endocrine precursors fail to differentiate appropriately, and the larvae are unable to maintain normal glucose levels. In summary, differential levels of Neurod are required to generate endocrine pancreas subtypes from precursors during both embryonic and larval stages, and Neurod function is in turn critical to endocrine function.
Keywords: zebrafish, pancreas, Neurod, glucagon, insulin, Notch, phlorizin, CRISPR
INTRODUCTION
The hormone producing pancreatic endocrine cells of the islets of Langerhans are crucial for proper regulation of blood glucose levels. The islets comprise insulin-expressing beta cells, glucagon-expressing alpha cells, somatostatin-expressing delta cells and ghrelin-expressing epsilon cells. As beta cell loss or failure causes diabetes, patients can potentially benefit from cell replacement therapies. Effective in vitro production of functional beta cells from stem cells for use in replacement therapies will likely depend on a fuller understanding of normal endocrine pancreas development. However, our understanding of the precise mechanisms through which multipotent endocrine progenitors differentiate into distinct endocrine cell types remains incomplete.
The vertebrate endocrine pancreas develops from dorsal and ventral pancreatic buds, which form as out-pockets of the endoderm-derived gut tube (Kinkel and Prince, 2009). In zebrafish, the dorsal bud gives rise to the initial primary endocrine islet, which is already established at 24 hours post fertilization (hpf), but complete endocrine pancreas development requires the contribution of ventral bud-derived cells. As the dorsal and ventral buds merge at around 52 hpf, endocrine cells from the ventral bud contribute to the primary islet (Field et al., 2003). Subsequently, during larval stages, duct cells provide a source of secondary endocrine cells that will ultimately produce the secondary islets (Parsons et al., 2009). In both mammals and zebrafish the duct-derived secondary endocrine progenitors are Notch responsive cells (NRCs) (Apelqvist et al., 1999; Jensen et al., 2000; Kopinke et al., 2011; Ninov et al., 2012; Parsons et al., 2009). Upon inhibition of Notch signaling, zebrafish intra-pancreatic duct (IPD)-NRCs precociously form secondary endocrine cells from as early as 6 days post fertilization (dpf) (Ninov et al., 2012; Parsons et al., 2009; Wang et al., 2011). While suppression of Notch signaling is sufficient to trigger endocrine cell production from the IPD, the molecular mechanisms guiding multipotent endocrine progenitor differentiation downstream of Notch suppression are not fully understood. In particular, it is unclear whether initial development of the primary endocrine cells and subsequent development of secondary endocrine cells from the IPD use equivalent mechanisms.
Endocrine pancreas cells in the mouse develop from precursors that transiently express the basic helix-loop-helix (bHLH) domain transcription factor Neurog3 (G. Gu et al., 2002; Mellitzer et al., 2004; Schonhoff et al., 2004), and Neurog3 mutant mice are unable to differentiate endocrine pancreas cells (Gradwohl et al., 2000). By contrast, there is no evidence that zebrafish endocrine precursors express neurog3, or other neurog homologs (Flasse et al., 2013), and neurog3 mutant zebrafish do not have any endocrine pancreas defects (Flasse et al., 2013). Although Neurog transcription factors do not appear to play a role in zebrafish pancreas development, Flasse and colleagues (2013) did uncover a role for the bHLH domain transcription factor Neurod; they showed that simultaneous knockdown of Ascl1b and Neurod blocks zebrafish endocrine cell differentiation (Flasse et al., 2013). In mice, Neurog3 activates expression of NeuroD1 (Huang et al., 2000), and importantly, NeuroD1 can substitute for Neurog3 in protocols to transform exocrine cells to beta cells (Zhou et al., 2008). Mice lacking NeuroD1 fail to form endocrine islets, develop diabetes and die shortly after birth (Naya et al., 1997). Beta cell specific deletion of NeuroD1 leads to glucose intolerance because the beta cells remain immature and fail to function properly (C. Gu et al., 2010). In humans, homozygous mutations in NEUROD1 are characterized by permanent neonatal diabetes (Rubio-Cabezas et al., 2010). Together, these data suggest a conserved role for Neurod homologs in endocrine pancreas development.
Here we have explored the role of zebrafish Neurod in the differentiation of endocrine pancreas cells. Analysis of specimens in which gRNA/cas9 genome editing was used to generate predicted null alleles has confirmed that Neurod plays a critical function in endocrine cell development. We have exploited a morpholino knockdown strategy to investigate the consequences of differential levels of Neurod knockdown, and report that different levels of zebrafish Neurod are required for the differentiation of particular endocrine cell types. Specifically, alpha cell differentiation is dependent on high levels of Neurod, while beta cell differentiation requires lower levels. Using endoderm-specific gain of function we confirm that high levels of Neurod promote differentiation of glucagon-expressing alpha cells. Although Neurod-deficient larvae produce precocious secondary endocrine precursors upon inhibition of Notch signaling, these cells remain undifferentiated, indicating that larval stage secondary endocrine cell differentiation is similarly dependent on Neurod. Remarkably, Neurod-deficient larvae initiate premature endocrine cell differentiation from the IPD, suggesting the presence of compensatory mechanisms to regulate endocrine cell numbers. Consistent with the inability of Neurod-deficient larvae to complete the endocrine pancreas differentiation program to produce appropriate numbers of hormone-expressing cells, these specimens are unable to maintain normal glucose levels.
MATERIALS AND METHODS
Zebrafish husbandry
Zebrafish (Danio rerio) were maintained as described (Westerfield, 1995). Embryos were obtained from wild-type AB, TgBAC(neurod:EGFP)nl1 [hereafter Tg(neurod:EGFP)] (Obholzer et al., 2008), Tg(mnx1:GFP) (Dalgin et al., 2011), Tg(ptf1a:EGFP) (Godinho et al., 2005) and Tg(−5.0sox17:EGFP)zf99 [hereafter Tg(sox17:EGFP)] (Mizoguchi et al., 2008) lines, raised and staged as described (Kimmel et al., 1995).
Microinjections of morpholino antisense oligonucleotides
Morpholino (MO) antisense oligonucleotides for Neurod: Neurod ATG MO: 5′ TTTCCTCGCTGTATGACTTCGTCAT and Neurod UTR MO: 5′ TGACTTCGTCATGTCGGAACTCTAG (Sarrazin et al., 2006), were purchased from Gene Tools, LLC. Mnx1 and Sox32 MOs were used as described (Dalgin et al., 2011). Tg(neurod:EGFP) embryos were microinjected at the one to two-cell stage with 1 nl of 1, 2 or 4 μg/μl Neurod ATG MO, or 2, 4 or 8 μg/μl Neurod UTR MO. Due to overlap of the target sites in the UTR of neurod and egfp transcripts Neurod UTR MOs were titrated away by the transgene, therefore higher Neurod UTR MO concentrations were used when injecting Tg(neurod:EGFP) embryos.
Whole mount in situ hybridization, immunohistochemistry, H2B-RFP mRNA injections and imaging
Whole mount in situ hybridization and immunohistochemistry were performed as described (Dalgin et al., 2011). The following antibodies were used: polyclonal rabbit anti-active Caspase-3 (1:100; Millipore AB3623), rabbit anti-GFP488 (1:500; Molecular Probes A21311), monoclonal mouse anti-glucagon (1:200; Sigma G2654), polyclonal rabbit anti-phospho-Histone H3 (Ser10) (1:100; Millipore 06-570), polyclonal guinea pig anti-insulin (1:100; Dako A0564), Neurod antibody (1:100, GST fusion epitope containing amino acids 1–57; a gift from Dr. Masahiko Hibi) (Kani et al., 2010), polyclonal rabbit anti-somatostatin (1:200; MP Biomedicals 11180). Plasmid to in vitro transcribe H2B-RFP mRNA was kindly provided by Dr. Ryan M. Anderson. H2B-RFP mRNA was in vitro transcribed (Ambion MEGAscript SP6 kit-AM 1330) and embryos were injected with 100 pg of capped synthetic mRNA at the one-cell stage. To obtain fluorescent images embryos were flat-mounted and imaged using a Zeiss LSM 710 confocal microscope with 25X or 40X objectives. To obtain bright-field images embryos were deyolked, flat-mounted, and photographed under a Zeiss Axioskop microscope. Cell counting for bright-field images were performed under the microscope; to enable visualization of single cells, chromogenic samples were monitored to avoid over-staining. Cell counts for confocal images (merged z-stacks) were performed using the Zeiss LSM 710 confocal microscope; z-stacks of optical sections (2–4 μm) were taken for each specimen and each stack was analyzed using the ImageJ cell counter plugin.
Real-Time qPCR
Total RNA was isolated from 20 embryos per group according to manufacturer’s instructions by removing genomic DNA contamination (Qiagen 74104). cDNA was prepared using iScript cDNA synthesis kit (Bio-Rad 170-8891). Primers were designed using PrimerQuest system (IDT) (Table S1). cDNA was amplified using StepOnePlus Real-Time PCR system (Applied Biosystems) with Power SYBR Green PCR Master Mix (Applied Biosystems 4367659). Relative expression of each sample was determined after normalization to beta-actin 1 (actb1) levels using the efficiency-corrected delta Ct method (Bookout et al., 2006) and displayed relative to an arbitrary value.
Cell transplantation
Transplantation was performed as described (Dalgin et al., 2011; Ho and Kane, 1990; Stafford et al., 2006). Host embryos were injected with 1 ng Sox32 MO at the one-cell stage. To achieve Neurod knockdown in the endoderm, donor embryos were injected at the one-cell stage with Neurod ATG MO (2 ng), together with synthetic capped sox32 mRNA (100–200 pg; Ambion MEGAscript SP6 kit-AM 1330) and 10 kD fixable rhodamine dextran (RD: Molecular Probes). To achieve overexpression of neurod in endoderm, donor embryos were injected at the one-cell stage with neurod mRNA (100 pg; Ambion MEGAscript SP6 kit-AM 1330), together with sox32 mRNA and 10 kD fixable RD. At 4 hpf 20–40 cells from a donor embryo were transplanted into the blastoderm margin of each host. Transplanted specimens with a fully reconstituted endoderm of normal morphology were assayed by whole-mount immunohistochemistry at 52 hpf.
