

Abstract
Osteoarthritis is a common and debilitating joint disease that affects up to 30 million Americans, leading to significant disability, reduction in quality of life, and costing the United States tens of billions of dollars annually. Classically, osteoarthritis has been characterized as a degenerative, wear-and-tear disease, but recent research has identified it as an immunopathological disease on a spectrum between healthy condition and rheumatoid arthritis. A systematic literature review demonstrates that the disease pathogenesis is driven by an early innate immune response which progressively catalyzes degenerative changes that ultimately lead to an altered joint microenvironment. It is feasible to detect this infiltration of cells in the early, and presumably asymptomatic, phase of the disease through noninvasive imaging techniques. This screening can serve to aid clinicians in potentially identifying high-risk patients, hopefully leading to early effective management, vast improvements in quality of life, and significant reductions in disability, morbidity, and cost related to osteoarthritis. Although the diagnosis and treatment of osteoarthritis routinely utilize both invasive and non-invasive strategies, imaging techniques specific to inflammatory cells are not commonly employed for these purposes. This review discusses this paradigm and aims to shift the focus of future osteoarthritis-related research towards early diagnosis of the disease process.
1. Introduction
Osteoarthritis (OA) is a painful and debilitative joint disease that commonly affects the hand, hip, and knee joints of aging adults. Disease progression is a leading cause of hospitalization and ultimately requires joint replacement surgery which costs the US healthcare industry over $42 billion in 2009 for the hip and knee joints alone [1]. Clinical OA affects up to 30 million Americans including one-third of seniors aged 65 or older and 13.9% of all adults at least 25 years of age [2]. While disease-modifying antirheumatic drugs (DMARDs) have been identified for rheumatoid arthritis (RA), an inflammatory joint disease often studied and characterized in comparison with OA, similar therapy for OA has yet to be identified [3, 4]. The classical definition of OA as a wear-and-tear, noninflammatory disease has recently transitioned to an inflammatory disease lying on a spectrum between normal control and RA. Despite the fact that the immune system plays a significant role in both diseases, DMARDs effective in the treatment of RA, including tumor necrosis factor α (TNFα) and interleukin-1 (IL-1) inhibitors, have so far proven unsuccessful in slowing disease progression and clinical deterioration of OA patients. This paper will characterize the key players in OA pathogenesis and identify disease-modifying therapeutic strategies which could be reasonably accommodated in the setting of a prevalent, high-morbidity, and costly disease in the United States of America.
Recent research has established that multiple cells, cytokines, chemokines, complement, and other aspects of the immune system are involved in the pathogenesis of OA, with the roles of integral cells and proteins summarized in Tables 1 and 2, respectively. There exists a continuum of inflammation along the spectrum of normal, OA, and RA, with progressive increases in cytokines and other mediators of inflammation along with leukocyte infiltration [5]. OA pathogenesis is multifactorial and complex with evidence pointing towards unique phenotypes and seemingly discrete stages: early, intermediate, and late. Numerous pathways exist and not all may be implicated in specific joints or individuals, but all eventually lead to the endpoint of joint degeneration.
Table 1.
Role of the essential cells implicated in OA pathogenesis.
| Cell type | Role | Comments |
|---|---|---|
| Macrophages | (i) Line intimal layer [5, 14] (ii) Required for production of MMPs and cartilage damage [5, 14] (iii) TNFα, IL-1, MMP, TGFβ, IL-10, IL-12, and chemokine production [29] (iv) Mediators of TGFβ induced osteophyte formation [76] (v) Possess TLRs [14] |
|
|
| ||
| T cell (TCR = T-cell receptor) | (i) Line subintimal layer [5] (ii) Present in different stages of activation: early (CD69+), intermediate (CD25,38+), and late (CD45RO+) [34] (iii) Increased CD4+/CD8+ ratio in OA knees versus control [35] (iv) Th1 > Th2 subset, as well as increased Th1 cytokine product IFNγ [34, 36] (v) Increased CD3ε + T cells and CD3ε +/CD3ζ + T cell ratio in OA synovium and decreased ratio of CD3ζ + T cells [65] (vi) Produce chemokines attractant to macrophages [65] (vii) CDR3 similarity in TCR [77] (viii) Several autoantigens including those on chondrocyte membrane have been identified [31, 78, 79] (ix) Cartilage linking protein and G1 domain of aggrecan have shown promise as potential autoantigens to follow [31, 78, 79] |
(i) Suggestive of chronic inflammation (ii) Suggestive of oligoclonal expansion and antigen-driven response |
|
| ||
| Mast cell (MC) | (i) Numbers are at least as high as those in RA synovium [5] (ii) Mostly in subintimal layer and around blood vessels [22] (iii) Levels positively correlated with total cellular infiltrate, however no correlation with ESR [22] (iv) Degranulated MCs found most commonly in intimal layer [23] (v) Higher ratio of tryptase to tryptase/chymase phenotype in OA than controls [24] (vi) Selective expansion of tryptase MC phenotype [24] |
(i) MCs lie around blood vessels and mediate vascular permeability hinting at crucial role of MCs, however not related to ESR and degranulated phenotype seen in intimal layer (ii) Tryptase phenotype suggestive of degranulation |
|
| ||
| B cells | (i) Not always present or may be present in small numbers [24] (ii) Mostly in subintimal layer [5] (iii) Undergo oligoclonal expansion with similar CDR3 regions [80, 81] (iv) Evidence of somatic hypermutation [80, 81] (v) In patients with moderate to strong infiltration, there were presence of germinal centers and increased T-cell populations [80, 81] (vi) Potential role as antigen presenting cell [80, 81] (vii) Several autoantigens have been identified including those on the chondrocyte membrane but multiple antigens have been discovered and patterns have yet to be characterized [31] (viii) Elevated antibody titer to cartilage membrane in OA patients versus control [82] (ix) Secrete IL-6 [83] |
(i) Suggestive of antigen-driven response |
|
| ||
| Fibroblast | (i) Activated by both IL-1β and TNFα and must neutralize both to decrease activation [29] (ii) Produces MMPs, IL-6, IL-8, ADAMTS-4,5, and MCP-1 [29] |
|
|
| ||
| NK cell | (i) May have role in early pathogenesis of OA: found to have CD16+CD56+ phenotype [19] positive for granzymes A and B [19, 21] and CD16−CD56+ phenotype negative for granzymes A and B [20] (ii) Poor in vitro IFNγ production upon stimulation in late OA patients [20] (iii) Stimulated by IL-15 [66] |
(i) Suggestive of activation/exhaustion phenotypes |
|
| ||
| Neutrophil (PMN) | (i) Generally not found in OA synovial tissue, but sometimes present [5] (ii) HNP1-3 (mainly produced by PMNs) found in synovia of OA patients, with levels inhibited by TNFα stimulation [27] (iii) NGAL (mainly produced by PMNs) found in OA synovia complexed with MMP-9, decreasing its degradation and increasing glycosaminoglycan levels released from cartilage explants [27] |
(i) PMNs may play a role in the earliest stages of OA and therefore might not be expected to be identified in most studies of established OA samples |
Table 2.
