

Abstract
Patients with proteinuric kidney diseases often experience symptoms of salt and water retention. It has been hypothesized that the dysregulated Na+ absorption is due to increased proteolytic cleavage of epithelial sodium channel (ENaC) and increased Na,K-ATPase expression. Microarray analysis identified a reduced corin mRNA expression in kidneys from rat models of puromycin aminonucleoside-induced nephrotic syndrome (PAN) and acute anti-Thy1 glomerulonephritis (GN). Corin has been shown to convert pro-atrial natriuretic peptide (ANP) to ANP. Because ANP resistance has been assumed to be a mechanism accounting for volume retention, experiments were undertaken to analyze the renal expression and function of corin. Immunohistochemistry revealed that corin co-localized with ANP. In PAN and GN, kidneys exhibited concomitant increased pro-ANP and decreased ANP protein expression levels consistent with low corin levels. Importantly, kidneys from corin −/− mice showed increased levels of renal β-ENaC, phosphodiesterase 5 (PDE5) and protein kinase G II (PKGII) when compared to wild-type mice. Similar expression profile was observed in cell culture experiments suggesting that the increase in PDE5 and PKGII could account for the increase in β-ENaC as observed in PAN and GN.
To conclude, our data provide novel insights into the mechanisms of volume retention in renal disease with corin as an important new mediator that acts through PKGII induction and ENaC activation.
INTRODUCTION
Clinical signs of volume retention are frequently observed in patients with acute and chronic glomerular disease. While acute glomerulonephritis (GN) results mostly in a nephritic syndrome, the nephrotic syndrome occurs mainly in minimal change disease, membranous glomerulonephritis and primary focal segmental glomerulosclerosis. Both share common symptoms such as proteinuria, as a result of glomerular damage, signs of salt and volume retention presented as hypertension and/or edema formation and hypercholesteremia. In addition, the nephrotic syndrome presents hypoalbuminemia and intravascular volume depletion as a cardinal feature.
It is generally agreed, that volume retention observed in nephrotic syndrome results primarily from a renal dysregulation. Early in vivo micropuncture studies using experimental animal models of GN or nephrotic syndrome localized the site of impaired sodium excretion to the connecting tubule and collecting duct (CD).1,2 The increase in Na+ absorption observed in nephrotic syndrome and GN was shown to be linked to aldosterone-independent activation of the epithelial sodium channel (ENaC),3–8 and concomitant upregulation of the basolaterally located sodium potassium ATPase (Na,K-ATPase)4. Recently, new regulatory mechanisms have emerged by the identification of proteolytical ENaC subunit cleavage, which increases the open probability of the channel 3 to 5 fold.9–12 Interestingly, the serine protease plasmin recovered in the urine of nephrotic patients was shown to activate ENaC. Proteolytical cleavage of ENaC may therefore represent a potential mechanism which could account for the increased Na+ reabsorption observed in proteinuric kidney diseases. Aldosterone13–14, vasopressin15, nitric oxide16, angiotensin II or insulin-like growth factor14 which regulate sodium reabsorption in the CD have also been proposed to play a role in volume retention in proteinuric kidney diseases. However, blockade of their specific pathways had no or only minimal beneficial effects on salt retention.14
Atrial natriuretic peptide (ANP) is another important factor proposed, which is highly produced in heart and in various organs such as the kidney. Once released, binding to its receptor, the membrane-bound guanylyl cyclase, stimulates intracellular cGMP production. cGMP then activates cGMP-dependent phosphodiesterase (PDEs) and cGMP-dependent protein kinases (PKGs).17 ANP target organs are the kidney and blood vessels leading to natriuresis, diuresis, and vasodilatation. Several studies have demonstrated a marked renal resistance to ANP18,19 which was hypothesized to be an underlying mechanism for volume retention in proteinuric disease. The observed dysregulation was suggested to be mediated through increased cGMP-specific PDE5 activity accelerating the degradation of cGMP.18,19
Recently, corin, a type II transmembrane serine protease, was found to be responsible for converting pro-ANP to active ANP.20 Generation of corin deficient (Cor−/−) mice confirmed corin as a rate limiting enzyme for ANP maturation.21 Similar to ANP and guanylyl cyclase-A deficient mice, Cor−/− mice developed hypertension which was exacerbated by salt load and pregnancy.21
Volume retention in proteinuric kidney diseases is thought to be multifactorial. In search of the origin microarray analysis comparing renal medullary gene expression levels in rats with PAN induced nephrotic syndrome and anti-Thy1 GN were performed and markedly reduced corin expression was identified in experimental rat models when compared to controls. Therefore we aimed to perform a detailed renal localization of corin, verified its expression levels in experimental models of PAN and GN, and explored its role in the kidney by analyzing the ANP signaling cascade using Cor−/− mice and cell culture experiments.
RESULTS
Renal function in PAN and GN
Rats injected either with the monoclonal antibody OX-7 (GN) or puromycin aminonucleoside (PAN) developed proteinuria (control: 15 ± 6 mg/24h; PAN: 539 ± 99* mg/24h; GN: 199 ± 24* mg/24h; P < 0.001) and PAN-injected rats also displayed a nephrotic syndrome with severe ascites. In both cases, plasma creatinine and urea levels significantly increased when compared to control rats (creatinine control: 0.17 ± 0.03 mg/dl; PAN: 0.53 ± 0.2* mg/dl; GN: 0.36 ± 0.13* mg/dl; urea control: 38 ± 6 mg/dl; PAN: 196 ± 88* mg/dl; GN: 79 ± 16* mg/dl; P < 0.01) whereas renal creatinine clearance and fractional sodium excretion significantly decreased (GFR control: 3.91 ± 1.3 ml/min; PAN: 1.14 ± 0.6* ml/min; GN: 2.31 ± 0.43* ml/min; FENa control: 0.22 ± 0.03 %; PAN: 0.07 ± 0.02* %; GN: 0.18 ± 0.03 %; P < 0.01).
Validation of monoclonal anti-corin anitbody specificity
A monoclonal anti-corin antibody was produced and its specificity was controlled by transfecting HEK 293 cells with His-tagged full-length corin and pcDNA3.1 (mock). Immunohistochemistry demonstrated identical signals using anti-corin double labeled with anti-His (Figure 1a and b). Preincubation of the antibody with the respective antigen blocked the signal (not shown). Western blot using the anti-corin and anti-His antibodies revealed a main labeled band at ~ 160 to 170 kDa only in corin construct transfected cells, but not in mock-transfected cells (Figure 1c). Corin was first identified in the human heart,22 atrial tissue was tested for further immunohistochemical verification of the specificity of our anti-corin antibody. Expression was found in the plasma membrane and vesicles of myocytes (Figure 1d).
Figure 1. Validation of monoclonal anti-corin antibody specificity.
