

Abstract
Defective oxidative phosphorylation has a crucial role in the attenuation of mitochondrial function, which confers therapy resistance in cancer. Various factors, including endogenous heat shock proteins (HSPs) and exogenous agents such as dichloroacetate, restore respiratory and other physiological functions of mitochondria in cancer cells. Functional mitochondria might ultimately lead to the restoration of apoptosis in cancer cells that are refractory to current anticancer agents. Here, we summarize the key reasons contributing to mitochondria dysfunction in cancer cells and whether and/or how restoration of mitochondrial function could be exploited for cancer therapeutics.
Keywords: mitochondria, apoptosis, Warburg effects and glycolysis, oxidative phosphorylation, dichloroacetate, cancer therapy
Introduction
Normal cellular growth and development require optimal function of all the regulatory physiological pathways, and any defect in any one or more of these pathways can have adverse effects on the entire cellular homeostasis. Growth and developmental signaling circuits are tightly regulated in normal cells, which tolerate, resist or die in response to external or internal stimuli [1]. Deregulation of these signaling pathways has been implicated in many human diseases, including diabetes, neurodegenerative disorders, developmental defects, and cancer [2]. Defects in pathways regulating cell growth and apoptosis contribute to the carcinogenesis process and are involved in the metastatic progression of tumors [3]. Advanced and metastatic malignancies are characterized by higher rates of cell proliferation and attenuated apoptosis or other cell death signaling [2, 4]. Therefore, current strategies in cancer management target these two processes to retard tumor growth and metastatic progression, resulting in a reduction of disease-related burden in patients.
Defects in apoptosis are among the primary cellular malfunctions observed in tumor initiation, growth, and metastatic progression [3]. Current conventional therapies are designed to induce cell death in cancer with minimal toxic effects to the surrounding normal tissues [5, 6]. Mitochondrial apoptosis is a complex process that is tightly regulated at multiple steps. It involves the release of cytochrome C from mitochondria and subsequent formation of the apoptosome and caspase activation followed by cell death induction [7]. Cancer cells exhibit strong metabolic ‘advantageous faults’, which include increased fatty acid synthesis, boosted glutamine metabolism, and dependence on aerobic glycolysis for energy needs [3, 8, 9] (Figure 1). These properties of cancer cells are normally referred to as the ‘Warburg effect’ [10–13]. All these metabolic adaptations contribute to the development of resistance to current cancer treatments, and subsequent apoptosis evasion in cancer cells [14, 15]. Therefore, exploiting and targeting such metabolic faults could be an attractive strategy in cancer control and management [9, 16]. The physiological status of mitochondria is directly correlated