Role of dominant effectors involved in OA pathogenesis.
| Protein | Role | Comments |
|
| ||
| IL-1β | (i) Produced by macrophages [29] (ii) Receptors upregulated in OA chondrocytes and fibroblasts [84] (iii) Stimulates production of MMPs [85], ADAMTS-4 [86], and chemokines [87] (iv) Inhibits proteoglycan and type II collagen via repressing GlcAT-1 [88] (v) Induces apoptosis in chondrocytes via upregulation of Bcl-2 family of proteins, mitochondrial depolarization [89], and perhaps ROS [90] and NO production [91] |
(i) GlcAT-1 is an important enzyme for production of glycosaminoglycan |
|
| ||
| TNFα | (i) Produced by macrophages [29] (ii) Promotes resorption and inhibits production of proteoglycan in cartilage [30] (iii) Stimulates MMP and chemokine production [32] (iv) Decreases collagen production [32] (v) May form a negative feedback loop with HNP1-3 [25] |
|
|
| ||
| IL-6 | (i) Produced by fibroblasts [14], chondrocytes [92, 93], and B cells [83] (ii) Production by chondrocytes induced by PGE2 [92], TNFα, and IL-1β [93] (iii) Found to be present in intimal layer and produced mostly by plasma cells when detected in high levels (>600 pg/mL) in synovial fluid [83] (iv) Activates JAK/STAT to inhibit aggrecan core and link protein and type II collagen gene expression; blocking STAT phosphorylation inhibits this downregulation [94] (v) After binding to its receptor, it binds and inactivates transcription factor for COL2A1 gene, which encodes procollagen chain of triple helix of type II collagen [95] (vi) Upregulates expression of MMPs in conjunction with IL-1 [96] |
|
|
| ||
| Complement | (i) Expression and activation abnormally high in OA synovium, significantly in early OA [17] (ii) MAC present around chondrocytes and in synovium in late OA [17] (iii) MAC stimulates MMP, ADAMTS, and chemokine production in chondrocytes [17] (iv) Cartilage ECM, fibromodulin, and aggrecan induced formation of C5b-9 [17] (v) C5− knockout mice showed no significant synovitis or cartilage loss versus control C5+ mice 8–12 weeks s/p medial meniscectomy [17] (vi) C6− mice developed roughly half the degeneration from synovitis as C6+ mice s/p medial meniscectomy [17] (vii) CD59− mice developed more severe OA [17] (viii) C1s cleaves IGFBP-5 which is chondroprotective [18] (ix) C1s inhibition shown to promote better joint architecture in dogs [18] |
|
|
| ||
| TLR | (i) Activated by DAMPs released from ECM in joint damage [14] (ii) Induce proinflammatory cytokine production (IL-1β, TNFα, MMP, etc.) by macrophages [14] (iii) Induce catabolic pathways in chondrocytes [15] (iv) Upregulated on chondrocytes in advanced OA [15] (v) TLR4 on OA chondrocytes more sensitive to S100 than control [16] |
(i) S100 is a DAMP |
|
| ||
| PGE2 | (i) Upregulated in OA joints [92] (ii) Inhibits proteoglycan synthesis by suppressing aggrecan gene transcription [92] (iii) EP2,4 receptors upregulated in joint cartilage as OA progresses [92] (iv) Decreases collagen type II/type I ratio [92] (v) When coupled with IL-1 stimulation, it greatly increases expression of IL-6 and iNOs [92] |
|
|
| ||
| ADAMTS | (i) ADAMTS-4 can be downregulated by inhibiting TNFα and/or IL-1β while ADAMTS-5 is constitutive in human [29] | (i) Uncertainty over which of the two is more significant in OA pathogenesis |
|
| ||
| TGFβ | (i) Osteophyte formation [76] | |
|
| ||
| VEGF | (i) Promotes angiogenesis and MMP production [14] | |
|
| ||
| IL-4,7,8,10,13,15,17,18, adipokines, and leukemia inhibitory factor | (i) Detected in synovium [5] (ii) IL-17 works synergistically with TNFα and IL-1 and is released by mast cells [97] (iii) Increased levels of IL-15 in early versus late OA [66] |
|
2. Immune Response
2.1. Early Innate Response
Both the innate and adaptive immune systems have been implicated in OA pathogenesis, but of particular interest is the role of the innate immune system in early OA. Pathogenesis begins with trauma to the joint, which may constitute repetitive microtrauma accumulated throughout a lifetime or a major traumatic event such as articular fracture. Trauma to the joint is absorbed by subchondral bone [6] and joint-associated fat pads [7], respectively. Subchondral bone releases cytokines while the fat pads release adipokines such as leptin, resistin, adiponectin, visfatin, and chemerin [7]. Although the role of adipokines in OA remains to be conclusively elucidated, many studies have implied that they may act as chemokines and increase matrix-degrading enzymes matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) [7–9], nitric oxide synthase (NOS) [10], Toll-like receptor (TLR) [7], and other cytokine production [7, 11]. Additionally, joint-associated fat pads are innervated by C-fiber neurons which release substance P, thereby increasing pain sensitivity, proinflammatory cytokine production, and vascular permeability [12, 13]. This series of events leads to the release of damage-associated molecular patterns (DAMPs), or alarmins, from the extracellular matrix (ECM) by both direct trauma and the action of MMPs and ADAMTS, as well as from neutrophils and monocytes. DAMPs stimulate TLRs on macrophages and chondrocytes, inducing a strong upregulation of catabolic markers (MMPs 1, 3, 9, and 13, IL-6, IL-8, and monocyte chemotactic protein 1) and cytokines TNFα and IL-1β by way of NFκB activation, which is the master regulator in immune response [14–16]. This chronic activation of TLRs leads to their upregulation in chondrocytes [15] and increased sensitivity [16].
The actions of complement are further demonstrating the significant role played by the innate immune system in early OA. Wang et al. reported that complement expression and activation were abnormally high in OA synovium, especially in early OA, seen in Figure 1. Additionally, the membrane attack complex (MAC, C5b-9) was present surrounding chondrocytes in late OA [17]. MAC directly damages the cell membrane but also stimulates MMP, ADAMTS, and chemokine production in chondrocytes, leading to increased chondrocyte destruction, catabolism of cartilage, and leukocyte infiltration. MMPs release components of the extracellular matrix, such as fibromodulin and aggrecan, which further induce MAC formation. To further assess the role of complement, Wang et al. knocked out C5 in mice and observed that, compared to C5+ controls, the C5− mice showed no significant synovitis or cartilage loss 8–12 weeks status post (s/p) medial meniscectomy, a surgery that can induce OA. Furthermore, C6− mice developed about half the synovial degeneration as C6+ mice s/p medial meniscectomy. CD59, a MAC inhibitor, was also knocked out in another mouse model, and these mice developed more severe OA compared to controls [17]. In another study, Busby Jr. et al. found that inhibiting C1s, a serine protease involved in the initiation of the classic activation pathway, promoted favorable joint architecture in dogs. One mechanism by which C1s exerts its effects is by cleaving chondroprotective IGFBP-5 [18].
Figure 1.
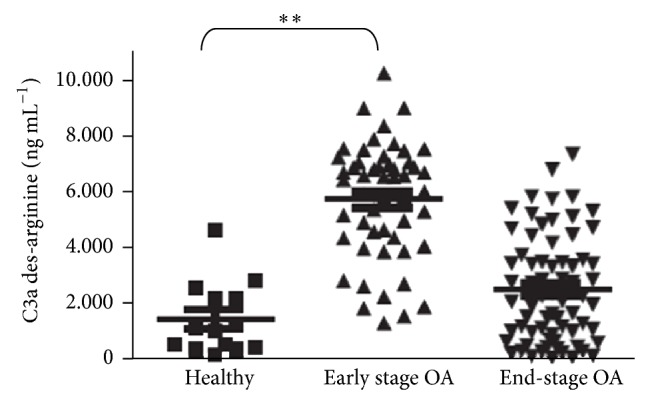
Complement synovial infiltration in the early pathogenesis of OA. ELISA quantification of C3a des-arginine in synovial fluid of healthy (n = 14), early-stage OA (n = 52), and end-stage OA (n = 69) patients. C3a des-arginine is a carboxypeptidase-cleaved, stable form of C3a that is generated from C3 during activation of the complement cascade. ** P ≤ 0.01 by one-way analysis of variance (ANOVA) and Dunnett's post hoc test (reproduction of image with permission and modified caption from Wang et al. [17]).
Other innate immune cells have also been found to play a role in pathogenesis. NK cells have been found in the synovium of OA patients, in one study exhibiting a CD16+CD56+ phenotype both with and without granzymes A and B [19]. Granzyme A and B expression correlates with cytolytic potency in vitro [19]. In another study, NK cells were identified within OA synovia with a CD16−CD56+ phenotype without granzyme expression. Additionally, these cells demonstrated poor production of interferon γ (IFNγ), a cytokine central to osteoclastogenesis, upon stimulation in vitro [20]. In yet another study, granzymes A and B could be identified in the synovia from OA, RA, and reactive arthritis patients [21]. These findings imply that, in OA joints, NK cells can be of an active, cytolytic phenotype, or of an exhaustive, postactivation versus immunoregulatory phenotype. Granzymes A and B, exclusively produced by cytolytic lymphocytes, were identified both intracellularly in NK cells and in the synovia of OA patients [19, 21]. While granzyme presence in the synovium could be explained by T cells, the exclusiveness of this is unlikely. The production and release of granzymes [19, 21] support the notion of an activation/postactivation phenotype theory of NK cell involvement [20]. Of note, Huss et al., who identified mostly CD16−CD56+ NK cells negative for granzymes and suggested that NK cells are of the immunoregulatory phenotype [20], performed their analysis on patients undergoing primary or revision joint replacement, indicative of late OA patients. Concordantly, IFNγ production and degranulation of NK cells were significantly lower after in vitro stimulation of synovial tissue taken from revision versus primary joint replacement patients (degranulation of 2% and 7%, resp., P < 0.05) [20]. The decreased sensitivity of synovial NK cells to stimulation in revision versus primary joint replacement patients demonstrates evidence for an exhaustive NK cell phenotype in late OA. Most likely there is a combination of both activating and immunoregulatory roles played by NK cells in OA pathogenesis.