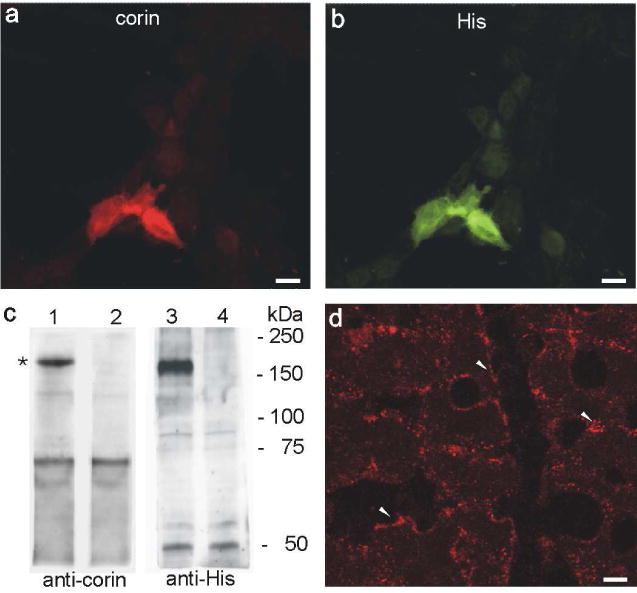
(a and b) Double labeling of anti-corin (red, a) and anti-His (green, b) on HEK 293 cells transiently transfected with a His-tagged full-length corin construct. Both signals are identical. (c) Western blot analysis of corin (lane 1 and 3) and mock (lane 2 and 4) transfected HEK 293 cells using anti-corin (lane 1 and 2) and anti-His antibodies (lane 3 and 4). Both show a strong band at ca. 170 kDa (asterisk). (d) Indirect immunohistochemistry revealed a highly abundant corin expression in heart tissue with a predominant localization at the plasma membrane (arrowheads). Additionally positive staining of intracellular vesicles near the plasma membrane is found. Magnification: calibration bar = 10 μm
Renal corin expression
Double staining procedures were then performed on rat kidney sections to analyze the intrarenal distribution of corin. Positive corin immunostaining was found in epithelial cells, with segmental expression in the proximal tubule, thick ascending limb (TAL), connecting tubule (CNT), and throughout the collecting duct (CD). Highest expression level was observed in the medulla. In the proximal tubule (identified by the brush border membrane stained with phalloidin) corin staining was found in the apical endocytic compartment (Figure 2a). In TAL (identified by NKCC2 staining), corin was expressed in vesicles around the nucleus and within the cytoplasm (Figure 2b). Similarly, in CD (identified by AQP2 staining) corin was localized evenly distributed in cytoplasmic vesicles (Figure 2c). Interstitial cells were also positive for corin.
Figure 2. Immunohistochemical localization of corin in rat kidney.
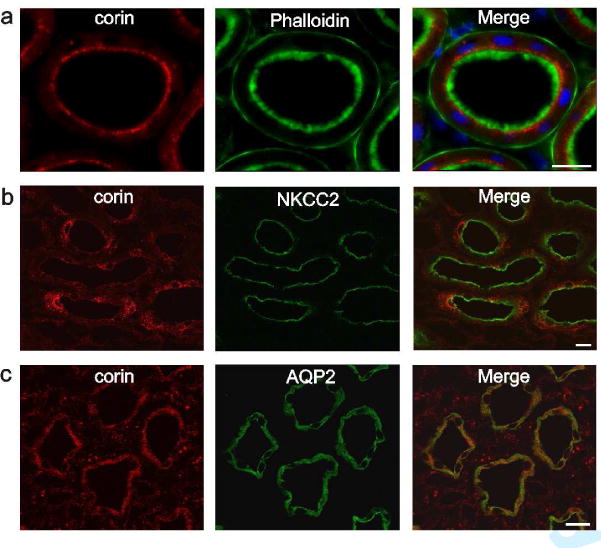
Double labeling experiments of corin (red) and nephron segments markers (green) using fluorescent phalloidin, anti-NKCC-2 and anti-aquaporin-2. (a) Corin is highly expressed in subapical vesicles of the proximal tubule. In the merge image nuclear DAPI stain is added. (b) Cytoplasmic vesicular expression is shown in the thick ascending limb and (c) collecting duct of the renal medulla. Magnification: calibration bar = 20 μm
Sites of renal ANP production
On rat kidney sections in situ hybridization was performed to establish the site(s) of renal ANP production. Within the glomerulus, ANP mRNA was found to be produced in podocytes (Figure 3a), while other glomerular cells remained negative. Along the nephron, the proximal tubule, identified by its brush border, showed weak staining for ANP mRNA mainly in the S3 segment. Strongest ANP mRNA signals were detected in TAL and in the CNT/CD system, as identified by their location within the kidney (Figure 3b). ANP mRNA labeling was also detected along the entire collecting duct until the papillary tip (Figure 3c). Interstitial cells of the renal medulla were stained for ANP as well. As controls, ANP cRNA sense probes were applied simultaneously; no signal was generated (Figure 3d–f). Immunohistochemical localization of ANP demonstrated the typical expression pattern in heart cardiomyocyte vesicles around the nucleus, confirming validity of the antibody used (Figure 3g, h). In the kidney however, ANP was only detected in the medullary CD of the papillary tip (Figure 3i – k). Additionally, CD cells exhibited an heterogenous pattern of cellular immunostaining, predominantly located in vesicular structures of the cytoplasm and of the subapical region in close to the plasma membrane.
Figure 3. ANP production and binding sites in the kidney and heart.
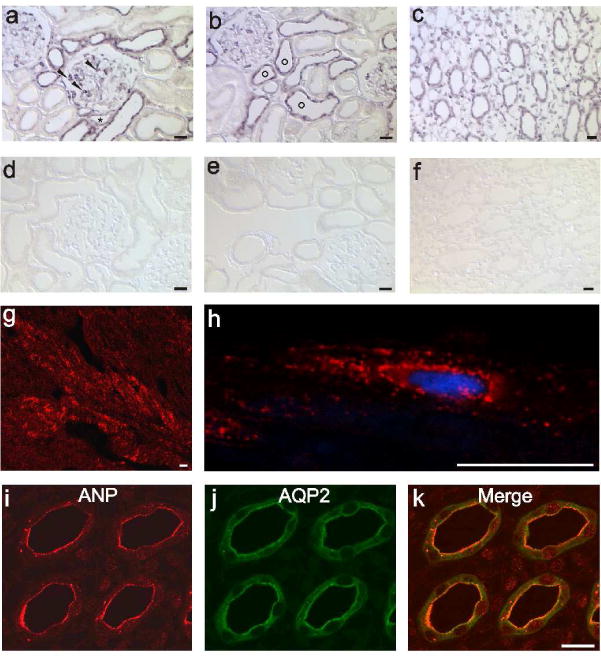
(a – c) ANP mRNA expression obtained by in situ hybridization. (a) In the renal cortex, ANP mRNA labeling is shown in podocytes (arrowheads) and distal tubules. (b) Typical formation of connecting tubules (circles) around an artery within a cortical region is presented. (c) Medullary collecting ducts and interstitial cells labeled for ANP mRNA. (d – f) ANP mRNA sense probes were applied as a negative control showing corresponding areas of the kidney. No signal is obvious. (g and h) Immunohistochemical detection of ANP protein in the heart atrium. ANP expression in (g) low and in (h) greater detail image demonstrates the typical perinuclear vesicle staining. (i – k) Double labeling of ANP with AQP2, localization of ANP protein is exclusively present in the medullary collecting duct within the papillary tip. Magnification: calibration bar = 20 μm
Changes in corin and ANP expression in PAN and GN
Microarray analysis of RNA isolated from the inner stripe and inner medulla revealed strongly reduced corin mRNA abundances in PAN and GN treated rats compared to that of controls (Table 1). Real time PCR revealed that the expression levels of corin mRNA in the renal cortex was slightly, but not significantly, reduced in PAN and GN rats when compared to that of control renal cortices. Concomitantly, levels of ANP mRNA expression were significantly increased in renal cortices from PAN and GN rats. A significant stronger reduction in corin mRNA and concomitant increase in ANP mRNA expression levels were detected in the medulla from PAN and GN rats when compared to that of control rats (Table 1).