with steady-state oxidative phosphorylation (OXPHOS) and has the potential to induce apoptosis in response to apoptotic stimulus. Defects in normal mitochondria function have strongly been linked to attenuation of apoptosis, and hence, increased cancer growth and progression (Figure 1) [17–19]. Studies suggest that OXPHOS defects in cancer are an important factor in apoptosis evasion [18]. Thus, changes in mitochondrial DNA (mtDNA), which encodes various proteins, including OXPHOS complex components, are linked to cancer [12, 13, 17].
Figure 1.
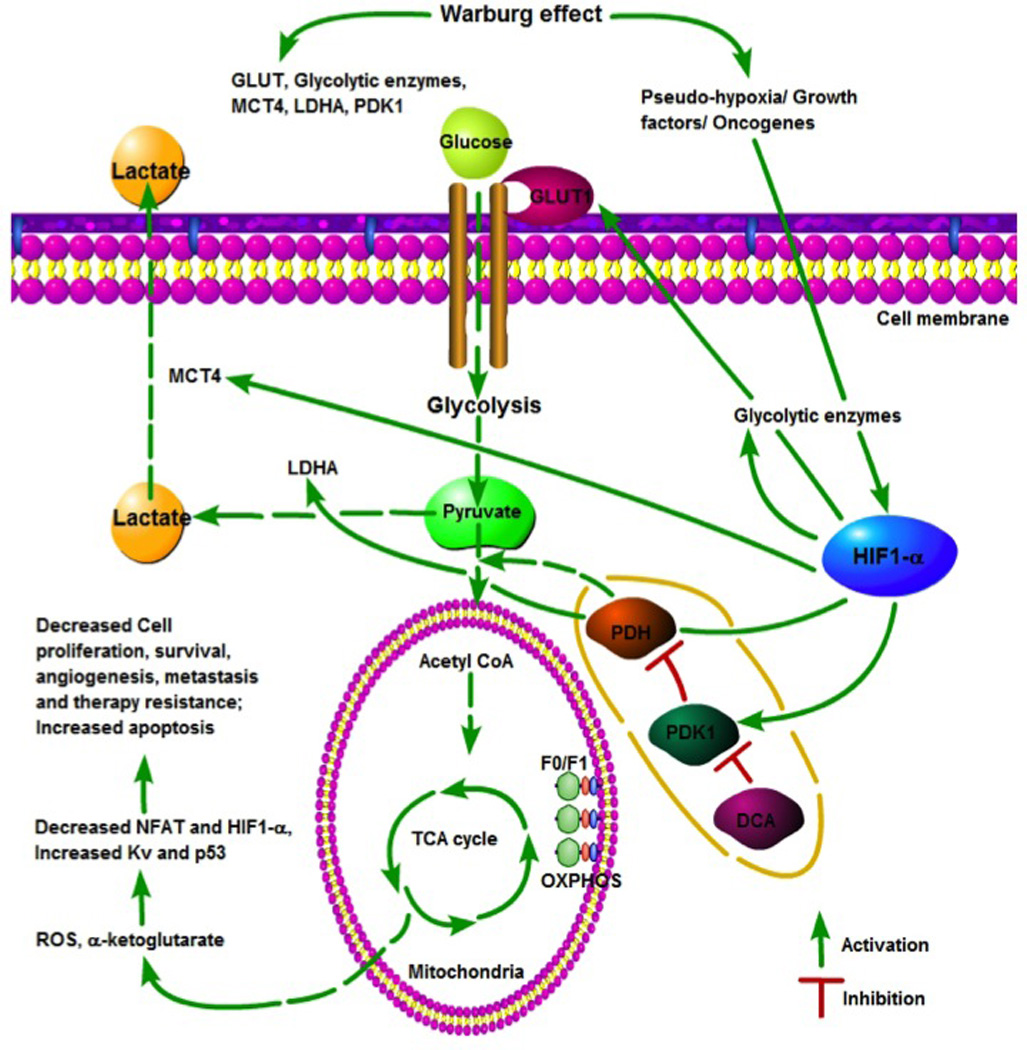
The Warburg effect in cancer cells. Aerobic glycolysis leads to pseudo hypoxic signals that upregulate hypoxia-inducible factor-1alpha (HIF-1α), which in turn regulates the expression of genes related to glucose metabolism. HIF-1α is induced by several factors, including hypoxia, growth factors, and oncogenes. HIF-1α induces the glucose transporter GLUT1, which results in an increase demand for glucose by cancer cells. It also enhances glycolysis via upregulation of glycolytic enzymes. HIF-1α-mediated upregulation of pyruvate dehydrogenase kinase (PDK) inhibits pyruvate conversion into acetyl-co-enzyme A (CoA) via suppression of pyruvate dehydrogenase (PDH). Lactate dehydrogenase A (LDHA) induction by HIF-1α leads to lactate production from pyruvate, which is transported to the external cell environment by monocarboxylate transporter-4 (MTC4), another target of HIF-1α. The anticancer agent dichloroacetate (DCA) suppresses PDK and restores PDH activity, leading to pyruvate entry into mitochondria to reactivate the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS). Subsequently, activation of OXPHOS produced reactive oxygen species (ROS) and α-ketoglutarate, which ultimately lead to the induction of apoptosis, suppression of cancer cell growth and survival, inhibition of angiogenesis and metastasis, and overcoming of tumor resistance.
Recently, there has been renewed interest in targeting and restoring mitochondrial steady state as an attractive strategy for cancer control and management [20]. Metabolic inhibitors, including 2-deoxy-D-glucose (2DG), dichloroacetate (DCA), hexokinase inhibitors, and lactate dehydrogenase (LDH) inhibitors, have been used to specifically block aerobic glycolytic pathway and restore steady-state OXPHOS, and have been shown to be effective against various cancers in vitro and in vivo (Figure 1) [16]. In this regard, DCA has shown positive outcomes by inhibiting growth and proliferation of various cancers in vitro and in vivo by inducing cell cycle arrest and apoptosis [21, 22]. DCA is a small molecule of 150 Da and can penetrate all major tissue types, including brain tissue [23]. Thus, targeting metabolic differences between normal and cancer cells is a rational approach in cancer control and management. Here, we discuss key metabolic alterations and their impact on cancer control, and whether restoration of mitochondrial function by small molecules such as DCA could be a viable approach for cancer management and control.