Mast cells have been identified in the synovium of OA patients [22–24], and in one study their counts were found to have a positive correlation with total cellular infiltrate (r s = 0.82, P = 0.0141) [19]. Interestingly, no correlation between ESR and mast cell count or total cellular infiltrate was found, suggesting only local effects in the joint microenvironment inconsistent with markers of systemic inflammation or disease process [22]. This point is a major barrier to diagnosing and monitoring OA and is expounded upon in later sections. Mast cells are a potent regulator of vascular permeability, and they may play a crucial role in leukocyte recruitment to OA joints. Degranulated mast cells have been found in OA synovium [23], and Buckley et al. discovered a selective expansion and higher ratio of mast cell tryptase phenotype in OA synovium, a phenotype consistent with degranulation [24].
While the significance of neutrophils in synovial disease is well characterized in RA, the role of neutrophils in OA is relatively unknown. Neutrophils are found in varying levels in the synovium of OA patients but generally are found only in small numbers if present at all [5]. However, human neutrophil peptides 1–3 (HNP1–3), α-defensins, were found in the synovial tissue of both OA and RA patients in one study [25]. Interestingly, stimulation with TNFα led to the inhibition of HNP1–3 levels in the synovium of OA patients but not RA patients. The authors concluded that this was most likely due to desensitization of TNF receptors in RA synovia. Paired with the finding that HNP1–3 stimulates macrophages to release TNFα [26], the authors concluded that TNF forms a negative feedback loop with HNP1–3 [25]. If HNP1–3 release does precede the actions of TNFα, this would suggest that neutrophils play a role in early OA pathogenesis, as TNFα is a central mediator of the disease process. In another study, neutrophil gelatinase-associated lipocalin (NGAL) was found in complex with MMP-9 in OA synovia. NGAL served to decrease degradation of MMP-9 [27], found to be the predominant gelatinase in actively resorbing cartilage [28]. In the presence of NGAL-MMP-9, increased levels of glycosaminoglycan were released from cartilage explants in vitro [27]. The role of the innate immune response in early OA pathogenesis is summarized in a stepwise fashion below.
Trauma to the joint is absorbed by subchondral bone and fat pads.
Cytokines, MMPs, and ADAMTS are released.
Direct trauma and MMP/ADAMTS activity release DAMPs which stimulate TLRs.
TLR activation stimulates NFκB, the release of cytokines (mainly TNFα and IL-1β), macrophages, complement, catabolic pathways in chondrocytes, other innate immune cells, and ultimately the adaptive immune response.
Chronic cascading increases TLR expression and receptor sensitivity, further increasing inflammation.
2.2. Adaptive Response
Actions of the innate immune system inevitably lead to activation of the adaptive immune system, increasing inflammation and damage to the joints. TNFα and IL-1β are the dominant and most abundant cytokines implicated in OA [5]. They act independently of each other and additively to shift synovial tissue homeostasis towards catabolism [29, 30]. Mechanisms of this shift include increased resorption and inhibition of proteoglycans in cartilage, production of MMPs and chemokines, endothelium activation, and induction of apoptosis in chondrocytes [31, 32]. This leads to increased macrophage and CD4+ T cell infiltration, blood vessel formation by increased vascular endothelial growth factor (VEGF), and increased cyclooxygenase-2 level [33]. Macrophages and T cells, specifically of the CD4+ Th1 subtype [34, 35], are the most abundant cell types found in the synovium of OA patients [5, 36]. Their activation initiates a repetitive cascade of events, activating both the innate and adaptive immune systems, and this propagating inflammation destroys increasing amounts of cartilage, decreasing function and increasing morbidity. T cells are responsible for enhanced stimulation of macrophages and the activation of B cells. Autoreactive B cells further damage cellular integrity and increase inflammation by producing autoantibodies specific for cartilage cell surface proteins such as osteopontin and collagen. Elevated titers of these autoantibodies were found in the sera from OA patients compared to controls [31]. The adaptive immune response is summarized in stepwise fashion below.
Cytokine release and increased vascular permeability lead to T-cell infiltration.
T cells release chemokines and cytokines including IFNγ, further stimulating macrophages.
Antigen presentation activates B cells.
B cells release IL-6, increasing acute phase reactants, and produce autoantibodies causing direct damage to cartilage.
Lymphocyte and macrophage activation in the joint microenvironment lead to a chronic, relapsing course of inflammation.
3. Early Diagnosis and Treatment
3.1. Imaging Techniques
Anatomic imaging techniques, such as radiography and magnetic resonance imaging (MRI), are currently used for epidemiological studies and clinical trials [37, 38]. Plain radiography is the traditional approach to monitoring progression of disease by clinicians; however, the drawbacks of this approach are apparent: insensitivity to change, nonspecificity, susceptibility to measurement error due to change in positioning, and inability to detect early stages of disease [39, 40]. MRI is regarded as sensitive, valid, and reproducible in that it can assess abnormalities of the whole-joint structure including cartilage degeneration [41], subchondral bone marrow lesions [42, 43], meniscal defects [44], and joint effusion and synovitis [45]. However, even MRI is not sensitive enough to detect the early immune cell infiltration of joints in OA, as inflammation far precedes cartilage destruction marked by radiographic change [46].
There is a substantial need to develop imaging techniques that can visualize the activity of the disease process itself, rather than measure structural changes that are a result of the disease process [47]. In this regard, a few reports have been published on the use of functional nuclear imaging techniques, such as positron emission tomography (PET) and planar or single-photon emission computed tomography (SPECT), for monitoring the inflammatory process of OA [48]. 18F-2-Fluoro-2-deoxy-D-glucose and 111In-diethylene triamine pentaacetic acid-folates have been explored as imaging tracers for OA because of respective increased metabolism of glucoses and elevated expression of folate receptors in activated immune cells [49]. Although these tracers have demonstrated some promise in clinical trials as well as in experimental OA models, they are likely not in use due to the lack of an inflammation-specific window of opportunity for imaging.
Alternatively, formyl peptide receptor (FPR) is primarily expressed on activated leukocytes as a defense mechanism to detect and trigger an immune cell response to inflammation caused by infections in a time and concentration dependent manner [50]. In the past years, based on a FPR-specific binding peptide, cFLFLF, we have successfully utilized the cFLFLF-PEG modules to build PET (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-64Cu, also known as DOTA-64Cu), SPECT (99mTc), and optical (cyanine-5 and cyanine-7) imaging probes and exhibited excellent imaging in a variety of animal inflammation models [51–55]. The cFLFLFK-Cy-7 probe is now commercially available (Kerafast, Inc.) and cFLFLF-based probes have been developed enthusiastically for animal imaging by the broader research community [56–58].
We are currently exploring if a cFLFLF-based SPECT imaging approach is feasible to monitor aseptic inflammation with a particular interest in OA. To this end, an acute model was created by intra-articular injection of monoiodoacetate for near-infrared fluorescence (NIRF) or PET imaging of inflammatory cells during OA development in rat knee joints. As shown in Figure 2, the inflamed joints were well imaged by either a NIRF probe cFLFLF-PEG-Cy 7 (Figure 2(a)) or a PET probe cFLFLF-PEG-DOTA-64Cu (Figure 2(b)). If available in the clinic, use of this SPECT technique can facilitate early detection and monitoring of the recruitment of innate leukocytes during OA development, allow correct characterization and diagnosis to direct early appropriate intervention, and improve long-term outcomes in OA patients [59].