Table 1. mRNA abundances of corin and ANP in renal cortex and medulla.
Medullary assessment of corin mRNA by Affymetrix microarray analysis.
| Calculations of mRNA abundance ± SE | |||
|---|---|---|---|
|
| |||
| Control | PAN | GN | |
|
| |||
| Microarray | |||
| corin | 6419# | 2531# | 3625# |
| Cortex | |||
| corin | 1.00 ± 0.20 | 0.66 ± 0.15 | 0.76 ± 0.20 |
| ANP | 1.00 ± 0.72 | 6.64 ± 1.75* | 6.51 ± 3.26* |
| Medulla | |||
| corin | 1.00 ± 0.06 | 0.33 ± 0.12* | 0.37 ± 0.12* |
| ANP | 1.00 ± 0.23 | 37.74 ± 12* | 20.75 ± 6.91* |
Normalized values are presented, detection P < 0.001. TaqMan analysis of cortical and medullary corin and ANP. Control levels are set as 1 = 100%. Means ± SE; n=6;
P < 0.005.
In the renal cortex, the amount of corin and pro-ANP protein did not differ between PAN and GN rats when compared to control rats. Values obtained for corin (control: 100 ± 16%; PAN: 78 ± 6%; GN: 92 ± 9%) and for pro-ANP (control: 100 ± 10%; PAN: 139 ± 24%; GN: 99 ± 12%). However, the amount of corin protein was significantly reduced in the renal medulla of PAN and GN rats (Figure 4a, control: 100 ± 10%; PAN: 35 ± 5%*; GN: 21 ± 8%*; P < 0.05) contrasting an increased level of pro-ANP (Figure 4b; control: 100 ± 23%; PAN: 214 ± 25%*; GN: 191 ± 13%*; *P < 0.05). Consistent with decreased pro-ANP into ANP conversion, PAN and GN rat kidneys exhibited significantly less ANP than control kidneys (Figure 4c; control: 5.6 ± 0.1 ng ANP/mg protein; PAN: 3.5 ± 0.3# ng ANP/mg protein; GN: 3.7 ± 0.2# ng ANP/mg protein ; #P < 0.001).
Figure 4. Altered corin, pro-ANP and ANP expression in proteinuric kidney diseases.
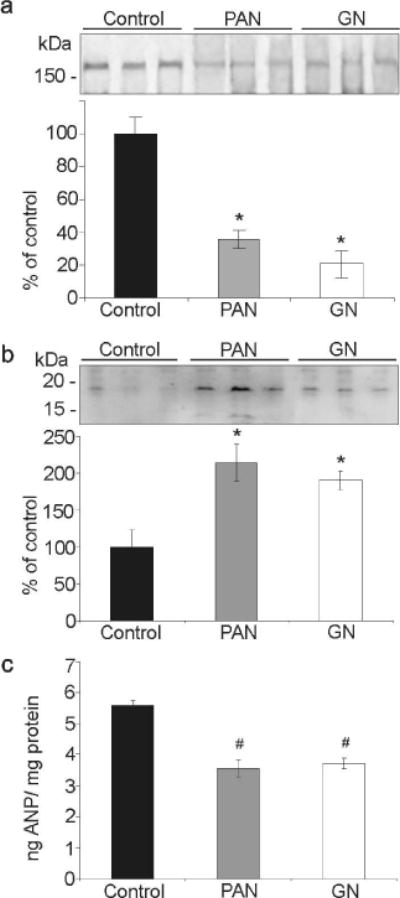
(a) Western blot analysis of medullary corin expression levels. Compared to controls strongly reduced expression levels are present in PAN and GN treated rats. (b) Increased medullary pro-ANP levels in PAN and GN treated rats are demonstrated and are in good agreement with reduced corin expression. (c) Tissue ANP levels were determined by ELISA. Significantly reduced ANP content is found in PAN and GN treated rats. Values are means ± SE; n=6 per group; *P < 0.05 and #P < 0.001.
Expression of ENaC, Na,K-ATPase, AQP2 and ANP signaling cascade in Cor−/− mice
Because corin was shown to play a key role in the processing of ANP, we next analyzed the protein levels of ENaC subunits, Na,K-ATPase, AQP2 and ANP signaling from Cor−/− mice. Western blot analysis of enriched plasma membrane fractions from whole kidney homogenate of Cor−/− mice demonstrated a significant increase in the amount of β-ENaC as compared to that of wild-type kidneys (WT vs. Cor−/−: β-ENaC 100 ± 27 vs. 296 ± 43%*; P < 0.001), whereas α- and γ-ENaC, as well as Na,K-ATPase, and AQP2 remained unchanged (Figure 5a and b). In accordance with these findings, immunohistochemistry revealed a strong medullary increase in β-ENaC expression in medullary kidney sections of Cor−/− mice (Figure 5c).
Figure 5. Alterations in the abundance of β-ENaC and ANP induced signaling components in Cor−/− mice compared to wild type mice.
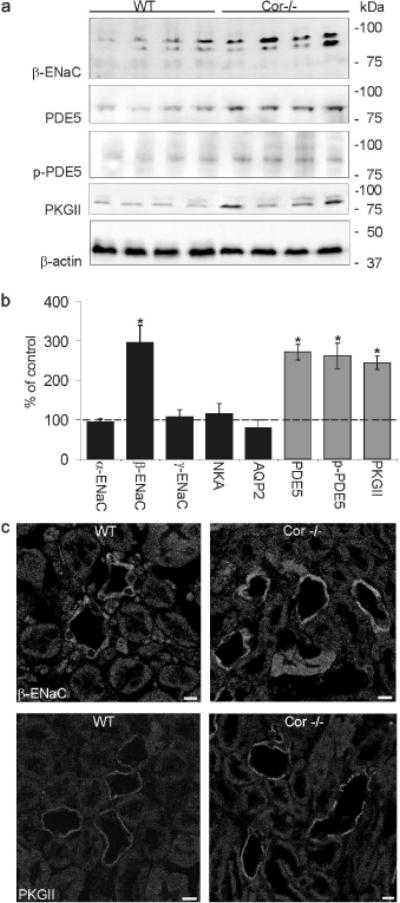
(a) Representative western blots of β-ENaC and PDE5, phospho-PDE5 (p-PDE5), PKGII and β-actin (as loading control) are shown. Strongly increased abundance is found for β-ENaC, PDE5, p-PDE5 and PKGII in Cor−/− mice compared to wild type mice. (b) Corresponding densitometric evaluations of western blot analysis on ENaC subunits, Na/K-ATPase (NKA), aquaporin-2 (AQP2) and on PDE5, p-PDE5 and PKGII. Values are means ± SE; n=5 per group; *P < 0.05. (c) Immunohistochemical labeling of kidneys from Cor−/− mice show increased signal intensity of β-ENaC and PKGII in medullary collecting duct cells compared to wild type mice. Magnification: calibration bar = 20 µm
Next, in order to identify mechanisms which could be responsible for the augmented β-ENaC abundance observed in Cor−/− mice, the expression levels of signaling components from the ANP transduction cascade were determined. Western blot analysis revealed a strong increase in PDE5, phospho-PDE5 and PKGII abundance in kidneys from Cor−/− mice as compared to that of wild-type counterparts (WT vs. Cor−/−; PDE5: 100 ± 14 vs. 272 ± 20%*; phospho-PDE5: 100 ± 25 vs. 262 ± 33%* and PKGII: 100 ± 7 vs. 245 ± 18%*; P < 0.05; Figure 5a and b). Immunohistochemical studies also showed a more intense immunostaining of PKGII in medullary CD from Cor−/− mice than in wild-type CDs (Figure 5c).