Metabolic differences between normal and cancer cells
Cancer cells differ from normal cells in various key metabolic aspects and are more dependent on aerobic glycolysis, glutaminolysis, and fatty acid synthesis for cellular proliferation, survival, and growth [10, 24]. To meet their energy needs, normal cells oxidize glucose via the tricarboxylic acid cycle (TCA) in mitochondria to generate 30 ATPs per glucose molecule. By contrast, cancer cells rely heavily on glycolysis to generate two ATPs per glucose molecule in the cytoplasm. Hence, cancer cells upregulate glucose transporters to increase glucose uptake into the cell and meet their energy needs [25–27]. Otto Warburg was the first to observe these effects and postulate that respiration dysfunction in cancer cells prevents glucose oxidation via the TCA in mitochondria [10]. In addition, increased glycolysis also provides metabolites for gluconeogenesis, lipid metabolism, and the pentose phosphate pathway to generate NADPH and macromolecules for anabolic reactions [24, 28]. Such bioenergetic differences in the metabolism of cancer cells versus normal cells provide a potential avenue for the development of cancer therapeutics.
Most cancers originate from hypoxic niches where glucose oxidation is hampered because of a lack of oxygen, and glycolysis remains the sole energy-generating mechanism [29, 30]. Hypoxia leads to induction of hypoxia-inducible factor-1alpha (HIF-1α), which upregulates several glucose transporters and enzymes required for glycolysis, including the gatekeeper pyruvate dehydrogenase kinase (PDK) [29, 30]. In the presence of activated PDK, pyruvate dehydrogenase (PDH) is inhibited, thus limiting the entry of pyruvate in to mitochondria. Activated PDK converts glucose to lactate via glycolysis, whereas inhibition of PDK restores glucose oxidation via mitochondrial respiration (Figure 1). Therefore, during carcinogenesis, enhanced aerobic glycolysis favors cancer growth and metastatic progression; such effects are considered to be one of the reasons for the development of apoptosis resistance in cancer cells (Figure 1) [16, 24, 31]. In conclusion, acquisition of the glycolytic phenotype as an adaptive response to hypoxia eventually confers apoptosis evading potential in cancer cells.
Glycolytic pathway and apoptosis resistance in cancer
Altered metabolism and active glycolytic pathway and its regulators have strongly been linked to apoptosis resistance in cancers. Hexokinase is upregulated and activated in many cancers and translocates to mitochondria, where it inhibits mitochondria-mediated apoptosis [32]. Many events, such as oncogenic activation (c-myc, Akt), tumor suppressor mutations [p53/ Phosphatase and tensin homolog (PTEN)], and hypoxic conditions have been reported to modulate several key glycolytic factors (e.g., hexokinase activation), which confer resistance to cancer cells [33]. Indeed, hexokinase II is upregulated in cancer via epigenetic events [34] or HIF-1, and dysregulated c-Myc [35] associates with chemoresistance and functions as a prognostic marker in various types of cancer [LM1][36, 37]. Given that hexokinase II expression and PDH inactivation by PDK1 cause inhibition of cellular respiration [35], the PDK–PDH circuit has an important role in regulating the energy metabolism switch between glycolysis and OXPHOS. PDK phosphorylates and inhibits the activity of PDH by using ATP, whereas PDH phosphatase dephosphorylates PDH to restore its activity. The activity of PDH converts pyruvate to acetyl-co-enzyme A (CoA), thus facilitating the entry of pyruvate into mitochondria. Thus, in the presence of activated PDK, PDH is inhibited and glucose is converted to lactate via glycolysis. When PDK is inhibited, PDH is activated and glucose oxidation via the TCA is favored in mitochondria [38]. Upregulation of PDK1–4 (natural inhibitors of PDH) inactivates PDH activity and favors aerobic glycolysis in cancer cells [16, 29, 39, 40]. Therefore, targeting the PDK–PDH signaling circuit could be an important anticancer strategy (Figure 1).
Overexpression of PDK3 in cancers enhances a metabolic switch towards glycolysis, and increases drug resistance and cancer recurrence [40]. In colorectal cancer cells, inhibition of PDK1–4 by DCA enhances PDH activity resulting in a flux towards OXPHOS and is accompanied by decreased cell proliferation, G2/M arrest, and increased apoptosis [40, 41]. Thus, a hyperactive glycolytic pathway results in apoptosis resistance in cancer cells, highlighting PDK–PDH signaling as a potential metabolic target in cancer therapy. Table 1 highlights some important targets exploited for metabolic targeting in cancer with the aim of achieving better cancer treatment outcomes.
Table 1.