Figure 2.

In vivo imaging of inflammation with two cFLFLF-derived probes in the rat knee joints treated with (right knee) or without (left knee) monoiodoacetate (MIA). (a) CFLFLF-PEG-Cy 7 probe with animal back down, at day 5 after MIA injection; (b) cFLFLF-PEG-DOTA-64Cu with animal back up, at day 5 after MIA injection (upper column: micro-CT; middle column: micro-PET; lower column: fused).
As a caveat, FPR expression in fibroblasts and mesenchymal stem cells (MSCs) has been demonstrated [60, 61]. However, these MSCs and fibroblasts likely serve to repair tissue, initiate tissue remodeling, and mediate leukocyte infiltration in response to the acute chemotactic stimuli of formyl peptide, thereby still reflecting early changes on imaging. The actions of fibroblasts are noted in Tables 1 and 2, respectively. MSCs decrease inflammation, and overexpression of FPR in these cells is currently being studied as potential therapy in chronic disease such as cystic fibrosis [61]. Additionally, FPR ligands have been shown to decrease inflammation in joints and have even been suggested as potential therapies for RA [62]. Regarding the utilization of FPR as an imaging target in early OA, the actions of MSCs, fibroblasts, and FPR ligands, while noteworthy, should have little to no effect or have not yet been discovered. Probes for many cell types and mediators of inflammation mentioned in this paper are displayed in Table 3 and Figure 3.
Table 3.
Probes for mediators of inflammation in modern imaging techniques.
| Cell type or protein | Probe |
|---|---|
| Macrophage | (i) 18F-FDG (PET) [98, 99] |
|
| |
| CD4+ T cell | (i) 64Cu-PTSM (PET) [100] (ii) 18F-FB-IL-2 (PET) [99] |
|
| |
| B cell | (i) 124I-rituximab (PET) [101] |
|
| |
| Neutrophil | (i) cFLFLF-PEG-Cy 7 (NIRF) (ii) cFLFLF-PEG-DOTA-64Cu (PET) |
|
| |
| Mast cell | (i) Ligand 1 (in vitro) [102] |
|
| |
| TNFα | (i) 64Cu-DOTA-etanercept (PET) [103] |
|
| |
| Complement | (i) USPIO-conjugated anti-C3mab (T2-MRI) [104] |
|
| |
| MMP | (i) 124I-HO-MPI (CGS 27023A) (PET) [105] |
Figure 3.
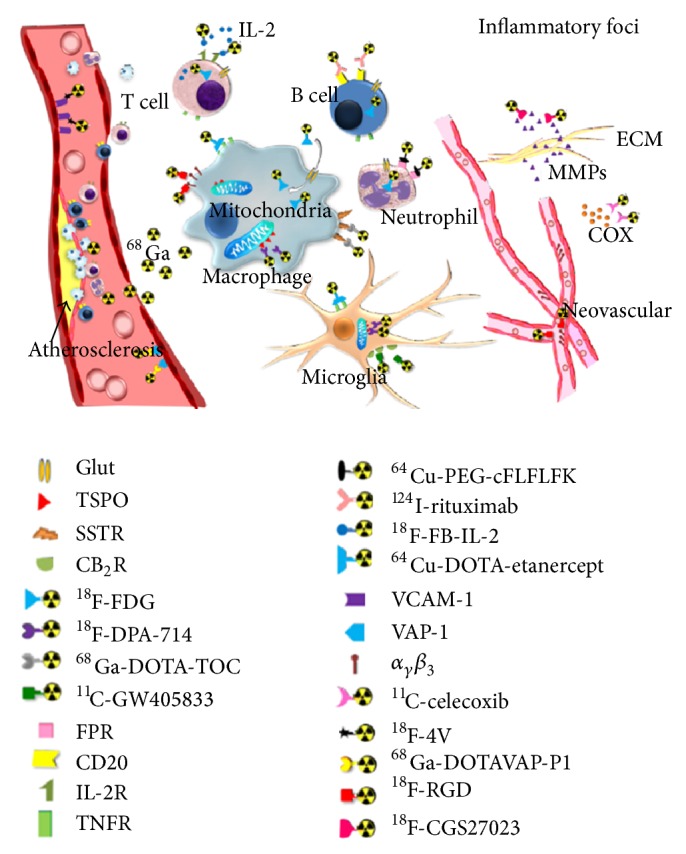
Inflammatory biomarkers in PET imaging (reproduced with permission from Wu et al. [99]).
3.2. Biomarkers
To date, many barriers exist in identifying biomarkers reflective of OA severity; histochemical findings have yet to be linked to clinical traits such as pain and function. Foremost, as evidenced in the next section, inflammation in OA is not only local but also systemic, making standard systemic measurements from individual to individual difficult. The confounding factors in systemic inflammation are immeasurable: age, genetics, diet, activity, kidney function, liver function, weight, and other comorbidities to name a few [63]. Numerous biomarkers have been thought to show promise in recent studies, such as serum cartilage oligomeric matrix protein and urine C-terminal cross-linked telopeptide type II collagen levels, but these are nonspecific to cartilage [63, 64]. Complicating the lack in specificity of inflammatory biomarkers is that measurements in OA patients are drawn once disease is already established. The ability is compromised to determine baseline patient values, cut-off values distinguishing normal from abnormal, and markers that are pathological rather than released naturally or concurrently. Another major barrier limiting the identification of both biomarkers and effective treatment is the unfortunate discrepancy between in vitro and in vivo studies. Decreasing specific mediators of inflammation has thus far not led to improved pain score or prognosis in vivo.
For these reasons testing biomarker levels in synovial fluid seems appropriate. However, the natural microenvironment between individual joints varies, making standard measurements difficult to implement [63]. This is evidenced by the unique infrapatellar fat pad of the knee, which greatly contributes to OA pathogenesis by way of adipokine release. Additionally, extracting synovial fluid is restricted to the larger joints and carries risks as compared to drawing blood. Finally, different phenotypes of disease presumably involve diverse biomarkers, pathways, and sequelae [63]. As evidenced by Table 1, T cells [34, 65] and NK cells [19, 20, 66] have been shown to possess exhaustive and chronic phenotypes, respectively, in late versus early disease, demonstrating that early disease is the primary mechanism responsible for changes in the joint microenvironment, underlining the importance of identifying these changes.
3.3. Hurdles to Treatment
The significance of the innate immune system in early OA becomes evident, as it leads to direct chondrocyte and cartilage destruction as well as NFκB activation with pronounced redundancy and perpetuation. As stated previously, NFκB is the master regulator of the immune response. It is involved in the activation of complement, defensins, adhesions, and caspase-1, as well as the production of cytokines, reactive oxygen species (ROS), and NO. Despite the attractiveness of targeting NFκB in disease-modifying therapy, it is an unreliable target in large part due to its universal role in normal cellular signaling. Its modulation has a significant side effect profile; however natural health products such as those found in grapes and green tea have shown promise but need further study [67].
The difficulty in treating OA is that once local and systemic inflammation is established, debilitative changes in affected joints are difficult to control. OA is both affected by and contributing to a baseline proinflammatory state, such as that seen in senescence, metabolic syndrome, and Alzheimer's disease amongst others (Figure 4) [4]. For example, Berenbaum et al. found that a high fat diet increased inflammation in the acute phase of OA [7]. In another study, Kyrkanides et al. found that inducing OA in mice genetically susceptible to Alzheimer's disease exacerbated and accelerated neuroinflammation, increasing the number and size of amyloid plaques [68]. Many therapies, including anti-TNFα and anti-IL-1β therapy, have been shown to decrease inflammation but fail to significantly improve function or prognosis in established OA patients [3, 4]. Pain levels have been shown to have a statistically significant correlation with level of change in synovitis (r = 0.21, P = 0.0003), but not cartilage destruction or baseline level of synovitis [69]. This correlation is only modest and does nothing but supporting the notion that a relative increase in inflammation will increase perception of pain. There is a disconnection between biomarkers of disease, radiographic change, and symptomology, complicating treatment. Degenerative change in OA can occur under two months following trauma [70], and epigenetics has been shown to play a role in mediating the acute inflammatory changes driven by the altered joint microenvironment [71]. It is for these reasons that we hypothesize that addressing early inflammatory change in the synovium consistent with OA is crucial in modulating disease progression and therefore patient disability. Therapies that have failed to show benefit to date may be effective when implemented at an appropriate stage of disease. Future research should be targeted toward identifying at-risk patients and early intervention.