ANP inhibits agonist-induced aldosterone synthesis in adrenal zona glomerulosa cells.23 In Cor−/− mice, defective ANP production could result in increased aldosterone synthesis, and therefore account for the increase in β-ENaC. Expression levels of components from the aldosterone-induced signaling cascade were determined. Western blot analysis revealed decreased Akt, phospho-Akt(S), phospho-Akt(T), phospho-serum- and glucocorticoid-inducible kinase 1 (p-SGK1) 1, and With No Lysine protein kinase1 (WNK1) in kidneys of Cor−/− mice whereas WNK4 and neuronal precursor cells expressed developmentally down-regulated (Nedd) 4–2 remained unchanged (Cor−/− in per cent reduction from WT mice; Akt: −74 ± 17%*; phospho-Akt(S) −47 ± 7%*; phospho-Akt(T) −48 ± 11%*; p-SGK1 −81 ± 7%*; WNK1 −68 ± 12%*; P < 0.05, Figure supplement 1). Collectively, these results argue against an aldosterone-dependent mechanism in Na+ retention and therefore hypertension observed in Cor−/− mice.
Effects of cGMP reduction in mpkCCDcl4 cells on ANP signaling
We hypothesized that the decline in cellular cGMP generation could account for the augmented β-ENaC, PDE5, phospho-PDE5 and PKGII abundance in Cor−/− mice. To validate this hypothesis, confluent mpkCCDcl4 cells were incubated with 500 μM 3-isobutyl-1-methylxanthine (IBMX) and 100 μM 8-bromo-cGMP (8-br-cGMP) for 24 h and then maintained under FCS-free medium for the next 0 to 24 hours. The reduction in cGMP content resulted in an increase in β-ENaC, PDE5, phospho-PDE5, and PKGII protein content (expressed in % increase from time 0: β-ENaC +111 ± 48%* (4h), +135 ± 62%* (8h), +108 ± 38%* (24h); PDE5 +275 ± 56%* (4h), +360 ± 72%* (8h), +212 ± 48%* (24h); phospho-PDE5 +363 ± 48%* (4h), +307 ± 42%* (8h), +287 ± 38%* (24h); and PKGII +117 ± 25%* (4h), +98 ± 8%* (8h), +173 ± 35%* (24h); P < 0.05, Figure 6a). Similar results were obtained when using IBMX only (data not shown). We next analyzed, whether augmented PDE5 or/and PKGII would result in increased phosphorylation of PDE5 and in increased β-ENaC expression level. mpkCCDcl4 cells were transfected with PDE5 or/and PKG II constructs. Cells transfected with PDE5 exhibited increases in β-ENaC, phospho-PDE5 and PKGII (in % increase from mock transfected cells: β-ENaC +134 ± 30%*; phospho-PDE5 +40 ± 5%*; and PKGII +107 ± 21%*; P < 0.05, Figure 6b); cells transfected with PKGII had increased β-ENaC and phospho-PDE5 (in % increase from mock transfected cells: β-ENaC +517 ± 48%*; phospho-PDE5 +78 ± 12%*; P < 0.05) and cells double-transfection with PDE5 and PKGII constructs exhibited increased levels of β-ENaC and phospho-PDE5 (in % increase from mock transfected cells: β-ENaC +548 ± 67%*; phospho-PDE5 +200 ± 15%*; P < 0.05).
Figure 6. Alterations in expression levels of β-ENaC and ANP signaling components after cGMP reduction and PDE5 and PKGII overexpression in mpkCCDcl4 cells.
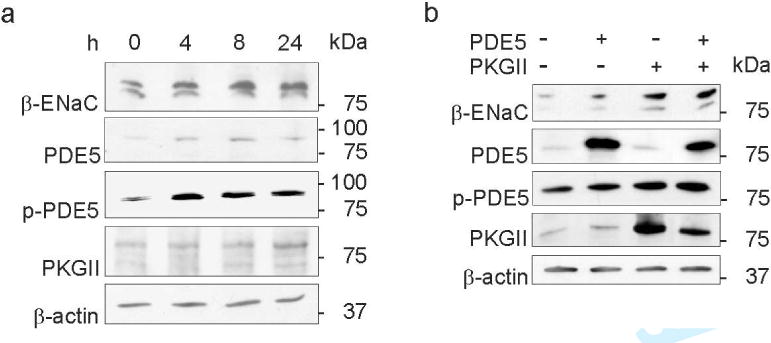
(a) Western blots of β-ENaC, PDE5, phospho-PDE5 (p-PDE5), PKGII and β-actin (as loading control) on mpkCCDcl4 cells underwent reduction in cGMP for 0, 4, 8 and 24 hours (h) are presented. Increases in β-ENaC, PDE5, p-PDE5 and PKGII abundance are shown. (b) Western blots of β-ENaC, PDE5, p-PDE5, PKGII and β-actin (as loading control) on transiently transfected mpkCCDcl4 cells using pcDNA3.1 (mock), PDE5 and PKGII constructs. Transient transfection of PDE5 and PKGII results in augmented β-ENaC abundance and transfection of PKGII phosphorylates PDE5. Values are means ± SE; n=8 per experiment; *P < 0.05.
Analysis of ANP signaling cascade in PAN and GN
Finally, western blot analysis of renal medulla-enriched plasma membrane fractions from control rat and PAN and GN rat renal medulla were undertaken to analyze the expression levels of components of the ANP signaling. Western blot analysis revealed a significant increased expression levels of β-ENaC, PDE5, phospho-PDE5 and PKGII proteins in the medulla from PAN and GN rat kidneys compared to that of control rat kidneys (Figure 7, control: 100 ± 14%; PAN: 256 ± 26%*; GN: 324 ± 7%*; P < 0.05, PDE5 control: 100 ± 19%; PAN: 354 ± 46%*; GN: 221 ± 10%*; P < 0.05, phospho-PDE5 control: 100 ± 20%; PAN: 185 ± 22%*; GN: 190 ± 5%*; P < 0.05 and PKGII control: 100 ± 19%; PAN: 267 ± 40%*; GN: 257 ± 20%*; P < 0.05).
Figure 7. Alterations in abundance of β-ENaC and ANP signaling components in proteinuric kidney diseases.