RO4929097, Phlorizin treatments and glucose fluorometric assay
RO4929097-gamma secretase inhibitor was purchased from Selleckchem (S1575) and dissolved in DMSO. Three day old Tg(neurod:EGFP) control or Neurod morphant embryos were incubated in embryo medium (E3) containing 3 μM RO4929097 or DMSO carrier (DMSO treated embryos were indistinguishable from untreated controls; data not shown) until 6 dpf. Phloridzin (Phlorizin) dihydrate (Phz) was purchased from Sigma-Aldrich (P3449). Phz was prepared fresh in E3 and filtered (Fisher 09-719A, 0.22 μm) to remove undissolved particles. Ten 5.5 dpf larvae were incubated in a 12 well plate (Becton Dickson 353043) in E3 medium containing 250 μg/ml Phz for 16 hours. We found that this treatment regimen was optimal; treatments starting prior to this stage and for longer time periods were toxic to larvae. Ten samples per group were frozen and stored at −80°C until analysis. For collections after 5 dpf, larvae were fed (ZM Fish Food, ZM000) twice daily; on the day of collection samples were harvested before the normal feeding time. Glucose levels were determined using a colorimetric/fluorometric assay kit (Biovision, K606-100). A glucose standard curve was prepared according to manufacturer’s instructions. Samples were hand-homogenized in PBS (20 μl/embryo). 20 μl of each sample was used to assemble reactions, using 96-well black flat bottom plates (Costar 3915) according to manufacturer’s instructions. Fluorescence (Ex/Em=535/590) was measured using a Beckman Coulter DTX 880 Multimode microplate reader.
Microinjections of gRNA and Cas9 mRNA
The CHOPCHOP website (Montague et al., 2014) was used to select genomic target sites on the zebrafish neurod locus. We designed an optimized single guide (sg) RNA scaffold, modified for efficient transcription and with an extended stem loop designed to improve interaction with the Cas9 protein (Chen et al., 2013) 5′ AAAAATTTAGGTGACACTATAGGACGACGAGGAAGAAGAAGGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGGCTAGTCCGTTATCAACTTGAAAAAG TGGCACCGAGTCGGTGCTTTTTTT containing an SP6 promoter (underlined) and genomic target site (bold and Fig. S12). This sequence was purchased as a gBlock from Integrated DNA Technologies and used as a template for transcribing sgRNA. The sgRNA was transcribed using the Ambion MEGAscript SP6 kit (AM 1330) with addition of 40U RNAsin (Promega N251A), and was incubated at 37°C for 90 minutes. RNA was then isolated according to manufacturer’s instructions with the addition of a phenol/chloroform/isoamyl alcohol extraction and 20 μg of glycogen carrier (Thermo Scientific R0551). Capped and polyadenylated 3xFLAG-NLS-SpCas9-NLS (hereafter Cas9, Addgene 51307) mRNA (Guo et al., 2014) was also transcribed using the Ambion MEGAscript SP6 kit (AM 1330). Tg(neurod:EGFP) embryos were microinjected at the one-cell stage with 60 pg sgRNA and 500 pg Cas9 mRNA per embryo.
Isolation of genomic DNA, T7 endonuclease I assay and identification of transient genomic mutations
Genomic DNA was isolated from sgRNA/Cas9 injected specimens and controls at 48 hpf. Dechorionated single embryos were transferred to microfuge tubes and rinsed with nuclease free H2O. Samples were incubated at 50°C for 1–2 hours in 50 μl of DNA extraction buffer (10 mM Tris pH 8.2, 10 mM EDTA, 200 mM NaCl, 0.5% SDS, 200 μg/ml proteinase K) until completely dissolved. DNA was extracted with 1:1 volume of phenol/chloroform/isoamyl alcohol and precipitated with 1:10 volume 3M sodium acetate (pH 5.2) and EtOH at −20°C overnight. DNA was resuspended in 10 μl TE buffer and stored at 4°C. 2 μl of genomic DNA was used to PCR amplify a 494 bp genomic region flanking the neurod target site (forward primer ATGACGAAGTCATACAGCGA, reverse primer TGCACAAAAGACATCAGGTC, Fig. S12). 1 μg of PCR amplicon was denatured and reannealed to facilitate heteroduplex formation (5-min denaturing step at 95 °C, followed by cooling to 85 °C at −1.5 °C per second and further to 25 °C at −0.1 °C per second). The reannealed amplicons were digested with 5U of T7 endonuclease I (New England Biolabs M0302S) at 37°C for 45 minutes. The reaction products were immediately resolved by electrophoresis through a 2% agarose gel. Selected PCR products were subcloned into pCR II-TOPO vector (Invitrogen 45-0640) according to manufacturer’s instructions and clones were sequenced to identify insertion/deletion mutant alleles.
RESULTS
Embryonic endocrine cell types display a differential sensitivity to Neurod knockdown
To investigate the function of neurod in zebrafish endocrine pancreas development we used morpholino (MO) knockdown. We used two different morpholinos (MOs), targeted to the 5′UTR (Neurod UTR MO) (Sarrazin et al., 2006) and to the translational start site (Neurod ATG MO) of the neurod mRNA. Our knockdown experiments were performed in Tg(neurod:EGFP) embryos; in these transgenic specimens EGFP recapitulates endogenous Neurod expression, and colocalizes with all pancreatic endocrine hormone markers (Dalgin et al., 2011). Tg(neurod:EGFP) embryos were injected with three different concentrations (see Methods) of either Neurod UTR MO or Neurod ATG MO, and the pancreatic region was analyzed at 30 hpf, by which stage the dorsal bud-derived endocrine primary islet is fully formed. Control and Neurod MO injected specimens (morphants) had normal gross morphology (Fig. S1, and data not shown). The Neurod UTR MO not only targets the 5′ UTR of endogenous neurod mRNA but also recognizes the 5′ UTR of the Tg(neurod:EGFP) transcript. As expected, injection of Neurod UTR MO reduced EGFP expression in Tg(neurod:EGFP) embryos, with increasing morpholino concentration (2–8 ng) causing a reduction in expression levels, indicative of dose-dependent knockdown (Fig. 1A–D and false colored images Fig. S2A–D). We confirmed a concomitant decrease in endogenous Neurod protein levels in response to increasing concentration of Neurod UTR MO by comparing immunoreactivity to Neurod antibody (Kani et al., 2010) in control and Neurod morphant embryos (Fig. 1E–H and false colored images Fig. S2E–H). We found that Neurod immunoreactivity was similarly depleted in response to increasing concentrations of Neurod ATG MO (1–4 ng; not shown). Immunolabeling also revealed heterogenous Neurod protein expression levels in the cells of the unmanipulated dorsal bud (Fig. 1E, inset and false colored image Fig. S2E). These results indicate that injection of low concentrations of Neurod MO causes partial knockdown, with a complete knockdown of Neurod, as assessed by inability to detect protein, achieved only at high Neurod MO concentrations. We conclude that the gradual decrease in Tg(neurod:EGFP) expression in response to the Neurod UTR MO provides a reliable readout of Neurod knockdown.
Fig. 1. Morpholino knockdown causes progressive depletion of Neurod.
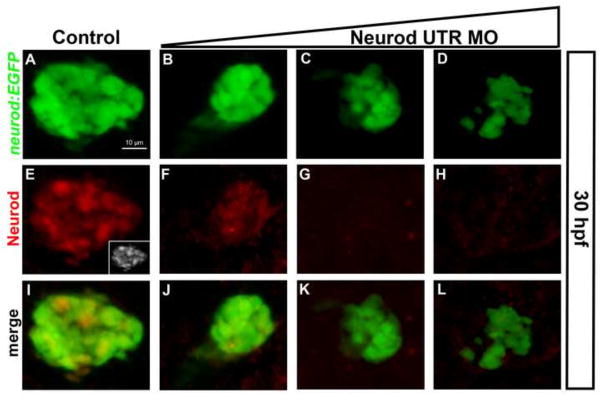
Whole mount immunolabeling for GFP (green) and Neurod (red) protein. Confocal images (merged z-stacks) of the dorsal pancreatic bud in Tg(neurod:EGFP) embryos at 30 hpf. (A, E, I) Control embryos; (E; inset) gray scale. Neurod UTR MO injected specimens, (B, F, J) 2 ng/embryo, (C, G, K) 4 ng/embryo, (D, H, L) 8 ng/embryo. Scale bar = 10 μm.
To analyze dorsal bud development in Neurod morphants we examined the expression of endocrine cell markers for alpha cells (glucagon; gcga), beta cells (insulin; insa) and delta cells (somatostatin; sst2) by whole mount in situ hybridization at 30 hpf. In Neurod UTR MO injected embryos, the number of gcga-expressing cells was decreased by 96% in response to the lowest MO concentration (2 ng; Fig. S3A–E), however the highest concentration of MO (8 ng) was required to obtain 51% and 67% decreases in the numbers of insa (Fig. S3F–J) and sst2-expressing (Fig. S3K–O) cells, respectively. High concentrations of MO were also required to obtain a significant decrease in epsilon cells (labeled with ghrelin; not shown). We observed similar results in response to injection of differing concentrations of Neurod ATG MO (not shown). To analyze changes in the number of endocrine cells in the context of a single islet we examined expression of gcga and insa by double whole mount in situ hybridization at 30 hpf. In response to the lowest MO concentration the number of gcga-expressing cells decreased significantly but the number of insa-expressing cells did not change (Fig. 2A,B), by contrast, a significant decrease in the number of insa-expressing cells was detected in response to a higher MO concentration (Fig. 2C,D). We next performed qRT-PCR analysis to quantify endocrine gene transcription in Neurod morphant specimens relative to sibling controls. The qRT-PCR results corroborated our in situ hybridization analysis, showing that expression of gcga decreased 7-fold in response to the lowest MO concentration, whereas the highest MO concentration was required to obtain 5- and 6-fold decreases in insa and sst2 expression, respectively (Fig. 2E). These data show that differentiation of all hormone-expressing endocrine cell types of the dorsal bud is dependent on Neurod, but also reveal that the glucagon-expressing alpha cells are the most sensitive to Neurod depletion.
Fig. 2. Quantitative analysis of Neurod morphant endocrine cell differentiation.
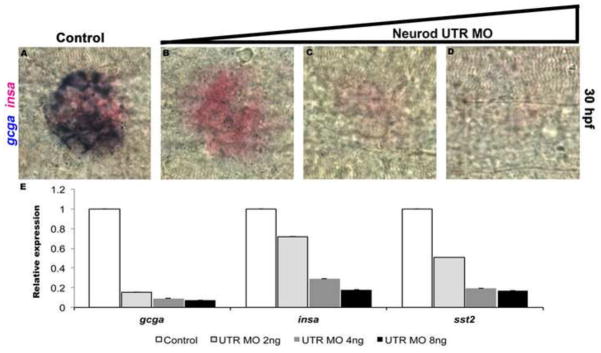
Double in situ hybridization for glucagon (gcga, blue) and insulin (insa, red) at 30 hpf. Control (A), Neurod UTR MO 2 ng (B), 4 ng (C) or 8 ng (D) injected specimens. Results are representative of two independent experiments and from a minimum of 20 embryos per group. (E) Relative levels of gcga, insa and sst2 by real-time qPCR. Results are from 2 independent experiments and from 3 technical replicates and confirmed by two independent primer sets per gene of interest. All values were normalized to beta-actin levels. Primer sequences are listed in Table S1.