Figure 4.

Model for role of systemic proinflammatory state and OA. Inflammatory mediators released into blood enter the joint exacerbating OA, which releases its own mediators of inflammation leading to increased systemic inflammation (reproduced with permission from Berenbaum [6]).
4. Perspectives
Pharmacological treatment to date has had varying effects on symptomology, but disease modulation has yet to be attained. Common modalities include NSAIDS, corticosteroids, chondroitin sulfate, and glucosamine [72]. These treatments are variably effective on an individual basis and often only provide temporary relief and are needed to be repeated chronically. Trials of anti-TNFα and anti-IL-1β therapy for disease modulation have been unsuccessful despite the dominance of TNFα and IL-1β in pathogenesis [4]. Chevalier et al. concluded that IL-1β antagonism may benefit patients with baseline high levels of pain if administered in low, frequent intra-articular (IA) injections to avoid neutropenia and serious infection [73]. One in vivo study revealed that IA injection of lubricin up to two weeks after injury reduced severity of OA in mice, while local antioxidants such as N-acetylcysteine after injury showed promise in vitro [74, 75]. The proposed benefit of these treatments administered soon after injury in injury-induced OA underlines the significance of early intervention in OA pathogenesis. Treatment with fibroblast growth factor 18, which is specific for the anabolic FGFR-3 versus the catabolic FGFR-1, is currently on trial [71].
We believe that regular screening is needed and is justified as OA is ubiquitous in seniors aged over 65, is clinically present in 13.9% of US adults aged 25 or above, and is a leading cause of disability and hospitalization in the USA [1, 2, 72]. However, further studies are needed in order to establish guidelines for screening. We recommend that regular screening for OA be implemented on an outpatient basis. Special attention should be given to patients of 65 years or above and patients with metabolic syndrome, Alzheimer's disease, or other systemic proinflammatory states. Candidate markers for screening should continue to be researched with particular attention paid to local articular levels. IL-6, complement, and ratio of FGFR-3/FGFR-1 should be considered.
Additionally, the role of physical, imaging, or combination diagnostic paradigms must be considered. Contrast-enhanced MRI and power Doppler ultrasound are the leading imaging modalities for synovitis [46]. Identifying early and specific changes in OA may best be visualized using PET, NIRF, or SPECT imaging. Many probes for cells and proteins involved in OA pathogenesis are listed in Table 3 and Figure 3. We are currently developing a Tc99m-cFLFLF/SPECT technique to visualize early leukocyte recruitment in OA joints based on a preliminary in vivo study (Figure 2). While our probe is not 100% specific for leukocytes, we are currently in the process of identifying more specific receptors.
As changes in the joint consistent with OA can occur rapidly following injury and are associated with inflammation, intervention should be aimed at the early reactive phase of OA pathogenesis [70]. Importantly, past therapeutic trials may have failed due to attempted intervention at irreversible stages of disease. Wang et al. showed that knocking out components of the complement cascade greatly reduced incidence of OA in mice [17] and this strategy for treatment management should be further researched. A study assessing whether there is an increased relative risk of OA diagnosis and severity in patients with paroxysmal nocturnal hemoglobinuria could be beneficial in this regard. Anticomplement therapy should initially be attempted locally to narrow the focus of treatment and lower the incidence of potential severe infection.
While disease modification in OA still eludes the medical community, recent advances in pathogenesis and understanding of the disease process beseech hope to solving the riddle of a ubiquitous, costly disease that significantly diminishes quality of life in millions of patients. With guided further research and international collaboration, we believe that early detection and intervention in OA are possible. Due to the lack of success and discrepancy of disease modulation between in vivo and in vitro studies, the significance of identifying patients in the early phase of disease becomes paramount in experimenting with detection and treatment of the disease process. Screening must be implemented in high-risk patients, and early, aggressive treatment is necessary and mandated to avoid substantial morbidity.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Murphy L., Helmick C. G. The impact of osteoarthritis in the United States: a population-health perspective: a population-based review of the fourth most common cause of hospitalization in U.S. adults. American Journal of Nursing. 2012;112(3):S13–S19. doi: 10.1097/01.naj.0000412646.80054.21. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence R. C., Felson D. T., Helmick C. G., et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis and Rheumatism. 2008;58(1):26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malemud C. J. Anticytokine therapy for osteoarthritis: evidence to date. Drugs and Aging. 2010;27(2):95–115. doi: 10.2165/11319950-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Calich A. L. G., Domiciano D. S., Fuller R. Osteoarthritis: can anti-cytokine therapy play a role in treatment? Clinical Rheumatology. 2010;29(5):451–455. doi: 10.1007/s10067-009-1352-3. [DOI] [PubMed] [Google Scholar]
- 5.de Lange-Brokaar B. J. E., Ioan-Facsinay A., van Osch G. J. V. M., et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthritis and Cartilage. 2012;20(12):1484–1499. doi: 10.1016/j.joca.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 6.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!) Osteoarthritis and Cartilage. 2013;21(1):16–21. doi: 10.1016/j.joca.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Berenbaum F., Eymard F., Houard X. Osteoarthritis, inflammation and obesity. Current Opinion in Rheumatology. 2013;25(1):114–118. doi: 10.1097/BOR.0b013e32835a9414. [DOI] [PubMed] [Google Scholar]
- 8.Yaykasli K. O., Hatipoglu O. F., Yaykasli E., et al. Leptin induces ADAMTS-4, ADAMTS-5, and ADAMTS-9 genes expression by mitogen-activated protein kinases and NF-κB signaling pathways in human chondrocytes. Cell Biology International. 2014 doi: 10.1002/cbin.10336. [DOI] [PubMed] [Google Scholar]
- 9.Hui W., Litherland G. J., Elias M. S., et al. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. Annals of the Rheumatic Diseases. 2012;71(3):455–462. doi: 10.1136/annrheumdis-2011-200372. [DOI] [PubMed] [Google Scholar]
- 10.Otero M., Lago R., Gómez R., Lago F., Gomez-Reino J. J., Gualillo O. Phosphatidylinositol 3-kinase, MEK-1 and p38 mediate leptin/interferon-gamma synergistic NOS type II induction in chondrocytes. Life Sciences. 2007;81(19-20):1452–1460. doi: 10.1016/j.lfs.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Francin P.-J., Abot A., Guillaume C., et al. Association between adiponectin and cartilage degradation in human osteoarthritis. Osteoarthritis and Cartilage. 2014;22(3):519–526. doi: 10.1016/j.joca.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Clockaerts S., Bastiaansen-Jenniskens Y. M., Runhaar J., et al. The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. Osteoarthritis and Cartilage. 2010;18(7):876–882. doi: 10.1016/j.joca.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Bohnsack M., Meier F., Walter G. F., et al. Distribution of substance-P nerves inside the infrapatellar fat pad and the adjacent synovial tissue: a neurohistological approach to anterior knee pain syndrome. Archives of Orthopaedic and Trauma Surgery. 2005;125(9):592–597. doi: 10.1007/s00402-005-0796-4. [DOI] [PubMed] [Google Scholar]