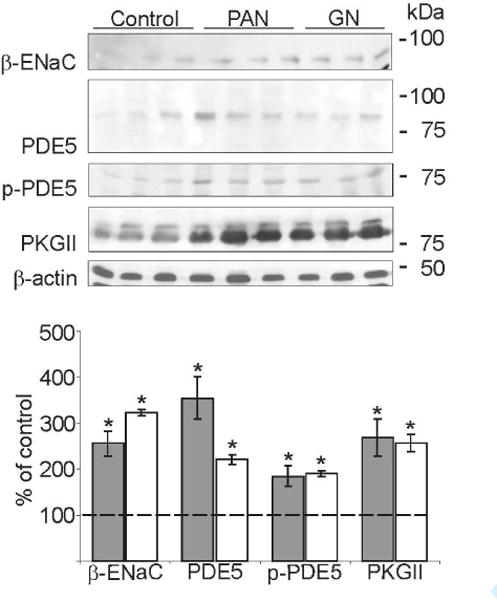
Western blots of medullary β-ENaC, PDE5, phospho-PDE5 (p-PDE5), PKGII and β-actin (as loading control). Compared to controls strongly increased expression levels are present in PAN (grey bar) and GN (white bar) rats for all parameters analyzed. Densitometric evaluations are presented below. Values are means ± SE; n=5 per group; *P < 0.05.
DISCUSSION
In search of possible causes for volume retention in proteinuric kidney disease gene expression profiling was performed in rat models of PAN-induced nephrotic syndrome and acute anti-Thy1 GN using Affymetrix microarray analysis. Corin, a transmembrane spanning protease responsible for converting pro-ANP into ANP, was identified to be markedly reduced in PAN and GN. Corin appears to be an interesting candidate since a number of studies proposed the hypothesis of a dysregulated renal ANP system which could account for the salt retention observed in PAN.18,19
We produced an anti-corin antibody which exhibited similar cardiac expression pattern as in the pioneer study of Yan et al.20 Additionally, we identified positive corin immunostaining in plasma membranes from rat cardiac myocytes, which is in agreement with the hypothesis of pro-ANP conversion at the cell surface. Corin was also detected in vesicles of the proximal tubule, TAL and CD and medullary interstitial cells. Similarly, ANP exhibited an analogous intra-renal pattern of mRNA expression but the ANP protein could only be detected in CD segments from the medulla. We have no clear explanation for the lack of ANP protein expression in nephron segments exhibiting significant ANP mRNA levels. Despite the high mRNA expression, it cannot be excluded that in contrast to cardiac myocytes, ANP protein processed in podocytes, proximal tubules and TAL is rapidly secreted, and therefore its expression remains under the detection limit. However, highest protein expression level of ANP was found in medullary CD and is in agreement with most prominent expression of the ANP receptor.24 Earlier studies demonstrated that the inner medullar CD is the main site of action of atrial peptides, although ANP actions were demonstrated on multiple nephron segments as well. Furthermore, the critical role of the distal nephron segments in ANP-induced natriuresis was nicely demonstrated in natriuretic peptide receptor overexpressing mice.25
Both, acute anti-Thy1 GN and PAN-induced nephrotic syndrome show strongly reduced corin expression, which resulted in augmented pro-ANP and diminished conversion into active ANP. These data suggest that, as in the heart,22 corin may function as a pro-ANP cleavage protease. Released ANP binds to the membrane-bound guanylyl cyclase and thereby modulates cellular functions mediated through the intracellular second messenger cGMP, which in turn regulates several target enzymes.17,26 Most of the known cGMP effects are mediated by the activation of cGMP-dependent protein kinases (PKGs), cGMP-regulated ion channels, or by the regulation of cGMP-dependent PDEs. The increase in cGMP is terminated by the action of cGMP-degrading PDEs. In many cell types, PDE5 plays a major role in cGMP hydrolysis.27 In intact cells, activation of PDE5 induced its phosphorylation at a conserved serine residue by PKG. Thus, cGMP response is determined by the ratio of cGMP-forming and -degradating enzymes.
We used Cor−/− mice to analyze the role of down-regulated corin observed in proteinuric kidney diseases. Cor−/− mice exhibited increased pro-ANP associated with deficient ANP levels as well as hypertension,24 both were shown to appear in proteinuric kidney diseases. Most effects were expected to occur in the medulla. Interestingly, Cor−/− mice exhibited a significant increase of the β-ENaC subunit expression in medullary CD. Similarly, aldosterone and vasopressin, differently upregulate the α-, and/or β- and γ-ENaC subunit level, respectively.28 Patients with Liddle syndrome, having a mutation in the PY-motif of the β-subunit, suffer from hypertension through decreased internalization of ENaC.29 Therefore, it can be assumed that the increase in plasma membrane β-ENaC following ANP deficiency could result in an increased cell surface ENaC expression, as has been reported in proteinuric animal models.3,5,6 Exploring the ANP-induced signaling cascade in Cor−/− mice, we found that PDE5, phospho-PDE5 and PKGII were shown to be unexpectedly increased, suggesting that the dysregulation in the signal transduction might be up-stream of both signalling proteins.
As a result of corin deficiency, plasma cGMP concentration and therefore urinary cGMP excretion are significantly reduced as has been shown for membrane-bound guanylyl cyclase A-deficient mice.30 Similarly, PAN rats exhibited reduced urinary cGMP excretion.18 Therefore we hypothesized that the reduction in cGMP could result in an increase of PDE5, PKGII and concomitant phosphorylation of PDE5. In accordance with this hypothesis, reducing the cGMP content in mpkCCDcl4 cells led to the upregulation of PDE5, PKGII and PKGII-induced phospho-PDE5. Similar paradoxical mechanisms are also observed in Cor−/− mice and in experimental models of proteinuric rats. However, further studies will be needed to elucidate the molecular mechanism underlying the cGMP regulatory pathway. Compartmentation and compartment-specific regulation of PDE5 by PKG was shown to allow selective cGMP-mediated regulation of platelet functions.31 Transient transfection of mpkCCDcl4 cells with PDE5 and PKGII constructs evidenced specific interaction between both proteins in terms of PDE5-phosphorylation by PKGII. Both, PDE5 and PKGII share similar effects on β-ENaC activation. Nie et al.32 reported that 8-pCPT-cGMP stimulates ENaC activity using ENaC transfected oocytes. However, early patch-clamp studies showed that cGMP and cGMP-induced PKG reduces Na+ absorption by inhibiting an amiloride-sensitive cation channel with a channel conductance of 28 pS,33 suggesting that different channels can be activated following ANP administration or reduction.
In addition to PKGII, elevated PDE5 itself contributes to salt retention by decreasing intracellular cGMP levels which is the mediator of natriuretic factors such as nitric oxide. PDE5 inhibition was shown to ameliorate renal damage and reduce systolic blood pressure.34 Therefore, a central role for corin, being at the beginning of the cascade must be suggested. In the past controversial results regarding ANP on sodium and fluid transport mechanisms were reported. Some studies demonstrated a decrease of Na+ transport and vasopressin stimulated water transport35–37 whereas others could not confirm these results.37,38 ANP may however influence salt- and water excretion also through its impact on aldosterone synthesis. ANP normally inhibits aldosterone-production in adrenal glands.39 In contrary to the assumed augmented aldosterone level, the observed diminished renal expression of aldosterone-induced signaling cascade in Cor−/− mice rather argue for a reduced aldosterone action that is associated with augmented ANP signaling components.
In summary, proteinuric kidney diseases, such as the nephrotic syndrome and acute GN, exhibit common dysregulated mechanisms mainly in the collecting duct system. One of them involves the reduction in corin expression level, a pro-ANP converting protease, leading to increased levels of ANP signaling components. Augmented PDE5 and PKGII in turn increase β-ENaC abundance and may also influence other renal transporter and channels. These findings therefore strongly suggest that corin, plays a key role in the control of Na+ and water absorption in proteinuric kidney diseases.