The homeobox transcription factor aristaless related homeobox (Arx) is critical for alpha cell fate in mice (Collombat et al., 2003) and in zebrafish (Djiotsa et al., 2012). To investigate if the decrease in gcga expression in Neurod morphants was due to a decrease in arxa (the zebrafish Arx homolog) expression, we analyzed the expression of arxa transcripts in Neurod morphants. Injection of a low concentration of Neurod MO, sufficient to severely reduce the number of gcga expressing cells, did not significantly affect the number of arxa-expressing cells, although at higher Neurod MO concentrations severe reductions were observed (Fig. S3P–T). These data suggest that expression of gcga depends on additional factors beyond Arxa.
Neurod-deficient endocrine precursors remain undifferentiated
The observed decrease in the number of hormone-expressing cells in Neurod morphants could be a consequence of increased cell death and/or decreased cell proliferation. To test these possibilities we used the Tg(neurod:EGFP) transgenic line, in which EGFP expression recapitulates endogenous neurod expression (Dalgin et al., 2011). Injection of the Neurod ATG MO, which does not block EGFP expression, allowed us to follow the fate of EGFP-expressing cells in Neurod-deficient specimens.
We detected cells undergoing cell death by immunohistochemistry with antibodies targeted against anti-active Caspase3 (Caspase3). Three different concentrations of Neurod ATG MO were injected into Tg(neurod:EGFP) embryos, and Caspase3-positive EGFP-expressing cells were counted in control and morphant specimens at 18 and 36 hpf. While we detected apoptotic cells in the ectoderm of normal and Neurod morphant specimens, the number of EGFP-expressing cells undergoing cell death in the endoderm was extremely low and did not differ between control and Neurod morphant specimens (data not shown). To investigate whether Neurod-deficiency might cause ectopic cell death more broadly we injected Neurod ATG MOs into Tg(Sox17:EGFP) embryos, which express EGFP throughout the endoderm. Control and morphant specimens were fixed at 36 hpf and cell death assayed. The morphology of the gut and dorsal pancreatic bud was grossly normal in morphants when compared to controls (Fig. S4A–D). Caspase3-labeling revealed that cell death was rare in the endoderm, with cell death rates equivalent in control and morphant specimens (Fig. S4A–D). Consistent with our findings, Flasse et al. (2013) also failed to detect cell death in the pancreatic region of Neurod morphant specimens. Together, these data establish that the decrease in number of hormone expressing cells in Neurod morphants is unlikely to be due to increased cell death.
We next tested whether the decrease in hormone expressing cells in Neurod morphants might be due to decreased endocrine cell proliferation. To establish proliferation rates we again used the Tg(neurod:EGFP) line. We first counted the total number of EGFP-expressing cells at 18 hpf in the dorsal bud precursors of controls and Neurod morphants. In controls the mean number of EGFP-expressing endocrine cells was ~36 (Fig. 3A, E and Fig. S5A). In Neurod morphants injected with 1 ng or 2 ng of Neurod ATG morpholino there was an insignificant (P<0.2 and P<0.067 respectively) decrease in the number of EGFP-expressing cells, with a mean value of ~33 and ~31 cells respectively (Fig. 3B, C, E and Fig. S5B, C). Specimens injected with 4 ng Neurod ATG MO showed a significant (21%) decrease in the number of EGFP-expressing cells (mean value ~28 cells, P<0.013) (Fig. 3D, E and Fig. S5D). We next identified proliferating cells by immunohistochemistry with antibodies against anti-phospho-histone H3 (pH3) (images co-labeled with TO-PRO3 to reveal the nuclei are shown in Fig. S5). Proliferation rates were low: only ~3.5% of endocrine cells were pH3-positive in control specimens (Fig. 3A, F and Fig S5A). Proliferation rates in Neurod ATG MO injected specimens were not significantly different (P>0.1); specimens injected with 1 ng, 2 ng or 4 ng of Neurod ATG MO averaged ~3%, ~3.7% or ~2.5% pH3-positive cells respectively (Fig. 3B–C, F and Fig S5B–C).
Fig. 3. Neurod-deficient endocrine cells remain undifferentiated and have normal proliferation rate.

Confocal images (merged z-stacks) of representative 18 hpf (A–D) and 36 hpf (G–H) Tg(neurod:EGFP) and 36 hpf Tg(sox17:EGFP) (M–P) embryos. Control (A, G, M), Neurod ATG MO 1 ng (B, H, N), 2 ng (C, I, O) or 4 ng (D, J, P) injected specimens. Whole mount immunolabeling for pH3 (red, A–D, G–J, M–P). Mean (± s.d.) number of cells expressing GFP (E, K, Q) and proliferation rate is shown as mean (± s.d.) percentage of cells expressing pH3 and GFP (F, L, R) from 2 independent experiments and from a minimum of 10 embryos per group. *, P<0.013, **, P<0.001; t-test, two-tailed distribution. NS, not significant. Embryos are oriented anterior to the left (A–D, G–J) and to the top (M–P). Arrows indicate liver and arrowheads indicate dorsal pancreas region. Yellow scale bar = 10 μm.
We next examined cell proliferation specifically within dorsal bud cells. Sibling specimens from the previous experiment were raised until 36 hpf and proliferating EGFP-expressing cells again identified using pH3. The average number of EGFP-expressing cells in the control dorsal bud was ~48, and a similar number of EGFP-expressing cells was found in the dorsal bud of 1 ng Neurod ATG MO injected specimens (Fig. 3G, H, K and Fig. S4E, F). In contrast, we found 20% and 25% decreases in the number of EGFP-expressing cells in the dorsal buds of 2 ng and 4 ng Neurod ATG MO injected specimens, respectively (P<0.001) (Fig. 3I, J, K and Fig. S4G, H). To determine the percentage of proliferating dorsal bud endocrine cells we counted pH3-positive EGFP-expressing cells. Proliferation rates were extremely low at this stage and did not differ significantly between control and Neurod-deficient specimens (P>0.7) (Fig. 3G–J, L). Together, these data demonstrated that although the number of Neurod:EGFP cells decreases in response to injection of 2 ng or 4 ng of Neurod ATG MO, the decrease is not a consequence of reduced proliferation of endocrine cells. It remained possible that Neurod knockdown reduces proliferation in endocrine precursors. To examine this possibility we injected Neurod ATG MO into Tg(Sox17:EGFP) embryos, and compared proliferation rates between controls and morphants of all dorsal bud cells at 36 hpf. Neurod-deficiency did not cause morphological defects and the dorsal bud was properly formed (Fig. 3M–P and Fig. S5I–L). We counted the total number of EGFP-expressing cells between the tip of the dorsal bud and the gut tube (Fig. S5M); the average number of cells in control dorsal buds was ~56, with similar numbers of EGFP-expressing cells present in 1 ng, 2ng and 4 ng Neurod ATG MO injected specimens (~55, ~52.5 and ~53, respectively). To determine the percentage of proliferating dorsal bud cells we counted pH3-positive Sox17:EGFP cells (Fig. S5M): ~1.5% of EGFP-expressing cells were pH3-positive in controls and this proliferation rate did not change significantly in Neurod ATG 1 ng, 2ng and 4 ng MO injected specimens (1.8%, 1.9% and 1.9%, respectively). Together, our data indicate that Neurod-deficiency does not cause detectable changes in rates of cell death or proliferation within the pancreatic region. We conclude that Neurod morphant cells are typically retained in the developing embryos, yet remain undifferentiated. Taken together with our analysis of hormone-expressing cell types, these data imply that partial Neurod knockdown causes alpha cell precursors to remain undifferentiated, whereas more extensive Neurod knockdown blocks differentiation of all endocrine cell types. While we do find a significant reduction in the number of total Neurod:EGFP cells present in specimens lacking most or all Neurod function, this reduction is nevertheless quite modest; there are several possible explanations for this reduction (discussed ahead).
Endocrine cells derived from dorsal and ventral bud are equally affected in Neurod morphants
The hormone-expressing cells of the primary islet are derived from a combination of dorsal bud and ventral bud-derived cells, with the two buds merging at around 52 hpf. We investigated whether the differential sensitivity to knockdown of Neurod observed for dorsal bud-derived endocrine cell differentiation also holds true for ventral bud-derived endocrine cells. We again injected Neurod UTR MO at three different concentrations, but analyzed the fate of Tg(neurod:EGFP) cells at 72 hpf, after the dorsal and ventral pancreatic buds have merged. Reduced EGFP expression in response to Neurod UTR MO injection confirmed that the morpholino continues to function during ventral bud-derived endocrine cell differentiation (Fig. 4A–D, F–I, K–N, single channels are shown in Fig. S6A–D, I–L, Q–T). Expression of hormone markers of alpha, beta and delta cells was detected by immunohistochemistry, and cells were counterstained with nuclear marker TO-PRO-3 to facilitate cell counting. Consistent with our analysis of dorsal bud-derived endocrine cell differentiation, specimens injected with low levels of Neurod UTR MO (2 ng) showed an 80% decrease in the total number of glucagon-positive cells (Fig. 4A–E; single channels Fig. S6E–H), whereas injection of the highest level of Neurod UTR MO (8 ng) was required to obtain 77% and 70% decreases in the total number of insulin (Fig. 4F–J; single channels Fig. S6M–P) and somatostatin-expressing (Fig. 4K–O; single channels Fig. S6U–X) cells, respectively. These data suggest that Neurod-deficiency affects dorsal and ventral bud-derived endocrine cells in a similar manner.
Fig. 4. Neurod is also required for differentiation of endocrine cells from the ventral bud.

Confocal images (merged z-stacks) of representative 72 hpf Tg(neurod:EGFP) embryos. Whole mount immunolabeling for glucagon (red, A–D), insulin (red, F–I), somatostatin (red, K–N), GFP (green), with nuclear staining TO-PRO-3 (blue). Control (A, F, K), Neurod UTR MO 2 ng (B, G, L), 4 ng (C, H, M) or 8 ng (D, I, N) injected specimens. Mean (± s.d.) number of cells expressing glucagon (E), insulin (J) and somatostatin (O) from 5 independent experiments and from a minimum of 55 embryos per group. (P) Relative levels of gcga, insa and sst2 by real-time qPCR. Results are from 2 independent experiments and from 3 technical replicas and confirmed by two independent primer sets per gene of interest. All values were normalized to beta-actin levels. Primer sequences are listed in Table S1. White scale bar = 10 μm.