- 14.Sohn D. H., Sokolove J., Sharpe O., et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Research and Therapy. 2012;14, article R7 doi: 10.1186/ar3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su S.-L., Tsai C.-D., Lee C.-H., Salter D. M., Lee H.-S. Expression and regulation of Toll-like receptor 2 by IL-1β and fibronectin fragments in human articular chondrocytes. Osteoarthritis and Cartilage. 2005;13(10):879–886. doi: 10.1016/j.joca.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 16.Schelbergen R. F. P., Blom A. B., van den Bosch M. H. J., et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on toll-like receptor 4. Arthritis & Rheumatism. 2012;64(5):1477–1487. doi: 10.1002/art.33495. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q., Rozelle A. L., Lepus C. M., et al. Identification of a central role for complement in osteoarthritis. Nature Medicine. 2011;17(12):1674–1679. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Busby W. H., Jr., Yocum S. A., Rowland M., et al. Complement 1s is the serine protease that cleaves IGFBP-5 in human osteoarthritic joint fluid. Osteoarthritis and Cartilage. 2009;17(4):547–555. doi: 10.1016/j.joca.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kummer J. A., Tak P. P., Brinkman B. M. N., et al. Expression of granzymes A and B in synovial tissue from patients with rheumatoid arthritis and osteoarthritis. Clinical Immunology and Immunopathology. 1994;73(1):88–95. doi: 10.1006/clin.1994.1173. [DOI] [PubMed] [Google Scholar]
- 20.Huss R. S., Huddleston J. I., Goodman S. B., Butcher E. C., Zabel B. A. Synovial tissue-infiltrating natural killer cells in osteoarthritis and periprosthetic inflammation. Arthritis & Rheumatism. 2010;62(12):3799–3805. doi: 10.1002/art.27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tak P. P., Spaeny-Dekking L., Kraan M. C., Breedveld F. C., Froelich C. J., Hack C. E. The levels of soluble granzyme A and B are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA) Clinical and Experimental Immunology. 1999;116(2):366–370. doi: 10.1046/j.1365-2249.1999.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Damsgaard T. E., Sørensen F. B., Herlin T., Schiøtz P. O. Stereological quantification of mast cells in human synovium. APMIS. 1999;107(3):311–317. doi: 10.1111/j.1699-0463.1999.tb01559.x. [DOI] [PubMed] [Google Scholar]
- 23.Dean G., Hoyland J. A., Denton J., Donn R. P., Freemont A. J. Mast cells in the synovium and synovial fluid in osteoarthritis. British Journal of Rheumatology. 1993;32(8):671–675. doi: 10.1093/rheumatology/32.8.671. [DOI] [PubMed] [Google Scholar]
- 24.Buckley M. G., Gallagher P. J., Walls A. F. Mast cell subpopulations in the synovial tissue of patients with osteoarthritis: selective increase in numbers of tryptase-positive, chymase- negative mast cells. The Journal of Pathology. 1998;186(1):67–74. doi: 10.1002/(SICI)1096-9896(199809)186:1<67::AID-PATH132>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 25.Riepl B., Grässel S., Wiest R., Fleck M., Straub R. H. Tumor necrosis factor and norepinephrine lower the levels of human neutrophil peptides 1–3 secretion by mixed synovial tissue cultures in osteoarthritis and rheumatoid arthritis. Arthritis Research and Therapy. 2010;12, article R110 doi: 10.1186/ar3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soehnlein O., Kai-Larsen Y., Frithiof R., et al. Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages. The Journal of Clinical Investigation. 2008;118(10):3491–3502. doi: 10.1172/jci35740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta K., Shukla M., Cowland J. B., Malemud C. J., Haqqi T. M. Neutrophil gelatinase-associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis & Rheumatism. 2007;56(10):3326–3335. doi: 10.1002/art.22879. [DOI] [PubMed] [Google Scholar]
- 28.Milner J. M., Rowan A. D., Cawston T. E., Young D. A. Metalloproteinase and inhibitor expression profiling of resorbing cartilage reveals pro-collagenase activation as a critical step for collagenolysis. Arthritis Research & Therapy. 2006;8(5, article R142) doi: 10.1186/ar2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bondeson J., Blom A. B., Wainwright S., Hughes C., Caterson B., van den Berg W. B. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis and Rheumatism. 2010;62(3):647–657. doi: 10.1002/art.27290. [DOI] [PubMed] [Google Scholar]
- 30.Saklatvala J. Tumour necrosis factor α stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature. 1986;322(6079):547–549. doi: 10.1038/322547a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haseeb A., Haqqi T. M. Immunopathogenesis of osteoarthritis. Clinical Immunology. 2013;146(3):185–196. doi: 10.1016/j.clim.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lefebvre V., Peeters-Joris C., Vaes G. Modulation by interleukin 1 and tumor necrosis factor α of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochimica et Biophysica Acta. 1990;1052(3):366–378. doi: 10.1016/0167-4889(90)90145-4. [DOI] [PubMed] [Google Scholar]
- 33.Benito M. J., Veale D. J., FitzGerald O., van den Berg W. B., Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Annals of the Rheumatic Diseases. 2005;64(9):1263–1267. doi: 10.1136/ard.2004.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakkas L. I., Scanzello C., Johanson N., et al. T cells and T-cell cytokine transcripts in the synovial membrane in patients with osteoarthritis. Clinical and Diagnostic Laboratory Immunology. 1998;5(4):430–437. doi: 10.1128/cdli.5.4.430-437.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito I., Koshino T., Nakashima K., Uesugi M., Saito T. Increased cellular infiltrate in inflammatory synovia of osteoarthritic knees. Osteoarthritis and Cartilage. 2002;10(2):156–162. doi: 10.1053/joca.2001.0494. [DOI] [PubMed] [Google Scholar]
- 36.Pessler F., Chen L. X., Dai L., et al. A histomorphometric analysis of synovial biopsies from individuals with Gulf War Veterans' Illness and joint pain compared to normal and osteoarthritis synovium. Clinical Rheumatology. 2008;27(9):1127–1134. doi: 10.1007/s10067-008-0878-0. [DOI] [PubMed] [Google Scholar]
- 37.Eckstein F., Wirth W., Nevitt M. C. Recent advances in osteoarthritis imaging—the Osteoarthritis Initiative. Nature Reviews Rheumatology. 2012;8(10):622–630. doi: 10.1038/nrrheum.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding C., Zhang Y., Hunter D. Use of imaging techniques to predict progression in osteoarthritis. Current Opinion in Rheumatology. 2013;25(1):127–135. doi: 10.1097/BOR.0b013e32835a0fe1. [DOI] [PubMed] [Google Scholar]
- 39.Guermazi A., Roemer F. W., Burstein D., Hayashi D. Why radiography should no longer be considered a surrogate outcome measure for longitudinal assessment of cartilage in knee osteoarthritis. Arthritis Research and Therapy. 2011;13, article 247 doi: 10.1186/ar3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kraus V. B. Waiting for action on the osteoarthritis front. Current Drug Targets. 2010;11(5):518–520. doi: 10.2174/138945010791011974. [DOI] [PubMed] [Google Scholar]
- 41.Roemer F. W., Kwoh C. K., Hannon M. J., et al. Risk factors for magnetic resonance imaging-detected patellofemoral and tibiofemoral cartilage loss during a six-month period: the Joints on Glucosamine study. Arthritis and Rheumatism. 2012;64(6):1888–1898. doi: 10.1002/art.34353. [DOI] [PubMed] [Google Scholar]
- 42.Tanamas S. K., Wluka A. E., Pelletier J.-P., et al. Bone marrow lesions in people with knee osteoarthritis predict progression of disease and joint replacement: a longitudinal study. Rheumatology. 2010;49(12):2413–2419. doi: 10.1093/rheumatology/keq286. [DOI] [PubMed] [Google Scholar]
- 43.Kwoh C. K. Osteoarthritis: clinical relevance of bone marrow lesions in OA. Nature Reviews Rheumatology. 2013;9(1):7–8. doi: 10.1038/nrrheum.2012.217. [DOI] [PubMed] [Google Scholar]