METHODS
Animals and experimental protocol
Male Wistar rats (n=30) weighing between 180–200g were obtained from Charles River. Animals were kept under standard conditions and had free access to standard chow and tap water. Anti-Thy1 acute GN and PAN- induced nephrotic syndrome was elicited by injecting intravenously a monoclonal antibody OX-7 (1mg/kg body weight in PBS, pH 7.5)5 or puromycin aminonucleoside (Sigma Aldrich, 150mg/kg body weight in 0.9% NaCl), respectively. For control rats, PBS was injected as a vehicle. To allow urine collection animals were kept in metabolic cages 24 h before and 6 days after injection. Two identical sets of animals were studied (n = 18) for biochemical and (n = 18) for immunohistochemical approaches. At the end of the experiment animals were anesthetized and kidneys were removed or perfusion-fixed. Additionally kidneys of five wild type and five Cor−/− mice21 were used for biochemical and immunohistochemical analysis. The experiments were conducted in accordance to the German Law for the protection of animals.
Cell culture and transfection studies
Human embryonic kidney (HEK) 293 cells were cultured at 37°C in 95% air/5% CO2 in DMEM supplemented with 4.5 g/L glucose, 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cortical collecting duct mpkCCDcl4 cells were grown in defined medium as described previously.40 In brief, growth medium was composed of equal volumes DMEM and Ham’s F12, 60 nM Na+ selenate, 5 μg/ml transferrin, 50 nM dexamethasone, 1 nM triiodothyronine, 10 ng/ml EGF, 5 μg/ml insulin, 2% FBS, and 100 μg/ml Pen/Strep. Cells were grown in a 5%CO2/95% air atmosphere at 37°C.
HEK 293 cells or mpkCCDcl4 cells were transfected either with the pcDNA3.1 alone (mock; Invitrogen) or pcDNA3.1-rcorin containing full-length rat corin, or the pcDNA3-Zeo-hPDE5A41 containing full-length human PDE5, pIRES2-EGFP-rPKGII containing full-length rat PKGII and/or pcDNA3-bPKGII42 containing full-length bovine PKGII. Transient transfection of plasmids was performed using FuGENE 6 transfection reagent (Roche) according to manufacturer′s instruction.
Cells were washed and lysed in RIPA buffer containing 25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and 5mM CaCl2 supplemented with protease inhibitor cocktail (Roche Diagnostics). After a clearing centrifugation step, lysates were loaded on SDS-Page and western blot analysis was performed.
mpkCCDcl4 cells were also incubated with either 500 μM IBMX (Sigma Aldrich) containing 100 μmol 8-br-cGMP (Sigma Aldrich) or IBMX alone for 24 hours. After washing, cells were incubated with FCS, insulin and dexamethsone free growth medium for 0, 4, 8 and 24 hours. Cells were harvested, lysed in RIPA buffer and finally western blot analysis was performed.
Urine analysis
Serum and urine electrolytes were determined by indirect ion-selective electrode measurements (Modular Analytics, Roche Diagnostics). Blood urea nitrogen was quantified enzymatically and serum creatinine concentration was measured by the kinetic Jaffé-method using routine automated methods (Modular Analytics). Creatinine clearance and fractional sodium excretion were calculated using standard equations. Total urinary protein concentrations were measured with standardized autoanalyzer methods (Hitachi 747, Hitachi 911, and STA analyzers; Roche Diagnostics). Urinary pro-ANP excretion was determined by western blot analysis.
Microarray Analysis
Total RNA was isolated from inner medulla and inner stripe of control, PAN and GN rats (n = 4 per group) using RNeasy Mini kit (Qiagen) and combined per group. Quality of total RNA was checked by analysis on a LabChip (Agilent Technologies). Then, RNA was transcribed into cDNA and used to synthesize biotin-labeled cRNA. Fifteen micrograms of fragmented cRNA was hybridized to the rat genome 230 2.0 Array (Affymetrix) for 16 h at 45°C. Raw gene expression data were processed with Microarray Suite 5.0 software (Affymetrix) according to the manufacturer’s recommendations. Text files were exported for the intensity information for each interrogating oligonucleotide and deposited in the NCBI database of gene expression data (GEO, accession number GSE 7103). The data were normalized to account for variability in hybridization and other hybridization artefacts.
Immunohistochemical studies
Immunohistchemical studies were performed on rat tissue sections or on HEK 293 cells grown on glass cover slips. Rats were anesthetized by an intraperitoneal injection of sodium pentobarbital (0.06 mg/g body weight). Kidneys were then in vivo-perfused via the abdominal aorta using 3% PFA dissolved in PBS. Kidneys of mice were removed and immersion fixed. For cryostat sectioning, tissues were protected from freezing artifacts by 800 mOsm sucrose/PBS, shock-frozen and stored at −80°C. For detection of corin monoclonal and polyclonal antibodies directed against the N-terminus were produced as described (procedure modified from Brodsky et al.)43 and characterized. Additionally we employed well-characterized antibodies to the following proteins: His-tag (MoBiTec), pro-ANP/ANP (Abcam and Phoenix Pharmaceuticals), β-actin (Sigma Aldrich), α, β, γENaC,6 αNa/K-ATPase (Upstate), anti-PDE5 and anti-Akt antibodies (Cell Signaling), anti-WNK1 and anti-WNK4 (Biotrend), anti-phospho-SGK1 (Epitomics), anti-Nedd4-2 (Upstate), anti-PKGII44 and anti-phospho-PDE5.41 In double staining experiments, segments were identified using phalloidin for actin filaments marking the brush border of the proximal tubule, anti-NKCC-2 antibody5 for TAL and anti-aquaporin-2 (Santa Cruz Biotechnology) for the CNT/CD system. Sections were blocked, incubated with the respective primary antibody followed by incubation with suitable cy-2- or cy-3-coupled secondary antibodies (Dianova). In double-labeling experiments the primary antibodies were administered consecutively. Nuclei were stained blue with 4′,6-diamino-2-phenylindole. Sections and cells were analyzed using a confocal scanning microscope (TCS SP-2, Leica Microsystems).
Western blot analysis
Preparations of cortical and medullary membrane fractions were performed as described.5 Total protein concentration was measured using a protein assay kit (BCA; Pierce). SDS gel electrophoresis was performed, and proteins were transferred to nitrocellulose membranes and stained with 0.1% Ponceau red as a loading control. After incubation with blocking reagent, membranes were probed with primary antibodies described above and then exposed to horseradish peroxidase-conjugated secondary antibodies (DAKO). Immunoreactive bands were visualized using an enhanced chemiluminescence kit, exposured to X-ray films (Amersham Pharmacia) and analyzed using BIO-PROFIL Bio-1D image software (Vilber Lourmat). Finally data of membrane fraction were normalized for β-actin expression. All experiments were replicated at least two times.
ELISA
Rat ANP was detected in whole kidney homogenate using an commercially available ANP ELISA kit (Assaypro). Assay was performed according to manufacturer’s instruction.