To confirm that Neurod is required in both dorsal and ventral bud-derived cells, we used a label-retaining method to distinguish dorsal from ventral bud-derived cells within the primary islet at 60 hpf, after the buds have merged. We injected one-cell stage embryos with mRNA encoding histone (H2B-RFP) fusion protein; this protein is retained in cells that become post-mitotic early in development, but due to dilution through subsequent cell divisions is below detection limits in cells that become post-mitotic at later stages. It was previously reported that at 52 hpf dorsal bud-derived cells are H2B-RFP-positive and ventral bud-derived cells are H2B-RFP-negative (Hesselson et al., 2009; Wilfinger et al., 2013). Recently, experiments using a transgenic fluorescent ubiquitylation-based cell cycle indicator (FUCCI) revealed that beta cells in the dorsal bud have a significant, and previously unappreciated, proliferation capacity at 36 hpf (Tsuji et al., 2014). This new finding implies that some dorsal bud-derived cells are likely to lack H2B-RFP labeling at 60 hpf; nevertheless any label retaining cells observed can safely be assumed to derive from the dorsal not the ventral bud. We therefore injected H2B-RFP mRNA alone or together with one of three different concentrations of Neurod ATG MO, and raised the specimens to 60 hpf. We used immunohistochemistry to detect insulin (Fig. S7A–D, I–L) and glucagon-expressing (Fig. S7M–P, U–X) H2B-RFP-positive and negative endocrine cells (A–H, M–T). Consistent with the findings described above, only high concentrations of Neurod MO significantly decreased the number of insulin-expressing cells (Fig. S7A–D, I–L). Importantly, Neurod knockdown decreased insulin expression in both H2B-RFP-positive and negative cells (Fig. S7A–D), confirming that Neurod plays similar roles in dorsal and ventral bud-derived cells. Again consistent with our previous findings, there was a dramatic reduction of glucagon expression in specimens injected with the low concentration of Neurod MO, and a near complete loss of glucagon expression in response to injection of higher Neurod MO concentrations (Fig. S7M–P, U–X). Both H2B-RFP positive and negative endocrine cells displayed this loss of glucagon expression in response to increasing levels of Neurod MO injections (Fig. S7M–P, Q–T). Together, these data confirm that Neurod is necessary for the differentiation of both dorsal and ventral bud-derived endocrine cells.
We next performed qRT-PCR analysis to quantify endocrine marker gene transcription in response to Neurod knockdown. We injected Tg(neurod:EGFP) embryos with Neurod UTR MO at three different concentrations and collected specimens at 72 hpf for qRT-PCR analysis. Consistent with our immunohistochemistry analysis (Fig. 4A–E) gcga transcript levels were decreased 5-fold in specimens injected with the low concentration of Neurod UTR MO (2 ng) and specimens injected with 4 ng and 8 ng of Neurod UTR MO displayed 9- and 11-fold decreases in gcga transcript levels, respectively (Fig. 4P). Again consistent with our immunohistochemistry analysis (Fig. 4F–J), injection of 2 ng Neurod UTR MO produced only a minimal decrease in insa transcript levels (1.8-fold; Fig. 4P), with 4 ng of Neurod UTR MO causing a 2.8-fold reduction. Interestingly, insa transcript levels decreased only 2.3-fold in specimens injected with the highest concentration (8 ng) of Neurod UTR MO (Fig. 4P). These data suggest that while the number of insulin-expressing cells decreases in response to increasing Neurod knockdown (Fig. 4F–J), the remaining insulin-expressing cells may try to compensate for the reduction in insulin levels by transcribing more insa. Somatostatin-expressing cells showed a similar response, with qRT-PCR analysis revealing that increasing concentrations of Neurod UTR MO did not progressively reduce sst2 transcript levels (Fig. 4P), despite reduction in the number of somatostatin- expressing cells.
We next examined if knockdown of Neurod disrupts acinar cell development. We injected Neurod ATG MO at three different concentrations into Tg(ptf1a:EGFP), which marks the developing acinar cells in zebrafish exocrine pancreas (Dong et al., 2008). Expression of hormone markers glucagon and insulin was analyzed by whole mount immunohistochemistry at 72 hpf. Injection of a low concentration of Neurod ATG MO (1 ng) caused a near complete loss of glucagon expression (Fig. S8A–D), and at higher Neurod MO concentrations insulin expression was decreased (Fig. S8E–H). However knockdown of Neurod at any concentration did not affect the expression of ptf1a:EGFP (Fig. S8I–P), indicating that Neurod does not have a function in acinar cell development. Taken together, our results demonstrate that Neurod does not play a role in acinar cell development, but that differential levels of Neurod are required for the differentiation of individual endocrine cell types, whether derived from dorsal or ventral bud, and that differentiation of alpha cells is particularly sensitive to reduction in Neurod levels.
High levels of Neurod promote differentiation of glucagon-expressing cells
Our data suggest that endocrine precursors express glucagon in response to high levels of Neurod. To investigate whether exogenous expression of Neurod is sufficient to increase the number of glucagon-expressing cells, we initially microinjected neurod mRNA into one-cell stage embryos, which leads to global overexpression. However, we found that overexpression of neurod mRNA throughout the embryo caused gross morphological defects (not shown). We therefore utilized an alternative approach that allowed Neurod function to be modulated specifically within the endoderm, using cell transplantation to generate chimeric embryos in which the endoderm derives from a donor embryo, while the other germ layers derive from a host embryo (Stafford et al., 2006). In these experiments we used Tg(neurod:EGFP) donor and host embryos to follow the fate of EGFP-positive pancreatic endocrine cells (Fig. S9A, arrow). Endoderm formation in host embryos was blocked using Sox32 knockdown (Fig. S9B). By contrast, donor embryos were injected with sox32 mRNA (together with rhodamine dextran (RD) lineage tracer), causing all mesendodermal cells to take on an endoderm fate. To generate chimeric embryos we transplanted RD-labeled donor endoderm into host embryos at the blastula stage. In successful transplants the endoderm of 52 hpf host embryos was fully reconstituted by donor cells, showed normal morphology (red cells in Fig. S9C), and developed Tg(neurod:EGFP)-positive pancreatic cells (Fig. S9C, arrow). Using this strategy we either knocked down or overexpressed Neurod specifically in the donor-derived endoderm cells.
We first used a knockdown approach to confirm that Neurod functions in the endoderm germ layer to promote endocrine pancreas cell differentiation. We analyzed the fate of Tg(neurod:EGFP) positive cells in transplants where the donor was either a Neurod-positive control (Fig. 5A,D) or a Neurod-deficient morphant embryo (Fig. 5G,J). To avoid knockdown of transgene-derived EGFP expression we used Neurod ATG MO (2 ng). In control chimeric embryos, in which endoderm cells expressed Neurod at endogenous levels, both glucagon and insulin-expressing cells differentiated (Figs 5B, C, E, F); approximately 18% of the total pancreatic endocrine cells (EGFP-expressing cells) were glucagon-positive (Fig. 5S) and 42% were insulin-positive (Fig. 5T). By contrast, in experimental chimeric embryos, in which the endoderm was rendered Neurod-deficient by morpholino injection, there was an almost complete failure to differentiate glucagon-expressing cells (Fig. 5H, I, S). The number of insulin-expressing cells was also decreased by approximately 60% relative to controls (Fig. 5K, L, T). These data confirm that Neurod functions directly in the endoderm germ layer to promote endocrine cell differentiation, and, importantly, are consistent with our whole embryo knockdown experiments, confirming that glucagon-expressing cells are more sensitive to knockdown of Neurod than insulin-expressing cells.
Fig. 5. Increased Neurod levels promote alpha cell differentiation; beta cells are less sensitive than alpha cells to Neurod knockdown.

(A–R) Confocal images (merged z-stacks) of representative 52 hpf chimeric specimens in which the entire endoderm is derived from control (A–F), Neurod morphant (G–L) or neurod mRNA injected (M–R) donor cell transplants. Embryos are immunolabeled for EGFP (green; A, D, G, J, M, P) for glucagon (blue; B, H, N) and for insulin (blue; E, K, Q). EGFP and Rhodamine Dextran (red) labels donor-derived cells (A, D, G, J, M, P). Merged images with all three colors are also shown (C, F, I, L, O, R). (S) Mean (± s.d.) average number of cells expressing glucagon and EGFP-expressing endocrine cells, from a minimum of 4 chimeric embryos per group. *, P<0.002, **, P<0.01; t-test, two-tailed distribution. (T) Mean (± s.d.) average number of cells expressing insulin and EGFP-expressing endocrine cells, from a minimum of 5 chimeric embryos per group. *, P<0.004; t-test, two-tailed distribution. Scale bar = 10 μm.
We next used cell transplantation to ask if elevated Neurod levels can promote the differentiation of glucagon-expressing cells. To test this hypothesis we performed transplants using donor embryos that had been microinjected with neurod mRNA (note that, at the blastula stages when the transplants were performed, these mRNA injected embryos remained healthy). We detected a normal number of Neurod EGFP expressing cells in chimeric embryos in which the endoderm overexpressed Neurod (Fig. 5M, P, S, T). However, the percentage of endocrine cells expressing glucagon increased significantly, from 18% to 29% (compare Fig. 5B, N, S). By contrast, the percentage of endocrine cells expressing insulin decreased, from 42% to 31% (Fig. 5E, Q, T). These data suggested that the beta cell precursors might differentiate as alpha cells in the presence of excess Neurod. To test this possibility we again used a transplantation approach (Fig. S10), but for this experiment utilized Tg(mnx1:GFP) embryos in which GFP provides a marker of beta cell precursors and beta cells, with GFP colocalizing with insulin-expressing cells (Dalgin et al., 2011 and Fig. S10A–C). As expected, in control chimeric embryos Tg(mnx1:GFP)-expressing cells colabeled with insulin but not glucagon (Fig. S10D–F). As in the previous experiment, chimeric embryos over-expressing endodermal Neurod showed an increased number of glucagon-expressing cells (compare Fig. S10D, G) and a decreased number of insulin-expressing cells (compare Fig. S10E, H). However, Tg(mnx1:GFP) did not colabel glucagon-expressing cells (Fig. S10G, I), suggesting that excess Neurod does not cause beta cell precursors to take on alpha cell fate. Furthermore, we noticed that chimeric embryos overexpressing Neurod had decreased numbers of Tg(mnx1:GFP) expressing cells (mean 18±3.5 s.d. p=0.08) compared to control chimeric embryos (mean 25±6 s.d.), suggesting that fewer beta cell precursors were specified in the presence of excess Neurod. Together, these data provide strong support for our hypothesis that differentiation of specific endocrine cell types requires differential levels of Neurod. Specifically, glucagon-expressing cells require high levels of Neurod to differentiate, and elevated Neurod levels can promote differentiation of glucagon-expressing cells.