Metabolic inhibitors targeting cancer cell energy metabolism
| Protein | Pathway | Drugs and/or agents | Function |
|---|---|---|---|
| Complex I | Mitochondrial respiration | Metformin | Inhibits mitochondrial respiration by suppressing complex1 activity |
| GLUT1 | Glycolysis | WZB117; RNAi | Pharmacological or genetic inhibition of GLUT1 exerts antineoplastic effects, both in vitro and in vivo |
| GLS1 | Glutamine metabolism | 968 BPTES RNAi | Malignant cells expressing mutant IDH1 can be particularly sensitive to GLS1-targeting agents |
| Hexokinases | Glycolysis | 2-DG; 3-BP; lonidamine; methyl jasmonate; RNAi | Has shown potential anticancer activities against many cancer models including[LM6] |
| PDK1 | Krebs cycle | DCA | DCA is a prescribed drug for lactic acidosis and has shown potential anticancer activities against many cancers in vitro as well as in vivo |
| PKM2 | Glycolysis | Small hairpin RNA and small-molecule inhibitors | Inhibition of PKM2 reverses the Warburg effect (at least in some tumor models), yet it might favor anabolism |
| LDHA | Glycolysis | FX11; oxamate | LDHA inhibition sensitizes breast cancer cells to cell killing by conventional agents |
Mitochondrial regulation of tumorigenesis
Among the hallmarks of cancer described by Hanahan and Weinberg, reprogramming energy metabolism is crucial and has a fundamental role in carcinogenesis [4]. The relation between mitochondria and overall cellular physiology is not restricted to ATP generation. Mitochondria regulate reactive oxygen species (ROS) production, Ca2+ homeostasis, and programmed cell death (apoptosis), which are important for normal cellular homeostasis [42]. Mitochondria sense the level of overall cellular physiological stress and regulate the release of various apoptosis-inducing factors (including cytochrome C) that lead to caspase activation and cell death induction [43]. Defects in the mitochondrial system, including mtDNA mutations, deletions, and alterations in nuclear-coded proteins that are crucial for mitochondrial function, have been strongly linked to cancer [12, 13, 44]. MtDNA mutations affect many aspects of mitochondrial physiology, including OXPHOS complexes, ROS-producing potential, Ca2+ homeostasis, and apoptosis-inducing potential, which are further aggravated by a high rate of glycolytic pathway [LM2]or via imposing the suppression of OXPHOS activity [44]. A hyperactive glycolytic pathway in cancer cells exerts an inhibitory effect on OXPHOS and has been linked to apoptosis resistance and enhanced cell survival [13, 16]. Mitochondria-mediated cell death induction is dependent on mitochondrial energy production, which is suppressed by excessive glycolysis in cancer cells [16, 18, 45].
Apoptosis-inducing factors are protected inside intact mitochondria and, upon opening of the mitochondrial transition pores (MTP), are released into the cytosol to induce cell death [43]. Opening of MTPs is governed by increased ROS and mitochondrial depolarization [46]. A flux of electrons down the electron transport chain (ETC) determines ROS production and redox state, causing the modulation of matrix metalloproteinases (MMPs). The global redox status depends on the production of NADH, and FADH2 electron donors from the TCA cycle. Acquisition of a glycolytic phenotype and blockade of pyruvate entry into the mitochondria suppress acetyl-CoA production, which in turn attenuates both Krebs’ cycle and the ETC, causing MTP closure and a suppression of apoptosis [16, 38]. Mitochondria are involved in apoptosis via other mechanisms, including intracellular Ca2+ uptake and H2O2 production via dismutation of mitochondrial superoxide by manganese superoxide dismutase. H2O2 diffuses freely and activates plasma membrane K+ channels, hence regulating the influx of Ca2+ and, thereby, caspase activation [22]. Furthermore, retrograde signaling from mitochondria to the nucleus regulates many aspects of cellular physiology other than cell death induction and energy metabolism. Mitochondrial retrograde signaling, including Ca2+ signaling, modulates genetic and epigenetic changes that promote cellular metabolism favoring tumorigenesis [47, 48].
HSP regulation of mitochondria function
Molecular chaperones, a superfamily of highly conserved proteins, maintain proper cellular homeostasis by assisting in protein folding and/or refolding processes as well as help to direct defective and/or misfolded proteins for degradation [49]. Thus, the maintenance of protein homeostasis has an important role in cell survival and functions. Protein homeostasis, also known as proteostasis, is an evolutionarily conserved process and involves a complex signaling pathway known as the unfolded protein response (UPR). UPR is a highly coordinated signaling process that is activated in response to stresses to maintain cellular homeostasis. UPR can independently, or in collaboration with folding machinery of other cellular compartments, regulate protein quality in the cytosol, endoplasmic reticulum (ER), and mitochondria [50, 51].