- 44.Sharma L., Chmiel J. S., Almagor O., et al. Significance of preradiographic magnetic resonance imaging lesions in persons at increased risk of knee osteoarthritis. Arthritis & Rheumatology. 2014;66(7):1811–1819. doi: 10.1002/art.38611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roemer F. W., Guermazi A., Felson D. T., et al. Presence of MRI-detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30-month follow-up: the MOST study. Annals of the Rheumatic Diseases. 2011;70(10):1804–1809. doi: 10.1136/ard.2011.150243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sokolove J., Lepus C. M. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Therapeutic Advances in Musculoskeletal Disease. 2013;5(2):77–94. doi: 10.1177/1759720x12467868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park H.-J., Kim S. S., Lee S.-Y., et al. A practical MRI grading system for osteoarthritis of the knee: association with Kellgren-Lawrence radiographic scores. European Journal of Radiology. 2013;82(1):112–117. doi: 10.1016/j.ejrad.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 48.Braun H. J., Gold G. E. Diagnosis of osteoarthritis: imaging. Bone. 2012;51(2):278–288. doi: 10.1016/j.bone.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura H., Masuko K., Yudoh K., et al. Positron emission tomography with 18F-FDG in osteoarthritic knee. Osteoarthritis and Cartilage. 2007;15(6):673–681. doi: 10.1016/j.joca.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 50.Wenham C. Y. J., Conaghan P. G. New horizons in osteoarthritis. Age and Ageing. 2013;42(3):272–278. doi: 10.1093/ageing/aft043. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y., Kundu B., Fairchild K. D., et al. Synthesis of novel neutrophil-specific imaging agents for Positron Emission Tomography (PET) imaging. Bioorganic and Medicinal Chemistry Letters. 2007;17(24):6876–6878. doi: 10.1016/j.bmcl.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Locke L. W., Chordia M. D., Zhang Y., et al. A novel neutrophil-specific PET imaging agent: cFLFLFK-PEG-64Cu. The Journal of Nuclear Medicine. 2009;50(5):790–797. doi: 10.2967/jnumed.108.056127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao L., Zhang Y., Liu Z., Yang M., Pu L., Pan D. Synthesis of the Cyanine 7 labeled neutrophil-specific agents for noninvasive near infrared fluorescence imaging. Bioorganic and Medicinal Chemistry Letters. 2010;20(12):3515–3517. doi: 10.1016/j.bmcl.2010.04.136. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y., Xiao L., Chordia M. D., et al. Neutrophil targeting heterobivalent SPECT imaging probe: CFLFLF-PEG-TKPPR-99mTc. Bioconjugate Chemistry. 2010;21(10):1788–1793. doi: 10.1021/bc100063a. [DOI] [PubMed] [Google Scholar]
- 55.Xiao L., Zhang Y., Berr S. S., et al. A novel near-infrared fluorescence imaging probe for in vivo neutrophil tracking. Molecular Imaging. 2012;11(5):372–382. [PubMed] [Google Scholar]
- 56.Massey S., Johnston K., Mott T. M., et al. In vivo bio luminescence imaging of Burkholderia mallei respiratory infection and treatment in the mouse model. Frontiers in Microbiology. 2011;2, article 174 doi: 10.3389/fmicb.2011.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stasiuk G. J., Smith H., Wylezinska-Arridge M., et al. Gd3+ cFLFLFK conjugate for MRI: a targeted contrast agent for FPR1 in inflammation. Chemical Communications. 2013;49(6):564–566. doi: 10.1039/c2cc37460a. [DOI] [PubMed] [Google Scholar]
- 58.Zhou J., Tsai Y.-T., Weng H., et al. Real-time detection of implant-associated neutrophil responses using a formyl peptide receptor-targeting NIR nanoprobe. International Journal of Nanomedicine. 2012;7:2057–2068. doi: 10.2147/ijn.s29961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorward D. A., Lucas C. D., Rossi A. G., Haslett C., Dhaliwal K. Imaging inflammation: molecular strategies to visualize key components of the inflammatory cascade, from initiation to resolution. Pharmacology and Therapeutics. 2012;135(2):182–199. doi: 10.1016/j.pharmthera.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 60.VanCompernolle S. E., Clark K. L., Rummel K. A., Todd S. C. Expression and function of formyl peptide receptors on human fibroblast cells. The Journal of Immunology. 2003;171(4):2050–2056. doi: 10.4049/jimmunol.171.4.2050. [DOI] [PubMed] [Google Scholar]
- 61.Viswanathan A., Painter R. G., Lanson N. A., Jr., Wang G. Functional expression of N-formyl peptide receptors in human bone marrow-derived mesenchymal stem cells. Stem Cells. 2007;25(5):1263–1269. doi: 10.1634/stemcells.2006-0522. [DOI] [PubMed] [Google Scholar]
- 62.Kao W., Gu R., Jia Y., et al. A formyl peptide receptor agonist suppresses inflammation and bone damage in arthritis. British Journal of Pharmacology. 2014;171(17):4087–4096. doi: 10.1111/bph.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lafeber F. P. J. G., van Spil W. E. Osteoarthritis year 2013 in review: biomarkers; reflecting before moving forward, one step at a time. Osteoarthritis and Cartilage. 2013;21(10):1452–1464. doi: 10.1016/j.joca.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 64.Attur M., Krasnokutsky-Samuels S., Samuels J., Abramson S. B. Prognostic biomarkers in osteoarthritis. Current Opinion in Rheumatology. 2013;25(1):136–144. doi: 10.1097/bor.0b013e32835a9381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakkas L. I., Koussidis G., Avgerinos E., Gaughan J., Platsoucas C. D. Decreased expression of the CD3ζ chain in T cells infiltrating the synovial membrane of patients with osteoarthritis. Clinical and Diagnostic Laboratory Immunology. 2004;11(1):195–202. doi: 10.1128/cdli.11.1.195-202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scanzello C. R., Umoh E., Pessler F., et al. Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis and Cartilage. 2009;17(8):1040–1048. doi: 10.1016/j.joca.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 67.Khalifé S., Zafarullah M. Molecular targets of natural health products in arthritis. Arthritis Research & Therapy. 2011;13, article 102 doi: 10.1186/ar3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kyrkanides S., Tallents R. H., Miller J.-N. H., et al. Osteoarthritis accelerates and exacerbates Alzheimer's disease pathology in mice. Journal of Neuroinflammation. 2011;8, article 112 doi: 10.1186/1742-2094-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hill C. L., Hunter D. J., Niu J., et al. Synovitis detected on magnetic resonance imaging and its relation to pain and cartilage loss in knee osteoarthritis. Annals of the Rheumatic Diseases. 2007;66(12):1599–1603. doi: 10.1136/ard.2006.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schenker M. L., Mauck R. L., Mehta S. Pathogenesis and prevention of posttraumatic osteoarthritis after intra-articular fracture. Journal of the American Academy of Orthopaedic Surgeons. 2014;22(1):20–28. doi: 10.5435/jaaos-22-01-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loeser R. F. Osteoarthritis year in review 2013: biology. Osteoarthritis and Cartilage. 2013;21(10):1436–1442. doi: 10.1016/j.joca.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goljan E. F. Rapid Review Pathology. 4th. Philadelphia, Pa, USA: Elsevier/Saunders; 2014. [Google Scholar]
- 73.Chevalier X., Conrozier T., Richette P. Desperately looking for the right target in osteoarthritis: the anti-IL-1 strategy. Arthritis Research and Therapy. 2011;13, article 124 doi: 10.1186/ar3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruan M. Z. C., Erez A., Guse K., et al. Proteoglycan 4 expression protects against the development of osteoarthritis. Science Translational Medicine. 2013;5(176) doi: 10.1126/scitranslmed.3005409.176ra34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martin J. A., McCabe D., Walter M., Buckwalter J. A., McKinley T. O. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. The Journal of Bone and Joint Surgery A. 2009;91(8):1890–1897. doi: 10.2106/jbjs.h.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Lent P. L. E. M., Blom A. B., Van Der Kraan P., et al. Crucial role of synovial lining macrophages in the promotion of transforming growth factor β -mediated osteophyte formation. Arthritis and Rheumatism. 2004;50(1):103–111. doi: 10.1002/art.11422. [DOI] [PubMed] [Google Scholar]
- 77.Nakamura H., Yoshino S., Kato T., Tsuruha J., Nishioka K. T-cell mediated inflammatory pathway in osteoarthritis. Osteoarthritis and Cartilage. 1999;7(4):401–402. doi: 10.1053/joca.1998.0224. [DOI] [PubMed] [Google Scholar]