Neurod function is required for the differentiation of IPD-derived endocrine cells
Our experiments so far have indicated that Neurod function is required for embryonic endocrine cell differentiation. We wished to test whether Neurod is similarly required at larval stages for differentiation of intra-pancreatic duct (IPD)-derived secondary endocrine cells that will ultimately contribute to the secondary islets. In unmanipulated specimens, larval secondary endocrine precursor cells do not begin to develop in significant numbers until approximately three weeks post fertilization (Parsons et al., 2009). However, IPD-derived endocrine precursors are Notch-responsive cells (NRCs), and upon γ-secretase inhibition precociously produce secondary endocrine cells expressing neurod (Fig. S11A–D). To test whether IPD-derived NRCs require Neurod function to complete their differentiation program, we treated control and Neurod morphant Tg(neurod:EGFP) larvae at 3 dpf with 3μM RO4929097 (Luistro et al., 2009) (γ-secretase inhibitor hereafter) and analyzed endocrine cell differentiation at 6 dpf. We used a low dose of Neurod MO (1 ng ATG or 2 ng UTR MO) because high doses caused mortality at 6 dpf. At the stage of analysis, untreated control embryos had a large primary islet composed of glucagon and insulin-expressing cells and rare secondary Tg(neurod:EGFP)-positive endocrine precursor cells (Fig. 6A, E, I, J). Consistent with previous reports (Ninov et al., 2012; Parsons et al., 2009; Wang et al., 2011), γ-secretase inhibitor treatment caused precocious development of large numbers of IPD-derived Tg(neurod:EGFP)-positive endocrine precursor cells (mean 23.76 ± 1.15 s.d), many of which also expressed glucagon or insulin, confirming that these cells subsequently differentiated into hormone-expressing secondary endocrine cells (Fig. 6B, F, I, J). As EGFP expression from Tg(neurod:EGFP) provides a convenient marker of endocrine precursors, we utilized the Neurod ATG MO (1 ng) that targets endogenous Neurod but has insufficient sequence overlap with Tg(neurod:EGFP) to block EGFP expression. In addition, as described above we established that specimens injected with 1 ng of Neurod ATG MO had similar numbers of EGFP-expressing cells compared to controls and showed no ectopic cell death or cell proliferation defects (Fig. 3 and Fig. S4). Comparison of γ-secretase inhibitor treated Neurod ATG morphants with γ-secretase inhibitor treated control specimens showed that the average cell number (mean 22.43 ± 1.58 s.d.), and the expression levels of Tg(neurod:EGFP)-derived EGFP protein (Fig. 6B, C, F,G) and neurod mRNA (Fig. S11B, C), were similar, indicating that Neurod knockdown does not disrupt the initial capacity of IPD-NRCs to produce new neurod-expressing endocrine precursors. By contrast, in γ-secretase inhibitor treated specimens injected with Neurod UTR MO, which does block translation of EGFP transcript, we observed a significant decrease in EGFP expression (but not neurod mRNA expression Fig. S11B, D), confirming that Neurod MO injection continues to effectively block translation up to 6 dpf (Fig. 6D, H).
Fig. 6. IPD-derived secondary endocrine cells, formed in response to inhibition of Notch signaling, are sensitive to Neurod knockdown.
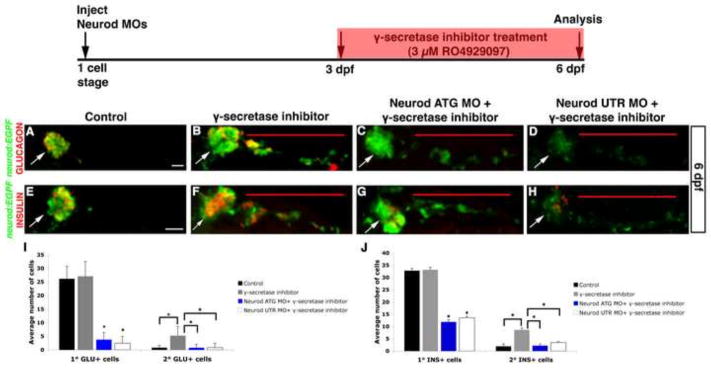
Experimental schedule (top); Tg(neurod:EGFP) control and Neurod morphant larvae were treated with 3 μM RO4929097 (γ-secretase inhibitor) from 3 to 6 dpf. Confocal images (merged z-stacks) of representative 6 dpf Tg(neurod:EGFP) untreated specimen (A, E), γ-secretase inhibitor treated specimen (B, F), γ-secretase inhibitor treated Neurod ATG morphant [1 ng] (C, G) and γ-secretase inhibitor treated Neurod UTR morphant [2ng] (D, H). Whole mount immunolabeling for GFP (green) and glucagon (red) (A–D) or for GFP (green) and insulin (red) (E–H). Mean (± s.d.) number of cells expressing glucagon (I) or insulin (J) from 4 independent experiments and from a minimum of 40 and 25 larvae per group, respectively. *, P<0.0001; t-test, two-tailed distribution. Primary endocrine islet (arrow), IPD-derived endocrine cells (red line). Magnification is different in panel A than in B–H. Scale bar = 20 μm.
Importantly, the differential sensitivity of glucagon and insulin-expressing cells to knockdown of Neurod was maintained in both the expanding primary islet and in the precociously forming secondary endocrine cells (Fig. 6C, D, G, H). While Neurod-deficient specimens retained the ability to produce precocious secondary endocrine precursors in response to Notch inhibition, these cells largely failed to express glucagon (Fig. 6C, D, I), although they did express arxa (Fig. S11K, L), and only a small number of cells expressed insulin (Fig. 6G, H, J). These data indicate that Neurod function is required for differentiation of IPD-derived endocrine cells upon inhibition of Notch signaling, and support the hypothesis that normal endocrine pancreas cell differentiation is dependent upon differential levels of Neurod at both embryonic and larval stages. We conclude that Neurod plays similar roles in the differentiation of early endocrine cells that contribute to the primary islet and in those that develop later from the IPD to contribute to secondary islets.
Neurod morphants show precocious endocrine precursor development yet cannot maintain normal glucose levels
As noted above, early larvae produce only a few IPD-derived secondary endocrine precursor cells in the absence of Notch signaling inhibition. However, we noted that Neurod-deficient 6 dpf Tg(neurod:EGFP) larvae showed a significant increase (approximately 4-fold) in the number of EGFP-positive secondary endocrine precursor cells when compared to unmanipulated controls (Fig. 7A, B, G, H, I). While in both unmanipulated and Neurod-deficient specimens a negligible number of secondary endocrine precursor cells have differentiated into hormone-expressing cells at 6 dpf (Fig. 7A–F), in unmanipulated specimens there is a significant increase in the number of differentiated endocrine cells within the primary islet between 3 and 6 dpf (compare Fig. 4E, J with Fig. 7I). Within the 6 dpf primary islet, as at 3 dpf, Neurod knockdown continues to cause a dramatic reduction in the number of glucagon-expressing cells (Fig. 7E, F, I), as well as a significant reduction in the number of insulin-expressing cells (Fig. 7C, D, I), again consistent with continuing efficacy of Neurod morpholino knockdown.
Fig. 7. Neurod knockdown increases production of secondary endocrine precursor cells.

Confocal images (merged z-stacks) of representative 6 dpf Tg(neurod:EGFP) Control (A, C, E, G) and Neurod ATG morphant (B, D, F, H) specimens. Whole mount immunolabeling for insulin (C, D), glucagon (E, F) and GFP (G, H). (I) Mean (± s.d.) number of cells expressing insulin, glucagon, GFP-positive secondary endocrine cells from 6 independent experiments and from a minimum of 20 larvae per group. *, P<0.0001; t-test, two-tailed distribution. Secondary endocrine precursor cells; arrowhead. Scale bar = 20 μm.
We hypothesized that the induction of unusually high numbers of Tg(neurod:EGFP)-expressing endocrine precursor cells from the IPD might be a response to functional impairment of the primary islet. To assess islet function we analyzed free glucose levels in control and Neurod-deficient specimens as previously described (Andersson et al., 2012; Gut et al., 2012; Jurczyk et al., 2011). The Neurod UTR MO was injected at three different doses into Tg(neurod:EGFP) embryos. At 3 dpf, the efficacy of Neurod knockdown was confirmed by screening for a decrease in EGFP expression, and specimens were then collected for free glucose analysis. Neurod-deficient specimens displayed higher levels of free glucose than control specimens (Fig. 8B). Free glucose levels increased in specimens injected with 4 ng versus 2 ng of Neurod UTR MO, but we found similar free glucose levels in specimens injected with 4 ng or 8 ng of morpholino. These measurements were consistent with our finding that although specimens injected with 8 ng Neurod UTR MO had fewer insulin-expressing cells than specimens injected with 4ng Neurod UTR MO their insulin expression levels remained similar (Fig. 4P). These findings support our hypothesis that the spared insulin-expressing cells might be producing more insulin to compensate for reduced beta cell numbers. To confirm a correlation between decreased beta cell number and elevated free glucose levels we made use of Mnx1-deficient embryos, which we have previously shown fail to express insulin and lack beta cells in the primary islet (Dalgin et al., 2011). We predicted that Mnx1-deficient specimens should have higher free glucose levels than Neurod-deficient specimens. Consistent with expectations, 3 dpf Mnx1-deficient specimens showed ~5 and ~2.5-fold higher free glucose levels than control and Neurod-deficient specimens, respectively (Fig. 8B). These results confirm previous findings that Mnx1-deficient zebrafish lack functional beta cells, and verify that at 3 dpf the primary endocrine islet is already functional. Together, these data strongly suggest that decreased insulin expression and decreased beta cell numbers in Neurod morphants (Fig. 4F–J) lead to impaired glucose homeostasis.
Fig. 8. Free glucose levels gradually increase in Neurod morphants.

(A) Experimental schedule. Morpholino injected Tg(neurod:EGFP) specimens were collected for free glucose assays at 3, 5, 7 and 9 dpf (red arrows). After 5 dpf, larvae were fed twice daily, and specimens were collected before feeding time on the appropriate days. (B) Free glucose levels in 3 dpf Tg(neurod:EGFP) control, Neurod morphant and Mnx1 morphant specimens (MO concentrations as indicated). Mean (± s.e.m) glucose levels from 4 independent experiments. (C) Free glucose levels from 5, 7 and 9 dpf Tg(neurod:EGFP) control and Neurod MO injected specimens. As free glucose levels were similar in Neurod ATG and UTR morpholino injected specimens these are presented together. Mean (± s.e.m) glucose levels from 4 independent experiments. *, P<0.15, **, P<0.04, ***, P<0.009; t-test, two-tailed distribution.
In the first five days of zebrafish development the yolk is the source of nutrition, after which feeding is required for normal development to proceed. Only those specimens injected with low levels of Neurod MO survived to feeding stages. To determine if Neurod function is required during feeding stages we raised specimens injected with Neurod MO (1 ng ATG MO or 2 ng UTR MO injected specimens) for up to 9 dpf. We confirmed Neurod knockdown by the expected decrease in EGFP, glucagon and insulin expression in Neurod MO injected specimens (data not shown). At 5 dpf, following the collection of some specimens for free glucose assays, we commenced twice daily feeding of the larvae (Fig. 8A). For subsequent analyses, specimens were collected at 7 or 9 dpf, before the normal feeding time, and free glucose levels were measured. Control larvae displayed an increase in free glucose levels between 5 and 7 dpf, however, free glucose levels dropped by 9 dpf (Fig. 8C). These data suggest that control larvae begin to efficiently regulate glucose by 9 dpf. In contrast, free glucose levels in Neurod-deficient specimens continued to increase after 7 dpf (Fig. 8C; ATG or UTR MO displayed similar results). We conclude that Neurod-deficient specimens fail to maintain normal glucose levels, consistent with decreased insulin expression and decreased beta cell numbers at feeding stages (Fig. 8 and data not shown).