RNA isolation, reverse transcription and real-time PCR
Total RNA was extracted from kidney cortices and medulla using RNeasy Mini kit (Qiagen), and treated with DNase I (Invitrogen). Reverse transcription was performed using Sensiscript RT kit (Qiagen). TaqMan Gene Expression Assay for corin Rn00711035_m1 and for ANP Rn00561661_m1 were used and the experiment was performed according to the manual provided by Applied Biosystem (Foster City, CA, USA). Threshold cycle (Ct) values were set in the linear phase of exponential amplification. The difference between values obtained for corin or ANP and that of the housekeeping gene GAPDH (∆ Ct) was calculated and compared between control, PAN and anti-Thy1 GN groups.
In situ hybridization
The mRNA of ANP was explored by in situ hybridization using digoxigenin-labeled riboprobes (Roche Diagnostics). The 458 bp coding sequence of ANP was cloned into the TOPO 2.1 vector (Invitrogen). Sense and antisense probes were generated by in vitro transcription of the corresponding cDNAs. In situ hybridization was performed on paraffin sections according to an established protocol.45
Presentation of data and statistical analysis
Quantitative data are presented as means ± SE. For statistical comparison, the Mann-Whitney rank sum test was employed. *P values of less than 0.05 and #P values of less than 0.001 were considered statistically significant.
Supplementary Material
Western blots of Akt, p-Akt(S), p-Akt(T), phospho-serum- and glucocorticoid-inducible kinase 1 (p-SGK1), and With No Lysin protein kinase1 (WNK1) demonstrate reduced expression levels in Cor−/− mice compared to wild type mice, whereas WNK4 and neuronal precursor cells expressed developmentally down-regulated (Nedd) 4–2 remained unchanged. Densitometric evaluations are presented below. Values are means ± SE; n=5 per group; *P < 0.05.
Acknowledgments
Support from the Deutsche Forschungsgemeinschaft (FOR 667; H.P., S.B., F.T.) is gratefully acknowledged. We thank Kerstin Riskwosky (FOR 667, Berlin) for technical expertise. W.W. and Q.W. were supported in part by grants from the NIH (R01HL089298) and Ralph Wilson Medical Research Foundation.
References
- 1.Ichikawa I, Rennke HG, Hoyer JR, et al. Role for intrarenal mechanisms in the impaired salt excretion of experimental nephrotic syndrome. J Clin Invest. 1983;71(1):91–103. doi: 10.1172/JCI110756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buerkert J, Martin DR, Trigg D, et al. Sodium handling by deep nephrons and the terminal collecting duct in glomerulonephritis. Kidney Int. 1991;39:850–857. doi: 10.1038/ki.1991.107. [DOI] [PubMed] [Google Scholar]
- 3.Kim SW, Wang W, Nielsen J, et al. Increased expression and apical targeting of renal ENaC subunits in puromycin aminonucleoside-induced nephrotic syndrome in rats. Am J Physiol. 2004;286(5):F922–F935. doi: 10.1152/ajprenal.00277.2003. [DOI] [PubMed] [Google Scholar]
- 4.Deschenes G, Doucet A. Collecting duct (Na+/K+)-ATPase activity is correlated with urinary sodium excretion in rat nephrotic syndromes. J Am Soc Nephrol. 2000;11:604–615. doi: 10.1681/ASN.V114604. [DOI] [PubMed] [Google Scholar]
- 5.Gadau J, Peters H, Kastner C, et al. Mechanisms of tubular volume retention in immunemediated glomerulonephritis. Kidney Int. 2009;75(7):699–710. doi: 10.1038/ki.2008.649. [DOI] [PubMed] [Google Scholar]
- 6.Kastner C, Pohl M, Sendeski M, et al. Effects of receptor-mediated endocytosis and tubular protein composition on volume retention in experimental glomerulonephritis. Am J Physiol. 296(4):F902–F911. doi: 10.1152/ajprenal.90451.2008. [DOI] [PubMed] [Google Scholar]
- 7.Audigé A, Yu ZR, Frey BM, et al. Epithelial sodium channel (ENaC) subunit mRNA and protein expression in rats with puromycin aminonucleoside-induced nephrotic syndrome. Clin Sci. 2003;104:389–395. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 8.Lourdel S, Loffing J, Favre G, et al. Hyperaldosteronemia and activation of the epithelial sodium channel are not required for sodium retention in puromycin-induced nephrosis. J Am Soc Nephrol. 2005;16:3642–3650. doi: 10.1681/ASN.2005040363. [DOI] [PubMed] [Google Scholar]
- 9.Carattino MD, Hughey RP, Kleyman TR. Proteolytic processing of the epithelial sodium channel γ subunit has a dominant role in channel actzivation. J Biol Chem. 2008;283(37):25290–25295. doi: 10.1074/jbc.M803931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughey RP, Carattino MD, Kleyman TR. Role of proteolysis in the activation of epithelial sodium channels. Curr Opin Nephrol Hypertens. 2007;16(5):444–450. doi: 10.1097/MNH.0b013e32821f6072. [DOI] [PubMed] [Google Scholar]
- 11.Svenningsen P, Bistrup C, Friis UG, et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20(2):299–310. doi: 10.1681/ASN.2008040364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol. 2009;71:361–379. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 13.Deschênes G, Wittner M, Stefano A, et al. Collecting duct is a site of sodium retention in PAN nephrosis: a rationale for amiloride therapy. J Am Soc Nephrol. 2001;12(3):598–601. doi: 10.1681/ASN.V123598. [DOI] [PubMed] [Google Scholar]
- 14.Doucet A, Favre G, Deschênes G. Molecular mechanism of edema formation in nephrotic syndrome: therapeutic implications. Pediatr Nephrol. 2007;22(12):1983–1990. doi: 10.1007/s00467-007-0521-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pyo HJ, Summer SN, Niederberger M, et al. Arginine vasopressin gene expression in rats with puromycin-induced nephrotic syndrome. Am J Kidney Dis. 1995;25(1):58–62. doi: 10.1016/0272-6386(95)90626-6. [DOI] [PubMed] [Google Scholar]
- 16.Ni Z, Vaziri ND. Downregulation of nitric oxide synthase in nephrotic syndrome: role of proteinuria. Biochim Biophys Acta. 2003;1638(2):129–137. doi: 10.1016/s0925-4439(03)00061-9. [DOI] [PubMed] [Google Scholar]
- 17.Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med. 1998;339(5):321–328. doi: 10.1056/NEJM199807303390507. [DOI] [PubMed] [Google Scholar]
- 18.Valentin JP, Ying WZ, Couser WG, et al. Extrarenal resistance to atrial natriuretic peptide in rats with experimental nephrotic syndrome. Am J Physiol. 1998;274(3 Pt 2):F556–F563. doi: 10.1152/ajprenal.1998.274.3.F556. [DOI] [PubMed] [Google Scholar]
- 19.Perico N, Delaini F, Lupini C, et al. Blunted excretory response to atrial natriuretic peptide in experimental nephrosis. Kidney Int. 1989;36(1):57–64. doi: 10.1038/ki.1989.161. [DOI] [PubMed] [Google Scholar]