- 78.Guerassimov A., Zhang Y., Cartman A., et al. Immune responses to cartilage link protein and the G1 domain of proteoglycan aggrecan in patients with osteoarthritis. Arthritis & Rheumatism. 1999;42(3):527–533. doi: 10.1002/1529-0131(199904)42:3<527::AID-ANR18>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 79.de Jong H., Berlo S. E., Hombrink P., et al. Cartilage proteoglycan aggrecan epitopes induce proinflammatory autoreactive T-cell responses in rheumatoid arthritis and osteoarthritis. Annals of the Rheumatic Diseases. 2010;69(1):255–262. doi: 10.1136/ard.2008.103978. [DOI] [PubMed] [Google Scholar]
- 80.Da R.-R., Qin Y., Baeten D., Zhang Y. B cell clonal expansion and somatic hypermutation of Ig variable heavy chain genes in the synovial membrane of patients with osteoarthritis. The Journal of Immunology. 2007;178(1):557–565. doi: 10.4049/jimmunol.178.1.557. [DOI] [PubMed] [Google Scholar]
- 81.Shiokawa S., Matsumoto N., Nishimura J. Clonal analysis of B cells in the osteoarthritis synovium. Annals of the Rheumatic Diseases. 2001;60(8):802–805. doi: 10.1136/ard.60.8.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mollenhauer J., von der Mark K., Burmester G., Gluckert K., Lutjen-Drecoll E., Brune K. Serum antibodies against chondrocyte cell surface proteins in osteoarthritis and rheumatoid arthritis. The Journal of Rheumatology. 1988;15(12):1811–1817. [PubMed] [Google Scholar]
- 83.Doss F., Menard J., Hauschild M., et al. Elevated IL-6 levels in the synovial fluid of osteoarthritis patients stem from plasma cells. Scandinavian Journal of Rheumatology. 2007;36(2):136–139. doi: 10.1080/03009740701250785. [DOI] [PubMed] [Google Scholar]
- 84.Smith M. D., Triantafillou S., Parker A., Youssef P. P., Coleman M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. The Journal of Rheumatology. 1997;24(2):365–371. [PubMed] [Google Scholar]
- 85.Kobayashi M., Squires G. R., Mousa A., et al. Role of interleukin-1 and tumor necrosis factor α in matrix degradation of human osteoarthritic cartilage. Arthritis and Rheumatism. 2005;52(1):128–135. doi: 10.1002/art.20776. [DOI] [PubMed] [Google Scholar]
- 86.Fan Z., Bau B., Yang H., Soeder S., Aigner T. Freshly isolated osteoarthritic chondrocytes are catabolically more active than normal chondrocytes, but less responsive to catabolic stimulation with interleukin-1β . Arthritis & Rheumatism. 2005;52(1):136–143. doi: 10.1002/art.20725. [DOI] [PubMed] [Google Scholar]
- 87.Akhtar N., Haqqi T. M. Epigallocatechin-3-gallate suppresses the global interleukin-1beta-induced inflammatory response in human chondrocytes. Arthritis Research and Therapy. 2011;13(3, article R93) doi: 10.1186/ar3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gouze J. N., Bordji K., Gulberti S., et al. Interleukin-1beta down-regulates the expression of glucuronosyltransferase I, a key enzyme priming glycosaminoglycan biosynthesis: influence of glucosamine on interleukin-1beta-mediated effects in rat chondrocytes. Arthritis & Rheumatism. 2001;44(2):351–360. doi: 10.1002/1529-0131(200102)44:2x0003C;351::aid-anr53x003E;3.3.co;2-d. [DOI] [PubMed] [Google Scholar]
- 89.Heraud F., Heraud A., Harmand M.-F. Apoptosis in normal and osteoarthritic human articular cartilage. Annals of the Rheumatic Diseases. 2000;59(12):959–965. doi: 10.1136/ard.59.12.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mathy-Hartert M., Hogge L., Sanchez C., Deby-Dupont G., Crielaard J. M., Henrotin Y. Interleukin-1β and interleukin-6 disturb the antioxidant enzyme system in bovine chondrocytes: a possible explanation for oxidative stress generation. Osteoarthritis and Cartilage. 2008;16(7):756–763. doi: 10.1016/j.joca.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 91.Pelletier J.-P., Mineau F., Ranger P., Tardif G., Martel-Pelletier J. The increased synthesis of inducible nitric oxide inhibits IL-1ra synthesis by human articular chondrocytes: possible role in osteoarthritic cartilage degradation. Osteoarthritis and Cartilage. 1996;4(1):77–84. doi: 10.1016/s1063-4584(96)80009-4. [DOI] [PubMed] [Google Scholar]
- 92.Li X., Ellman M., Muddasani P., et al. Prostaglandin E2 and its cognate EP receptors control human adult articular cartilage homeostasis and are linked to the pathophysiology of osteoarthritis. Arthritis & Rheumatism. 2009;60(2):513–523. doi: 10.1002/art.24258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guerne P.-A., Zuraw B. L., Vaughan J. H., Carson D. A., Lotz M. Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. The Journal of Clinical Investigation. 1989;83(2):585–592. doi: 10.1172/JCI113921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Legendre F., Dudhia J., Pujol J.-P., Bogdanowicz P. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of Sox9 expression. The Journal of Biological Chemistry. 2003;278(5):2903–2912. doi: 10.1074/jbc.M110773200. [DOI] [PubMed] [Google Scholar]
- 95.Porée B., Kypriotou M., Chadjichristos C., et al. Interleukin-6 (IL-6) and/or soluble IL-6 receptor down-regulation of human type II collagen gene expression in articular chondrocytes requires a decrease of Sp1.Sp3 ratio and of the binding activity of both factors to the COL2A1 promoter. The Journal of Biological Chemistry. 2008;283(8):4850–4865. doi: 10.1074/jbc.M706387200. [DOI] [PubMed] [Google Scholar]
- 96.Cawston T. E., Curry V. A., Summers C. A., et al. The role of oncostatin M in animal and human connective tissue collagen turnover and its localization within the rheumatoid joint. Arthritis & Rheumatology. 1998;41:1760–1771. doi: 10.1002/1529-0131(199810)41:10<1760::AID-ART8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 97.Suurmond J., Dorjee A. L., Boon M. R., et al. Mast cells are the main interleukin-17-positive cells in anti-citrullinated protein antibody-positive and -negative rheumatoid arthritis and osteoarthritis synovium. Arthritis Research & Therapy. 2011;13(5, article R150) doi: 10.1186/ar3466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Brown T. L. Y., Spencer H. J., Beenken K. E., et al. Evaluation of dynamic [18F]-FDG-PET imaging for the detection of acute post-surgical bone infection. PLoS ONE. 2012;7(7) doi: 10.1371/journal.pone.0041863.e41863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu C., Li F., Niu G., Chen X. PET imaging of inflammation biomarkers. Theranostics. 2013;3(7):448–466. doi: 10.7150/thno.6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Griessinger C. M., Kehlbach R., Bukala D., et al. In vivo tracking of th1 cells by PET reveals quantitative and temporal distribution and specific homing in lymphatic tissue. The Journal of Nuclear Medicine. 2014;55(2):301–307. doi: 10.2967/jnumed.113.126318. [DOI] [PubMed] [Google Scholar]
- 101.Tran L., Huitema A. D. R., Van Rijswijk M. H., et al. CD20 antigen imaging with 124I-rituximab PET/CT in patients with rheumatoid arthritis. Human Antibodies. 2011;20(1-2):29–35. doi: 10.3233/hab-2011-0239. [DOI] [PubMed] [Google Scholar]
- 102.Wang Q., Shi X., Zhu X., Ehlers M., Wu J., Schmuck C. A fluorescent light-up probe as an inhibitor of intracellular β-tryptase. Chemical Communications. 2014;50(46):6120–6122. doi: 10.1039/c4cc02208d. [DOI] [PubMed] [Google Scholar]
- 103.Cao Q., Cai W., Li Z.-B., et al. PET imaging of acute and chronic inflammation in living mice. European Journal of Nuclear Medicine and Molecular Imaging. 2007;34(11):1832–1842. doi: 10.1007/s00259-007-0451-0. [DOI] [PubMed] [Google Scholar]
- 104.Girardi G., Fraser J., Lennen R., Vontell R., Jansen M., Hutchison G. Imaging of activated complement using ultrasmall superparamagnetic iron oxide particles (USPIO)—conjugated vectors: an in vivo in utero non-invasive method to predict placental insufficiency and abnormal fetal brain development. Molecular Psychiatry. 2014 doi: 10.1038/mp.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hartung D., Schäfers M., Fujimoto S., et al. Targeting of matrix metalloproteinase activation for noninvasive detection of vulnerable atherosclerotic lesions. European Journal of Nuclear Medicine and Molecular Imaging. 2007;34(supplement 1):S1–S8. doi: 10.1007/s00259-007-0435-0. [DOI] [PubMed] [Google Scholar]