Normalization of free glucose level in Neurod morphants is not sufficient to prevent precocious endocrine precursor development
Our data suggest that decreased levels of endocrine hormone expression in the primary islet and/or increased free glucose levels in the larva may be the primary cause of precocious endocrine precursor development in the IPD. To investigate the role of elevated free glucose levels we treated control and Neurod morphant larvae with the competitive sodium-glucose-transporter (sglt1 and 2) inhibitor phlorizin. Phlorizin blocks reabsorption of glucose from the kidney and small intestine, thereby facilitating the excretion of glucose in the urine (Ehrenkranz et al., 2005). Tg(neurod:EGFP) control and 1 ng Neurod ATG MO injected specimens were treated with 250 μg/ml of phlorizin at 5.5 dpf for 16 hours (Fig. 9A). Phlorizin treated control specimens had similar free glucose levels to untreated controls (Fig. 9B). In contrast, while Neurod morphants showed a significant increase in free glucose levels compared to controls, phlorizin treatment normalized these glucose levels back to control levels (Fig. 9B). We next analyzed IPD-derived cell differentiation, to determine if normalized glycemic conditions would prevent precocious precursor development in Neurod morphants treated with phlorizin. Untreated and phlorizin treated control specimens had similar numbers of secondary endocrine cells (Fig. 9C–E), and the expression of glucagon and insulin in the primary islet of phlorizin treated control specimens was normal (Fig. 9H, I, L, M, P, Q). In contrast, both untreated and phlorizin treated Neurod morphant specimens had decreased glucagon and insulin expression in the primary islet (Fig. 9J, K, N, O, R, S) and the number of secondary endocrine cells in these specimens increased ~4 fold (Fig. 9C, F, G). These data establish that phlorizin treatment is able to normalize glucose levels in Neurod morphants, yet the normal glycemic conditions are not sufficient to prevent precocious precursor development.
Fig. 9. Neurod morphants treated with sodium glucose cotransporter inhibitor retain increased production of secondary endocrine cells.

(A) Experimental schedule. 1 ng of Neurod ATG MO injected Tg(neurod:EGFP) specimens were treated with 250 μg/ml Phloridzin (Phz) at 5.5 dpf for 16 hours and collected for free glucose assay. (B) Free glucose levels in 6 dpf Tg(neurod:EGFP) untreated and Phz treated control and Neurod morphant specimens. Phz treatment normalized glucose levels in Neurod morphants. Mean (± s.e.m) glucose levels from 4 independent experiments. (C) Mean (± s.d.) number of cells expressing GFP-positive secondary endocrine cells from 4 independent experiments and from a minimum of 16 larvae per group. Confocal images (merged z-stacks) of representative 6 dpf Tg(neurod:EGFP) control (D, H, L, P), control treated with phlorizin (Phz) (E, I, M, Q), Neurod ATG morphant (F, J, N, R) and Neurod ATG morphant treated with Phz (G, K, O, S). Whole mount immunolabeling for GFP (green, D–G), glucagon (red, H–K), insulin (blue, L–O) and merged images with nuclear marker DAPI (P–S). *, P<0.007, **, P<0.0005; t-test, two-tailed distribution. Secondary endocrine precursor cells; arrowhead. White scale bar = 10 μm.
gRNA/Cas9 induced disruption of the endogenous neurod locus significantly reduces endocrine hormone expression
Recently, clustered regularly interspaced short palindromic repeats (CRISPR) associated (Cas) has been shown to function as a powerful mutagenesis system in many species, including zebrafish (Doudna and Charpentier, 2014). We used this genome editing technology to confirm the critical role of Neurod in zebrafish endocrine cell development. We selected two different neurod target sites for single guide (sg) RNA/Cas9 binding (Fig. S12), and found that the first of these sites (indicated in blue, Fig. 10D and Fig. S12) efficiently generated mutations.
Fig. 10. Neurod sgRNA/Cas9 mediated transient mutagenesis phenocopies the endocrine cell defects found in Neurod morphants.

(A) Control and (B) neurod sgRNA/Cas9 injected specimens at 48 hpf. No gross morphological defects were observed. (C) T7 endonuclease I assay (T7EI) assay. (Lane 1) marker, (lane 2) untreated and (lane 3, 4) T7EI treated amplicons from control embryos. (Lane 5–15) Amplicons from embryos injected with neurod sgRNA/Cas9 were digested in varying ratios by T7EI enzyme. Asterisks indicate the PCR product that was TOPO cloned and sequenced. (D) The sequence alignment of wild type (wt), insertion (18/20, red) and deletion (2/20, green) mutations recovered from sequenced clones. Neurod sgRNA genomic target sequence (blue), protospacer adjacent motif-PAM sequence (pink). (E) Relative levels of gcga, insa, sst2, neurod and cp by real-time qPCR. Results are from 3 independent experiments and from 2 technical replicas. All values were normalized to beta-actin levels. Primer sequences are listed in Table S1.
The neurod sgRNA/Cas9 injected specimens were morphologically indistinguishable at 48 hpf from uninjected controls (Fig. 10A, B), indicating that the reagents were not toxic. We randomly selected two control and 11 experimental embryos injected with neurod sgRNA/Cas9 (blue sgRNA sequence; Fig. 10D and Fig. S12) from three independent experiments for genomic DNA extraction at 48 hpf, followed by PCR amplification of a 494 bp region flanking the neurod sgRNA/Cas9 target sites (Fig. S12). PCR products were then assayed with T7 endonuclease I (T7EI) to identify mismatches diagnostic of insertion-deletion (indel) mutations. As expected the amplicons obtained from control specimens were not cleaved by T7EI enzyme, however, nearly all amplicons obtained from neurod sgRNA/Cas9 injected specimens were cleaved by T7EI enzyme, consistent with sgRNA/Cas9 injection effectively generating transient indel mutations (Fig. 10C). To determine the range of indel mutations generated in a single embryo, we subcloned PCR products (asterisk, Fig. 10C) into pCR II-TOPO vector and sequenced multiple individual neurod amplicons. All the sequenced neurod alleles (20/20) had insertions and/or deletions (Fig. 10D) confirming the results of our T7EI assay. In comparison, the second sgRNA we designed against the neurod locus (green, Fig. S12) only produced four mutant alleles out of 44 sequenced amplicons (data not shown).
We next performed qRT-PCR analysis to quantify gene transcription in neurod sgRNA/Cas9 (blue sequence) injected specimens relative to sibling controls. qRT-PCR from three independent experiments showed that expression of neurod decreased 2.5 fold (Fig. 10E), suggesting that sgRNA/Cas9 induced mutations caused nonsense mediated decay (Chang et al., 2007). Consistent with this prediction, 16/20 mutant alleles generated premature stop codons (data not shown). Analysis of endocrine hormone markers showed that expression of gcga, insa and sst2 decreased ~2.5, 1.9 and 2-fold respectively. To investigate possible off-target effects of the neurod gRNA/Cas9 mRNAs we assayed expression of the liver gene ceruloplasmin (cp) in injected specimens. qRT-PCR analysis from the same three independent experiments showed similar levels of cp expression in control and neurod sgRNA/Cas9 injected specimens (Fig. 10E), consistent with specific gene targeting. We note that injections with high doses of Neurod morpholino caused more dramatic decreases in endocrine gene expression than observed in the neurod sgRNA/Cas9 generated transient mutants, but suggest that this likely reflects the incomplete and variant nature of sgRNA/Cas9 generated mutations in the F0 generation. Overall, our genome editing data corroborate our morpholino knockdown experiments and confirm that Neurod function is necessary for endocrine gene transcription.
Discussion
We have shown that transcription factor Neurod plays a critical role in zebrafish endocrine pancreas cell differentiation, and that different levels of Neurod are required for differentiation of distinct hormone-expressing endocrine cell types. Specifically, differentiation of alpha versus beta cells requires higher versus lower levels of Neurod. Morpholino knockdown findings were corroborated by two additional experimental approaches. First, we used endoderm-specific gene modulation (both knockdown and overexpression) to confirm that different levels of Neurod function directly in the endoderm to promote alpha versus beta cell fates. Second, we generated sgRNA/Cas9 mediated transient mutations in neurod to confirm the requirement for Neurod in endocrine cell differentiation. We went on to show that Neurod function is required for the differentiation of both primary and secondary islet cells. Our analysis further suggests that decreased endocrine hormone expression in the primary islet of Neurod-deficient specimens triggers premature endocrine precursor production from the IPD. However, these precociously formed endocrine precursors do not complete their differentiation and are therefore unable to normalize free glucose levels in larval zebrafish.
The neurod transgenic reporter line Tg(neurod:EGFP) (Obholzer et al., 2008), contributed to this study in two important ways. First, EGFP transgene expression provided a convenient proxy for levels of target protein expression. Second, by using an alternative morpholino that did not target the transgene, we were able to track the fate of Neurod-expressing cells when functional Neurod protein was depleted. The continued expression of Tg(neurod:EGFP) in Neurod morphants implies that Neurod function is not required for specification of endocrine precursors, but rather provides a marker for these precursors, consistent with previous reports (Dalgin et al., 2011). Consistent with this model, expression of Neurod throughout all endoderm does not cause ectopic Tg(neurod:EGFP) expression, confirming that Neurod function is not sufficient to specify ectopic endocrine cells.
Complete zebrafish pancreas development requires the contribution of cells from both the dorsal and ventral buds. We have shown that differentiation of endocrine cells from either of these buds requires Neurod function. In both cases, differentiation of glucagon-positive alpha cells is more sensitive to reduced function of Neurod than differentiation of insulin-positive beta cells. Our analysis of Neurod-deficient Tg(neurod:EGFP) and Tg(sox17:EGFP)-expressing cells in the dorsal bud revealed that neither cell death nor proliferation rates are detectably affected by Neurod deficiency. We therefore conclude that the reduced numbers of hormone-positive cells present in Neurod-deficient specimens are primarily consequence of a block to the differentiation of endocrine precursors: alpha cell precursors remain undifferentiated in response to partial Neurod knockdown, whereas other endocrine cell types remain undifferentiated only in response to a more complete knockdown. In this study we used pH3 immunohistochemistry to reveal a snap shot of the proliferating endocrine cells in the dorsal bud of control and Neurod morphants. Our results are consistent with those of Hesselson et al., 2009, who detected similar low proliferation rates using EDU and genetic labeling of dorsal bud cells. Recently, two distinct stages of beta cell proliferation were described at 36 hpf and 120 hpf using live imaging with the transgenic fluorescent ubiquitylation-based cell cycle indicator (FUCCI) (Tsuji et al., 2014). Beta cell proliferation rates detected with this advanced system are elevated in comparison to those we detected with pH3 labeling; in the future it would be interesting to use this new technology to confirm endocrine cell proliferation rates are unaffected by Neurod levels.