Cytosolic UPR is called a heat shock response (HSR) and is regulated by HSP70 and HSP90 proteins. Cytosolic HSP70 and HSP90 bind with heat shock protein factor 1 (HSF1) under normal physiological condition, whereas, in response to stress, these two chaperons dissociate from HSF1. The released HSF1 then translocates to the nucleus and enhances the expression of HSPs that attenuate the stresses [50, 52]. HSF1 remodels energy metabolism by promoting cancer cell glycolysis, leading to tumor cell survival and growth by upregulating LDH-A [53]. HSF1 increases glucose uptake, LDH activity, and lactate production, and is involved in tumor resistance to current therapies; therefore, it has been identified as an important therapeutic target in cancer [16]. Recently, HSPs have also been shown to target aerobic glycolysis and to suppress the growth of cancer cells. HSP40 binds to and destabilizes the pyruvate kinase muscle isozyme 2 (PKM2) isoform, which leads to the downregulation of PKM2-mediated PDK1 expression followed by cancer cell growth inhibition [54]. Mitochondrial HSPs, such as HSP70, protect mitochondria against damage from simulated ischemia and/or reperfusion that could occur as a result of decrease in ROS, leading to the preservation of mitochondrial complex activities and, hence, ATP formation [55].
In addition to participating in HSR in the cytosol, HSP70 also participates in mitochondria UPR (UPRmt). Although the mitochondrial HSP70-containing presequence translocase-associated import-motor (PAM) complex directly folds the incoming proteins in the matrix, the HSP60–HSP10 folding machinery in the matrix also has an important role in protein folding, homeostasis, and mitochondria quality control. Defects in this folding machinery are recognized by protein quality control (PQC) proteases, leading to the activation of UPRmt, which can be activated by multiple activities within or around mitochondria, such as increased ROS production or OXPHOS defects resulting from a protein imbalance encoded by mtDNA or nuclear DNA [56–58]. Sustained UPRmt can ultimately lead to activation of the cell death program. Given that HSP60 and other HSPs have important roles in the maintenance of mitochondria function and quality control, the modification of HSPs could be a new target for cancer therapy. Indeed, hyperacetylation-mediated removal of HSP60 from mitochondria was found to be associated with increased cancer cell death [59]. Thus, HSPs offer a new therapeutic window for cancer intervention and could be novel anticancer metabolic targets.
Potential mitochondrial targets for cancer therapy
A normal physiological state of mitochondria is crucial for cellular homeostasis and cell death induction during stress responses [20]. Apoptotic cell death induction requires mitochondrial membrane permeabilization (MMP), which may include both outer and inner MMP. Agents modulating MMP affect the outer MMP or the inner MMP or both [20, 60]. The integrity of the outer mitochondrial membrane is regulated by evolutionarily conserved B-cell lymphoma 2 (Bcl-2) family proteins. Bcl-2 and Bcl-xL proteins prevent the release of apoptogenic factors from mitochondria, whereas proapoptotic Bcl-2 members such as Bcl2-associated X protein (Bax) and Bcl2-antagonist/killer (Bak), induce outer MMP, causing the release of proapoptotic factors from mitochondria. Bcl-2 family members act either alone or in combination with other factors to regulate the outer MMP [61–64]. Therefore, the decision to induce cell death or survival depends on the balance and competition between these two processes. Novel agents that push this balance towards inducing MMP causing the release of proapoptotic factors and subsequent cell death induction are highly desired in cancer control and management. Given that Bcl-2 is overexpressed in many solid tumors and undermines the potential of conventional therapies to induce apoptosis in cancer cells by imparting resistance, many strategies have been used to overcome Bcl-2-induced apoptosis resistance in cancer cells [65–67].
In addition, because mtDNA encodes crucial OXPHOS proteins, the regulation of mtDNA copy number could also be a target for cancer therapy. Indeed, mtDNA has been shown to have a role in determining the sensitivity of cancer cells in response to multiple chemotherapeutic agents [68]. Many agents cause a depletion of mtDNA in mammalian cells, which could be tested for their potential anticancer activities. For instance, 4-quinolone drugs, such as ciprofloxacin, mediate mtDNA loss, which associates with the disruption of mitochondrial function [69]. Lipophilic cations, such as the intercalating anticancer drug ditercalinium, causes selective mtDNA depletion by inhibiting DNA polymerase gamma activity in cultured mammalian cells, which could be more selective than ethidium bromide because it differentially accumulates in the mitochondria [70]. It has been shown that resveratrol depletes mtDNA during apoptosis induction in cancer cells [71]. Additionally, cisplatin shows preferential binding with mtDNA compared with genomic DNA, causing impairment of mitochondrial function and cell death [72]. Together, depletion of mtDNA by anticancer agents or binding of cancer agents with mtDNA suggests the involvement of mtDNA in regulating cancer cell death.