- 20.Yan W, Wu F, Morser J, et al. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. PNAS. 2000;97:8525–8529. doi: 10.1073/pnas.150149097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan JCY, Knudson O, Wu F, et al. Hypertension in mice lacking the proatrial natriuretic peptide convertase corin. PNAS. 2005;102:785–790. doi: 10.1073/pnas.0407234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan W, Sheng N, Seto M, et al. Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J Biol Chem. 1999;274(21):14926–14935. doi: 10.1074/jbc.274.21.14926. [DOI] [PubMed] [Google Scholar]
- 23.Barrett PQ, Isales CM. The role of cyclic nucleotides in atrial natriuretic peptide-mediated inhibition of aldosterone secretion. Endocrinology. 1988;122(3):799–808. doi: 10.1210/endo-122-3-799. [DOI] [PubMed] [Google Scholar]
- 24.Terada Y, Moriyama T, Martin BM, et al. RT-PCR microlocalization of mRNA for guanylyl cyclase-coupled ANF receptor in rat kidney. Am J Physiol. 1991;261(6 Pt 2):F1080–F1087. doi: 10.1152/ajprenal.1991.261.6.F1080. [DOI] [PubMed] [Google Scholar]
- 25.Zhao D, Pandey KN, Navar LG. ANP-mediated inhibition of distal nephron fractional sodium reabsorption in wild-type and mice overexpressing natriuretic peptide receptor. Am J Physiol. 2010;298(1):F103–F108. doi: 10.1152/ajprenal.00479.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenner BM, Ballermann BJ, Gunning ME, et al. Diverse biological actions of atrial natriuretic peptide. Physiol Rev. 1990;70:665–699. doi: 10.1152/physrev.1990.70.3.665. [DOI] [PubMed] [Google Scholar]
- 27.McAllister-Lucas LM, Sonnenburg WK, Kadlecek A, et al. The structure of a bovine lung cGMP-binding, cGMP-specific phosphodiesterase deduced from a cDNA clone. J Biol Chem. 1993;268(30):22863–22873. [PubMed] [Google Scholar]
- 28.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflügers Arch. 2009;458(1):111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- 29.Lu C, Pribanic S, Debonneville A, et al. The PY motif of ENaC, mutated in liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic. 2007;8:1246–1264. doi: 10.1111/j.1600-0854.2007.00602.x. [DOI] [PubMed] [Google Scholar]
- 30.Shi SJ, Vellaichamy E, Chin SY, et al. Natriuretic peptide receptor A mediates renal sodium excretory responses to blood volume expansion. Am J Physiol. 2003;285(4):F694–F702. doi: 10.1152/ajprenal.00097.2003. [DOI] [PubMed] [Google Scholar]
- 31.Wilson LS, Elbatarny HS, Crawley SW, et al. Compartmentation and compartment-specific regulation of PDE5 by protein kinase G allows selective cGMP-mediated regulation of platelet functions. Proc Natl Acad Sci U S A. 2008;105(36):13650–13655. doi: 10.1073/pnas.0804738105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nie HG, Zhang W, Han DY, et al. 8-pCPT-cGMP stimulates {alpha}{beta}{gamma}-ENaC activity in oocytes as an external ligand requiring specific nucleotide moieties. Am J Physiol. 2010;298(2):F323–F334. doi: 10.1152/ajprenal.00307.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Light DB, Corbin JD, Stanton BA. Dual ion-channel regulation by cyclic GMP and cyclic GMP-dependent protein kinase. Nature. 1990;344(6264):336–339. doi: 10.1038/344336a0. [DOI] [PubMed] [Google Scholar]
- 34.Rodríguez-Iturbe B, Ferrebuz A, Vanegas V, et al. Early treatment with cGMP phosphodiesterase inhibitor ameliorates progression of renal damage. Kidney Int. 2005;68(5):2131–342. doi: 10.1111/j.1523-1755.2005.00669.x. [DOI] [PubMed] [Google Scholar]
- 35.Nonoguchi H, Sands JM, Knepper MA. Atrial natriuretic factor inhibits vasopressin-stimulated osmotic water permeability in rat inner medullary collecting duct. J Clin Invest. 1988;82(4):1383–1390. doi: 10.1172/JCI113742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nonoguchi H, Sands JM, Knepper MA. ANF inhibits NaCl and fluid absorption in cortical collecting duct of rat kidney. Am J Physiol. 1989;256(1 Pt 2):F179–F186. doi: 10.1152/ajprenal.1989.256.1.F179. [DOI] [PubMed] [Google Scholar]
- 37.Rouch AJ, Chen L, Troutman SL, et al. Na+ transport in isolated rat CCD: effects of bradykinin, ANP, clonidine, and hydrochlorothiazide. Am J Physiol. 1991;260(1 Pt 2):F86–F95. doi: 10.1152/ajprenal.1991.260.1.F86. [DOI] [PubMed] [Google Scholar]
- 38.Schlatter E, Cermak R, Forssmann WG, et al. cGMP-activating peptides do not regulate electrogenic electrolyte transport in principal cells of rat CCD. Am J Physiol. 1996;271(6 Pt 2):F1158–F1165. doi: 10.1152/ajprenal.1996.271.6.F1158. [DOI] [PubMed] [Google Scholar]
- 39.Gambaryan S, Butt E, Marcus K, et al. cGMP-dependent protein kinase type II regulates basal level of aldosterone production by zona glomerulosa cells without increasing expression of the steroidogenic acute regulatory protein gene. J Biol Chem. 2003;278(32):29640–29648. doi: 10.1074/jbc.M302143200. [DOI] [PubMed] [Google Scholar]
- 40.Bens M, Vallet V, Cluzeaud F, et al. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol. 1999b;10:923–934. doi: 10.1681/ASN.V105923. [DOI] [PubMed] [Google Scholar]
- 41.Mullershausen F, Russwurm M, Koesling D, et al. In vivo reconstitution of the negative feedback in nitric oxide/cGMP signaling: role of phosphodiesterase type 5 phosphorylation. Mol Biol Cell. 2004;15(9):4023–4030. doi: 10.1091/mbc.E03-12-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koltes JE, Mishra BP, Kumar D, et al. A nonsense mutation in cGMP-dependent type II protein kinase (PRKG2) causes dwarfism in American Angus cattle. Proc Natl Acad Sci U S A. 2009;106(46):19250–19255. doi: 10.1073/pnas.0904513106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brodsky FM. Clathrin structure characterized with monoclonal antibodies. II. Identification of in vivo forms of clathrin. J Cell Biol. 1985;101(6):2055–2062. doi: 10.1083/jcb.101.6.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eigenthaler M, Nolte C, Halbrügge M, et al. Concentration and regulation of cyclic nucleotides, cyclic-nucleotide-dependent protein kinases and one of their major substrates in human platelets. Estimating the rate of cAMP-regulated and cGMP-regulated protein phosphorylation in intact cells. Eur J Biochem. 1992;205(2):471–481. doi: 10.1111/j.1432-1033.1992.tb16803.x. [DOI] [PubMed] [Google Scholar]
- 45.Theilig F, Bostanjoglo M, Pavenstädt H, et al. Cellular distribution and function of soluble guanylyl cyclase in rat kidney and liver. J Am Soc Nephrol. 2001;12(11):2209–2220. doi: 10.1681/ASN.V12112209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blots of Akt, p-Akt(S), p-Akt(T), phospho-serum- and glucocorticoid-inducible kinase 1 (p-SGK1), and With No Lysin protein kinase1 (WNK1) demonstrate reduced expression levels in Cor−/− mice compared to wild type mice, whereas WNK4 and neuronal precursor cells expressed developmentally down-regulated (Nedd) 4–2 remained unchanged. Densitometric evaluations are presented below. Values are means ± SE; n=5 per group; *P < 0.05.