We also observed a modest decrease in the number of Tg(neurod:EGFP)-expressing cells in response to injection of high doses of Neurod-ATG-MO. Possible explanations for this include a difficult to detect brief episode of cell death, or a requirement for a low level of Neurod function in order to maintain neurod expression (auto-regulation). Consistent with the hypothesis of auto-regulation, neurod transcript level, as detected by in situ hybridization, are reduced in response to high doses of Neurod-UTR-MO injection (data not shown). Ultimately, a full answer to this question will require additional experimental approaches, including the establishment of neurod null mutant lines, for example by using our sgRNA/Cas9 mutagenesis approach.
Alpha cell differentiation in mice and zebrafish requires the homeobox transcription factor Arx (Collombat et al., 2003; Djiotsa et al., 2012). However, while partial knockdown of zebrafish Neurod is sufficient to abrogate glucagon expression in the dorsal bud, arxa continues to be expressed at normal levels. We similarly observed that arxa expression is retained in IPD-derived endocrine cells (Fig. S10I–L), where again glucagon expression is abrogated. These findings suggest either that arxa expression is insufficient to activate glucagon expression in zebrafish, or alternatively, that arxa is initially expressed in multiple endocrine lineages, with only later expression becoming restricted to glucagon-expressing cells. Detailed molecular and genetic analysis of arxa will be required to address these possibilities in zebrafish.
By using cell transplantation to generate chimeric embryos we confirmed that high levels of endodermal Neurod expression promote differentiation of alpha cells. Chimeric embryos overexpressing Neurod in the endoderm produce normal numbers of Tg(neurod:EGFP) expressing endocrine precursor cells, but the relative proportions of differentiated hormone-expressing cells are altered. Specifically, elevated Neurod expression increases the number of glucagon-expressing cells but decreases the number of insulin-expressing cells. A similar role has recently been reported for differential levels of Neurod within the developing zebrafish posterior lateral line (PLL) (Sato and Takeda, 2013). In this group of migrating neurons there are two classes of cells, leaders and followers, which express high and low levels of Neurod protein, respectively. Overexpression of Neurod in a single PLL neuron precursor is sufficient to promote its differentiation into the leader cell type (Sato and Takeda, 2013). Our immunohistochemistry has shown that the individual cells of the dorsal endocrine pancreatic bud similarly display heterogeneous Neurod protein levels (Fig. 1E inset and Fig. S2E). Our observed loss of glucagon-expressing alpha cells in response to partial depletion of Neurod protein, and the reciprocal gain of glucagon-expressing alpha cells from precursors in which Neurod expression has been elevated, are consistent with a model in which glucagon expression is dependent on high Neurod protein levels.
Differential levels of Neurod expression in specific precursor cell populations might be achieved in a variety of ways. For example, all endocrine cell types may transcribe similar levels of neurod mRNA, with post-transcriptional regulation leading to varying levels of Neurod protein in distinct cells. Such post-transcriptional regulation could be microRNA dependent: overexpression of the murine beta cell microRNA (miR)-30a-5p directly suppresses expression of NeuroD to induce beta cell dysfunction (Kim et al., 2013). An alternative possibility is that activation of different enhancers controlling neurod transcription could allow differential Neurod levels to arise in distinct endocrine cell types. Our analysis of phylogenetically conserved sequences has revealed multiple putative enhancer regions that might regulate Neurod transcription (not shown). In preliminary experiments we have identified a regulatory region that is first activated in beta-cells (GD unpublished, to be discussed elsewhere). In the future, a thorough analysis will be necessary to address how differential levels of Neurod are activated in the endocrine precursors.
Notch-responsive cells (NRCs) in the duct are a source of endocrine precursor cells in both mouse and zebrafish (Apelqvist et al., 1999; Jensen et al., 2000; Kopinke et al., 2011; Ninov et al., 2012; Parsons et al., 2009; Wang et al., 2011). Our results reveal that cell fate decisions are determined at least in part by differential levels of Neurod expression after Notch signaling is inhibited in the newly formed endocrine precursors. Notch mediated suppression of cellular differentiation occurs through activation of Hes1, which in turn inhibits expression of bHLH proteins (Davis and Turner, 2001; Jarriault et al., 1998). Both neurog3 and neurod encode bHLH proteins and are thus potential Notch targets in the developing pancreas. Consistent with this prediction we have shown that Notch signaling inhibition increases neurod expression (Fig. S11A–D). Similarly, zebrafish mind bomb mutants, which have disrupted Delta-mediated Notch signaling, show strongly elevated neurod expression (Zecchin et al., 2007). The human NEUROG3 gene has multiple HES1 binding sites and is directly repressed by HES1 (Lee et al., 2001). As the zebrafish neurog3 mutant does not have endocrine pancreatic defects (Flasse et al., 2013), it will be especially important to address in future studies whether zebrafish neurod is directly regulated by Hes1 in the IPD.
Our free glucose analysis indicated that Neurod morphants are unable to maintain normal glucose levels. This may indicate that the reduced number of beta cells that differentiate in Neurod morphants is insufficient to produce sufficient insulin, or alternatively, that those beta cells that do form lose functionality as they mature. Consistent with the latter hypothesis, mouse NeuroD1 is required to maintain functional maturity of beta cells (C. Gu et al., 2010). We have also found that in Neurod-deficient larvae there is a robust increase in progenitor maturation from the IPD. It has recently been shown that beta cell differentiation from the zebrafish larval IPD also increases in response to high nutrient conditions, with mTOR signaling implicated in the activation of IPD-Notch responsive cells (Ninov et al., 2012). Glucose metabolism within beta cells has been proposed as a signal to induce beta cell replication in mammals (Dadon et al., 2012), and in vitro experiments using cultured rodent pancreatic rudiments have shown that glucose can control beta cell differentiation by regulating expression of NeuroD (Guillemain et al., 2007). While these previous reports suggest that elevated glucose levels might stimulate progenitor maturation from the IPD, our experiments using phlorizin to normalize glucose levels do not support this hypothesis: normalizing glucose levels in Neurod morphants does not prevent precocious endocrine precursor production from the IPD. In these experiments, in which we manipulated Neurod levels at larval zebrafish stages, our partial knockdown likely recapitulates a hypomorphic mutant condition. Thus we propose that reduced primary islet hormone levels and/or sustained defects caused by reduced levels of Neurod, promote new endocrine precursor production from the IPD at stages when beta cells begin to function in metabolism.
In conclusion, our results have revealed that activation of differential levels of Neurod plays an important role in the determination of endocrine cell fate choice. Flasse et al. (2013) recently reported that Neurod and another Notch-regulated bHLH protein, Ascl1b, play complementary roles in zebrafish endocrine cell development. This previous work (Flasse et al. 2013) used a relatively low dose of Neurod MO (3 ng), which we would expect to cause loss of only glucagon expression from the dorsal bud. However, when Neurod and Ascl1b genes were simultaneously knocked down a complete loss of dorsal bud-derived endocrine cell types resulted. Such findings indicate that multiple factors are involved in endocrine cell differentiation, with Neurod functioning as one component of a broader gene network comprising both cell-intrinsic factors and cell-extrinsic signals that ultimately establish specific endocrine fates. We previously showed that Mnx1 is required for differentiation of beta cell fate. Thus, we predict that while low levels of Neurod together with Mnx1 are required for beta cell differentiation, high levels of Neurod together with as yet unidentified factors might be required for alpha cell differentiation. Similarly, Sussel and colleagues have suggested that the combined functions of transcription factors Nkx2.2 and NeuroD1 regulate the fate of multiple endocrine cell types in mice (Mastracci et al., 2013). Our results show a role for Neurod in both primary and secondary endocrine cell differentiation, with Neurod functioning in an equivalent fashion in both instances. Equivalent mechanisms do not always act in primary and secondary cells, as recently shown for retinoic acid (RA) signaling, which promotes primary endocrine cell development from the dorsal pancreatic bud yet inhibits secondary endocrine cell development from the IPD (Rovira et al., 2011). Recently, Matsuda and colleagues additionally showed that RA activity controls IPD cell commitment and maturation (Matsuda et al., 2013). However, our data reveal that subsequent to endocrine cell commitment Neurod activity is required to differentiate distinct endocrine cell types. A more complete understanding of the molecular processes that underlie both primary and secondary endocrine cell differentiation will be helpful in the establishment of in vitro protocols for the differentiation of endocrine cells for use in cell-based therapies.
Supplementary Material
Highlights.
Alpha versus beta cell differentiation requires higher versus lower Neurod levels
Neurod deficiency promotes precocious endocrine precursor development in the IPD
Neurod-deficient larvae cannot maintain normal glucose levels
Precocious endocrine cell development is not due to increased glucose levels
sgRNA/Cas9 mediated transient neurod mutagenesis confirms morpholino results
Acknowledgments
We thank members of the Prince laboratory for helpful advice and discussions, Anita Ng for expert fish care, Dr. Masahiko Hibi for Neurod antibody, Dr. Ryan M. Anderson for H2B-RFP plasmid and Dr. Philipp Gut for technical suggestions on the free glucose assay. We are also grateful to Drs Graeme Bell, Barton Wicksteed, Robert K Ho, Stefani Eames Nalle, Devorah Goldman, Sarah Wanner and Alana Beadell for helpful comments on the manuscript. This work is supported by the National Institutes of Health [grant DK064973 to V.E.P]; and in part by a P&F award from the University of Chicago Diabetes Research Center (P30 DK020595).