OXPHOS is another promising mitochondrial target in cancer therapy. In this regard, two approaches with opposing mechanisms have been suggested and utilized to achieve cell death induction in cancer cells. One approach focuses on activating OXPHOS with subsequent apoptosis induction via mechanisms such as ROS production [22]. Another approach suppresses OXPHOS activity along with inhibition of the glycolytic pathway to generate a global energy crisis, resulting in the induction of cell death in highly proliferating tumor cells [73]. Under low glucose conditions, mitochondrial activity stimulation by forskolin rendered cancer cells more sensitive to very low levels of OXPHOS inhibitors and, consequently, cell death [74]. Cancer cells develop resistance to existing treatment strategies because of defective mitochondrial functions. Indeed, restoration of OXPHOS function by DCA induced ROS production with subsequent apoptotic cell death [22]. Further studies support the notion that the restoration of OXPHOS function can overcome tumor cell resistance to current therapeutics [75], leading to the development of multiple agents targeting OXPHOS. These agents induce cell death and suppress cell proliferation [22, 75, 76]. For example, rosamines and their derivatives target OXPHOS to exert toxic effects in various cancer cell types [77]. Furthermore, in hepatocellular carcinomas, OXPHOS activation by PDK suppression overcomes sorafenib resistance, leading to stronger tumor regression [75].
Restoration of mitochondrial function by DCA as a potential anticancer strategy
Given that mitochondrial dysfunction is common in cancer cells, restoring mitochondrial function in cancer control and management will be both selective and beneficial. Cancer cell mitochondria are hyperpolarized compared with those of normal cells, manifesting as reduced ROS levels, increased intracellular Ca2+, closure of Kv channels (Kv 1.5) and MTP, activation of nuclear factor of activated T cells (NFAT), upregulation of antiapoptotic Bcl-2, and increased aerobic glycolysis [22, 78]. Increased glycolysis in cancer cells results in an increase in the hexokinase level, which translocates to mitochondria and leads to hyperpolarization of mitochondria and apoptosis inhibition [79]. These events in cancer cells ultimately lead to increased proliferation, enhanced survival, and decreased apoptosis, causing resistance to cell death induction in response to existing anticancer regimens. Overall, reactivating mitochondrial functions will reverse these elements and lead to apoptosis in cancer cells concomitant with the suppression of aerobic glycolysis. The decision either to follow aerobic glycolysis or undergo glucose oxidation depends on the overall PDH–PDK and LDH regulatory circuit interactions. Both activating PDH by inhibiting PDK, and/or suppressing LDH would increase the pumping of pyruvate into the mitochondria, reactivating OXPHOS and ETC and leading to ROS production, Ca2+ flux into mitochondria, and induction of apoptosis.
Agents that increase glucose oxidation by pumping pyruvate to fuel the TCA cycle reactivate OXPHOS and induce ROS production, leading to apoptotic cell death in cancer cells. DCA induces forced glucose oxidation, and reactivates OXPHOS and mitochondrial activity. DCA is inexpensive and has been in human use for more than 40 years. Since the seminal report on the use of DCA to inhibit cancer, multiple studies emphasizing its potential to treat many cancers have been published [22, 80[LM3]].
DCA was found to be less effective in neuroblastoma cells that harbor fewer mitochondrial abnormalities and lack mitochondrial hyperpolarization [22, 78]. However, it has recently been shown that DCA offers potential anticancer effects against highly malignant neuroblastoma cells compared with more differentiated and less malignant cancer cells [81]. Such outcomes warrant further investigations to demonstrate that DCA could be used as a novel therapeutic agent against neuroblastoma. Glycolytic phenotype reversal by DCA has been shown to suppress the growth of metastatic breast cancer cells and to inhibit lung metastasis of rat mammary adenocarcinoma cells [79]. The antitumor action of DCA against T cell lymphoma has also been shown to be associated with the alterations in pH homeostasis and glucose metabolism that lead to suppression of tumor cell survival [82].
DCA activates PDH, resulting in the increased delivery of pyruvate into mitochondria followed by an increase in glucose oxidation leading to restoration of mitochondrial function. DCA mediates its apoptosis-inducing effects via production of ROS and activation of NFAT, leading to the release of apoptogenic factors such as cytochrome C and apoptosis-inducing factor (AIF) from mitochondria into the cytosol [22, 83]. DCA disrupts the HIF-1α-dependent adaptive response under hypoxia in tumors, thus increasing the effectiveness of chemotherapy designed to kill hypoxic tumor cells without posing a toxic threat to oxygenated normal tissue [83]. DCA induces apoptosis in non-small cell lung, breast, and glioblastoma cancer cell lines by restoring glucose oxidation by inhibition of PDK [22]. Subsequently, it was also reported that DCA induces apoptosis in endometrial and prostate cancer cells via a similar mechanism [84].