Footnotes
Author contributions
G.D. conceived, designed and performed experiments, collected and interpreted data, and wrote the manuscript. V.E.P. conceived experiments, interpreted data, and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson O, Adams BA, Yoo D, Ellis GC, Gut P, Anderson RM, German MS, Stainier DYR. Adenosine Signaling Promotes Regeneration of Pancreatic β Cells In Vivo. Cell Metab. 2012;15:885–894. doi: 10.1016/j.cmet.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelqvist A, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol. 2006;Chapter 15(Unit 15.8) doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- Chang Y-F, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B. Dynamic Imaging of Genomic Lociin Living Human Cells by an Optimized CRISPR/Cas System. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Mansouri A, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadon D, Tornovsky-Babaey S, Furth-Lavi J, Ben-Zvi D, Ziv O, Schyr-Ben-Haroush R, Stolovich-Rain M, Hija A, Porat S, Granot Z, Weinberg-Corem N, Dor Y, Glaser B. Glucose metabolism: key endogenous regulator of β-cell replication and survival. Diabetes Obes Metab. 2012;14(Suppl 3):101–108. doi: 10.1111/j.1463-1326.2012.01646.x. [DOI] [PubMed] [Google Scholar]
- Dalgin G, Ward AB, Hao LT, Beattie CE, Nechiporuk A, Prince VE. Zebrafish mnx1 controls cell fate choice in the developing endocrine pancreas. Development. 2011;138:4597–4608. doi: 10.1242/dev.067736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Turner DL. Vertebrate hairy and Enhancer of split related proteins: transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene. 2001;20:8342–8357. doi: 10.1038/sj.onc.1205094. [DOI] [PubMed] [Google Scholar]
- Djiotsa J, Verbruggen V, Giacomotto J, Ishibashi M, Manning E, Rinkwitz S, Manfroid I, Lvoz M, Peers B. Pax4 is not essential for beta-cell differentiation in zebrafish embryos but modulates alpha-cell generation by repressing arx gene expression. BMC Dev Biol. 2012;12:37. doi: 10.1186/1471-213X-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong PDS, Provost E, Leach SD, Stainier DYR. Graded levels of Ptf1a differentially regulate endocrine and exocrine fates in the developing pancreas. Genes Dev. 2008;22:1445–1450. doi: 10.1101/gad.1663208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science (New York, NY) 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Ehrenkranz JRL, Lewis NG, Ronald Kahn C, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- Field HA, Dong PDS, Beis D, Stainier DYR. Formation of the digestive system in zebrafish. II. Pancreas morphogenesis. Dev Biol. 2003;261:197–208. doi: 10.1016/s0012-1606(03)00308-7. [DOI] [PubMed] [Google Scholar]
- Flasse LC, Pirson JL, Stern DG, Von Berg V, Manfroid I, Peers B, Voz ML. Ascl1b and Neurod1, instead of Neurog3, control pancreatic endocrine cell fate in zebrafish. BMC Biol. 2013;11:78. doi: 10.1186/1741-7007-11-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho L, Mumm JS, Williams PR, Schroeter EH, Koerber A, Park SW, Leach SD, Wong ROL. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 2005;132:5069–5079. doi: 10.1242/dev.02075. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Stein GH, Pan N, Goebbels S, Hornberg H, Nave KA, Herrera P, White P, Kaestner KH, Sussel L. Pancreatic β Cells Require NeuroD to Achieve and Maintain Functional Maturity. Cell Metab. 2010;11:298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Guillemain G, Filhoulaud G, Da Silva-Xavier G, Rutter GA, Scharfmann R. Glucose is necessary for embryonic pancreatic endocrine cell differentiation. J Biol Chem. 2007;282:15228–15237. doi: 10.1074/jbc.M610986200. [DOI] [PubMed] [Google Scholar]
- Guo X, Zhang T, Hu Z, Zhang Y, Shi Z, Wang Q, Cui Y, Wang F, Zhao H, Chen Y. Efficient RNA/Cas9-mediated genome editing in Xenopus tropicalis. Development. 2014;141:707–714. doi: 10.1242/dev.099853. [DOI] [PubMed] [Google Scholar]
- Gut P, Baeza-Raja B, Andersson O, Hasenkamp L, Hsiao J, Hesselson D, Akassoglou K, Verdin E, Hirschey MD, Stainier DYR. Whole-organism screening for gluconeogenesis identifies activators of fasting metabolism. Nat Chem Biol. 2012 doi: 10.1038/nchembio.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselson D, Anderson RM, Beinat M, Stainier DYR. Distinct populations of quiescent and proliferative pancreatic beta-cells identified by HOTcre mediated labeling. Proceedings of the National Academy of Sciences. 2009;106:14896–14901. doi: 10.1073/pnas.0906348106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho RK, Kane DA. Cell-autonomous action of zebrafish spt-1 mutation in specific mesodermal precursors. Nature. 1990;348:728–730. doi: 10.1038/348728a0. [DOI] [PubMed] [Google Scholar]
- Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol. 2000;20:3292–3307. doi: 10.1128/mcb.20.9.3292-3307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Le Bail O, Hirsinger E, Pourquié O, Logeat F, Strong CF, Brou C, Seidah NG, Isra l A. Delta-1 activation of notch-1 signaling results in HES-1 transactivation. Mol Cell Biol. 1998;18:7423–7431. doi: 10.1128/mcb.18.12.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- Jurczyk A, Roy N, Bajwa R, Gut P, Lipson K, Yang C, Covassin L, Racki WJ, Rossini AA, Phillips N, Stainier DYR, Greiner DL, Brehm MA, Bortell R, diIorio P. Dynamic glucoregulation and mammalian-like responses to metabolic and developmental disruption in zebrafish. General and Comparative Endocrinology. 2011;170:334–345. doi: 10.1016/j.ygcen.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kani S, Bae YK, Shimizu T, Tanabe K, Satou C, Parsons MJ, Scott E, Higashijima SI, Hibi M. Proneural gene-linked neurogenesis in zebrafish cerebellum. Dev Biol. 2010;343:1–17. doi: 10.1016/j.ydbio.2010.03.024. [DOI] [PubMed] [Google Scholar]
- Kim J-W, You Y-H, Jung S, Suh-Kim H, Lee I-K, Cho J-H, Yoon K-H. miRNA-30a-5p-mediated silencing of Beta2/NeuroD expression is an important initial event of glucotoxicity-induced beta cell dysfunction in rodent models. Diabetologia. 2013 doi: 10.1007/s00125-012-2812-x. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kinkel MD, Prince VE. On the diabetic menu: zebrafish as a model for pancreas development and function. Bioessays. 2009;31:139–152. doi: 10.1002/bies.200800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D, Brailsford M, Shea JE, Leavitt R, Scaife CL, Murtaugh LC. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development. 2011;138:431–441. doi: 10.1242/dev.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Smith SB, Watada H, Lin J, Scheel D, Wang J, Mirmira RG, German MS. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes. 2001;50:928–936. doi: 10.2337/diabetes.50.5.928. [DOI] [PubMed] [Google Scholar]
- Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, Roberts J, Cai J, Berkofsky-Fessler W, Hilton H, Linn M, Flohr A, Jakob-Røtne R, Jacobsen H, Glenn K, Heimbrook D, Boylan JF. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Research. 2009;69:7672–7680. doi: 10.1158/0008-5472.CAN-09-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastracci TL, Anderson KR, Papizan JB, Sussel L. Regulation of neurod1 contributes to the lineage potential of neurogenin3+ endocrine precursor cells in the pancreas. PLoS Genet. 2013;9:e1003278. doi: 10.1371/journal.pgen.1003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H, Parsons MJ, Leach SD. Aldh1-Expressing Endocrine Progenitor Cells Regulate Secondary Islet Formation in Larval Zebrafish Pancreas. PLoS ONE. 2013;8:e74350. doi: 10.1371/journal.pone.0074350.s002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellitzer G, Martín M, Sidhoum-Jenny M, Orvain C, Barths J, Seymour PA, Sander M, Gradwohl G. Pancreatic islet progenitor cells in neurogenin 3-yellow fluorescent protein knock-add-on mice. Molecular Endocrinology. 2004;18:2765–2776. doi: 10.1210/me.2004-0243. [DOI] [PubMed] [Google Scholar]
- Mizoguchi T, Verkade H, Heath J, Kuroiwa A, Kikuchi Y. Sdf1/Cxcr4 signaling controls the dorsal migration of endodermal cells during zebrafish gastrulation. Development. 2008:2521–2529. doi: 10.1242/dev.020107. [DOI] [PubMed] [Google Scholar]
- Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–7. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninov N, Borius M, Stainier DYR. Different levels of Notch signaling regulate quiescence, renewal and differentiation in pancreatic endocrine progenitors. Development. 2012;139:1557–1567. doi: 10.1242/dev.076000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obholzer N, Wolfson S, Trapani JG, Mo W, Nechiporuk A, Busch-Nentwich E, Seiler C, Sidi S, Söllner C, Duncan RN, Boehland A, Nicolson T. Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J Neurosci. 2008;28:2110–2118. doi: 10.1523/JNEUROSCI.5230-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MJ, Pisharath H, Yusuff S, Moore JC, Siekmann AF, Lawson N, Leach SD. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mechanisms of Development. 2009;126:898–912. doi: 10.1016/j.mod.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira M, Huang W, Yusuff S, Shim JS, Ferrante AA, Liu JO, Parsons MJ. Chemical screen identifies FDA-approved drugs and target pathways that induce precocious pancreatic endocrine differentiation. Proceedings of the National Academy of Sciences. 2011;108:19264–19269. doi: 10.1073/pnas.1113081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Minton JAL, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous Mutations in NEUROD1 Are Responsible for a Novel Syndrome of Permanent Neonatal Diabetes and Neurological Abnormalities. Diabetes. 2010;59:2326–2331. doi: 10.2337/db10-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin AF, Villablanca EJ, Nuñez VA, Sandoval PC, Ghysen A, Allende ML. Proneural gene requirement for hair cell differentiation in the zebrafish lateral line. Dev Biol. 2006;295:534–545. doi: 10.1016/j.ydbio.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Sato A, Takeda H. Neuronal Subtypes Are Specified by the Level of neurod Expression in the Zebrafish Lateral Line. J Neurosci. 2013;33:556–562. doi: 10.1523/JNEUROSCI.4568-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhoff SE, Giel-Moloney M, Leiter AB. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol. 2004;270:443–454. doi: 10.1016/j.ydbio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Stafford D, White RJ, Kinkel MD, Linville A, Schilling TF, Prince VE. Retinoids signal directly to zebrafish endoderm to specify insulin-expressing beta-cells. Development. 2006;133:949–956. doi: 10.1242/dev.02263. [DOI] [PubMed] [Google Scholar]
- Tsuji N, Ninov N, Delawary M, Osman S, Roh AS, Gut P, Stainier DYR. Whole Organism High Content Screening Identifies Stimulators of Pancreatic Beta-Cell Proliferation. PLoS ONE. 2014;9:e104112. doi: 10.1371/journal.pone.0104112.s002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rovira M, Yusuff S, Parsons MJ. Genetic inducible fate mapping in larval zebrafish reveals origins of adult insulin-producing β-cells. Development. 2011;138:609–617. doi: 10.1242/dev.059097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book: A guide for the laboratory use of zebrafish (Danio rerio) 3. University of Oregon; 1995. [Google Scholar]
- Wilfinger A, Arkhipova V, Meyer D. Cell type and tissue specific function of islet genes in zebrafish pancreas development. Dev Biol. 2013;378:25–37. doi: 10.1016/j.ydbio.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchin E, Filippi A, Biemar F, Tiso N, Pauls S, Ellertsdottir E, Gnugge L, Bortolussi M, Driever W, Argenton F. Distinct delta and jagged genes control sequential segregation of pancreatic cell types from precursor pools in zebrafish. Dev Biol. 2007;301:192–204. doi: 10.1016/j.ydbio.2006.09.041. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.