Although the underlying molecular mechanisms of the anticancer effects of DCA on cancer cells are still not fully defined, the restoration of mitochondrial respiration is considered to be one of the key molecular events causing inhibition of cancer cell proliferation and induction of cell death in cancer. DCA induces Forkhead box O3 (FOXO3) and p53, which trigger activation of BH3-only proteins, such as Bim, Bad, Puma, and Noxa [85, 86]. Thus, increased expression of BH3-only proteins upon DCA treatment leads to Bax activation and cytochrome C release, causing the induction of apoptosis in cancer cells. DCA enhances ROS production, resulting in the induction of autophagy via suppression of Akt-mammalian target of rapamycin (mTOR) signaling [87, 88]. In addition to cell death induction, the anticancer effects of DCA also include inhibition of cancer cell proliferation [89].
DCA overcomes resistance and shows synergistic effects during cancer therapy
Studies have shown that DCA can sensitize cancer cells and, thus, could be used to improve current therapies. For instance, DCA enhances the cytotoxicity of platinum compounds, and such combinatorial strategies could have potential clinical applications for tumors, including in small cell lung cancers and Ewing’s sarcoma, as well as in cancers refractory to cisplatin chemotherapy, such as ovarian cancer [90]. DCA and cisplatin synergistically inhibit the growth of HeLa cells by shifting metabolism from aerobic glycolysis to OXPHOS [91]. Furthermore, a case study reported that DCA in combination with omeprazole (a proton pump inhibitor) and/or tamoxifen exhibited synergistic antiproliferative effects on malignant tumors [92]. DCA enhances adriamycin-induced hepatoma cell killing by inducing ROS levels in vitro as well as in vivo [93]. Interestingly, enhancement in sensitization to various anticancer agents in the presence of DCA has been attributed to the modulation of tumor microenvironment, which requires reprogramming of glucose metabolism, perturbations in pH levels, induction of ROS, and modulation of survival pathways [82]. In a multiple myeloma model, DCA enhanced sensitivity to bortezomib by inhibiting aerobic glycolysis, inducing superoxide production and apoptosis, and suppressing proliferation via G0/G1 and G2/M cell cycle arrest [94]. In hepatocellular carcinoma, cancer cells develop resistance to the drug sorafenib by modulating the bioenergetic environment. DCA activates OXPHOS via inhibition of PDK to overcome such resistance [75]. Similarly, DCA overcame PDK1-mediated taxol-resistance in oral cancer cells under hypoxic conditions [95].
The combination of DCA with capecitabine induced cancer cell apoptosis in a B16 melanoma allograft and human non-small lung cancer cell A549 xenograft tumors [96]. DCA in combination with 5-fluorouracil synergistically inhibited cell proliferation and induced apoptosis in colorectal cancer in vitro [97]. Recently, a co-drug, Bet-CA, an ester derivative of betulinic acid and DCA, has shown promising anticancer effects against a variety of cell types, and also reduced melanoma tumor growth and pulmonary metastasis [98]. DCA sensitizes metformin cytotoxicity by reprogramming glucose metabolism in cancer cells leading to apoptosis induction [73] under high glucose conditions. This suggests that inhibition of global energy metabolism by targeting both glycolysis and OXPHOS leads to cancer cell death. Additionally, DCA in combination with metformin synergistically induced apoptosis in breast cancer cells via increased ROS production as well as diminished metformin-dependent lactate production [99].
DCA-dependent restoration of mitochondria function leads to suppression of angiogenesis
Solid tumors grow up to ~2–3 mm diameter in size and stop growing beyond this because of the limited diffusion of nutrients and gaseous exchange. The Warburg effect in cancer cells or inhibition of OXPHOS leads to induction of pseudohypoxic signals upregulating HIF-1α, and promotes angiogenesis by recruiting nascent blood vessel growth in the tumor. Upregulation of HIF-1α under normoxic conditions leads to the expression of glucose transporter, glycolytic enzymes, and LDHA, causing induction of glycolytic phenotypes [100–102]. In addition, HIF-1α induces expression of PDK1 under hypoxic conditions, which leads to the inhibition of PDH function and causes a shift in metabolism towards aerobic glycolysis [29, 103]. Interestingly, HIF-1α regulates the function of OXPHOS complex IV as well as reducing ROS levels to avoid their toxic effects [104]. This overall scenario leads to tumor growth, progression, and metastasis [105]. Tumor angiogenesis has been implicated as an important target in cancer therapy [106]. In patients with glioblastoma who were given oral DCA for up to 15 months, DCA inhibited HIF-1α and suppressed angiogenesis both in vivo and in vitro by suppressing PDK II [107]. Recently, it was reported that DCA suppressed pseudohypoxia-induced HIF-1α-mediated tumor angiogenesis in non-small cell lung and breast cancer xenotransplant models [83]. Although resistance to current therapies has been attributed to varied mechanisms and hypoxic adaptation, inhibitory tumor angiogenesis ultimately leads to the acquisition of a hypoxic microenvironment in tumors that upregulates HIF-1α, providing resistance to future therapy. Therefore, reversing the hypoxic tumor environment could be an exciting strategy in overcoming such resistance even with existing therapies. DCA has the potential to reverse the hypoxic adaptation to bevacizumab by inducing mitochondrial respiration and enhancing its antitumor effects against glioblastoma xenograft models [108]. Targeting the HIF-1α/PDK III signaling axis using DCA and other silencing strategies has been reported to reactivate oxidative metabolism in melanoma, thus potentiating the therapeutic activity of pro-oxidants such as elesclomol [109].
Inhibition of radiation-induced senescence by DCA associates with glycolysis suppression
Radiation-induced senescence in tumor cells is one of the mechanisms that create a pro-oncogenic niche via a senescence-associated secretory phenotype. Lactate production in the tumor microenvironment during this process induces tumor survival and growth [110]. DCA inhibits radiation-induced senescence via suppressing glycolysis, and enhances radiation-induced apoptosis in tumor cells but not in normal cells. However, it also induces hypoxia in tumor tissues, which might contribute to the reversal of radiation-induced suppression of tumor growth in vivo [111]. Figure 1 summarizes the anticancer effects of DCA.
Clinical implications of DCA in cancer
As a result of tremendous public and scientific acknowledgement of the efficacy of DCA in cancer treatment, several clinical trials have been initiated to evaluate its toxicity and dosage. One such trial with recurrent malignant brain tumors suggested that oral DCA at dosage range established for metabolic diseases was feasible and well tolerated in patients with recurrent malignant gliomas and tumor metastasis to the brain [112]. This highlights the promise of using DCA in clinical trials for various cancer types using doses already established for metabolic diseases. Another clinical trial in glioblastoma either with DCA alone or in combination with standard treatment including radiotherapy reported suppression of angiogenesis and induction of apoptosis in cancer cells both in vivo and in vitro [107]. Oral administration of DCA at a high dose (1 g/day) to a relapsed non-Hodgkin’s lymphoma showed complete remission over 4 years [113]. Treatment of patients with advanced breast and lung cancer with DCA were terminated prematurely because of disease progression and associated complications, which might not have been associated with DCA administration per se [114]; however, oral administration of DCA was not beneficial for patients with advanced-stage non-small cell lung cancer who had been previously treated [114].
DCA inhibits PDK at micromolar concentrations, but higher doses are needed to realize antitumor effects in vitro and in vivo. Such a discrepancy has been associated with the lack of expression of DCA transporters (SLC5A8) in colon and breast tumor cells compared with normal cells, which was the result of methylation-dependent silencing of such transporters in cancer cells [115]. It has been proposed that expressing DCA transporters in tumor cells by using DNA methylation inhibitors would reduce the therapeutic doses of DCA as well as help to alleviate the detrimental effects of higher doses without altering its antitumor mechanisms or efficacy. Indeed, breast, colon, and prostate cancer cells become sensitive to low doses of DCA upon ectotopic expression of SLC5A8 in these cell types [115].
Concluding remarks and future perspectives
Acquisition of aerobic glycolysis and mitochondrial dysfunction in cancers are strongly linked to cancer cell survival, apoptosis evasion, and resistance to current therapies. These outcomes might be associated with the sequestration of proapoptotic proteins by defective mitochondria or the nonapoptotic function of various proapoptotic proteins [116] causing inhibition of cancer cell apoptosis during therapy. Hexokinase and PDK activation inhibit the proapoptotic function of Bax and Bak, thus inhibition of PDK will enable Bax/Bak channel formation, causing cytochrome C release and enhanced cancer cell apoptosis [16, 32]. Additionally, mitochondrial HPS folding machinery has been implicated in regulating mitochondrial function [117, 118], which can be defective in cancer cells, causing uncontrolled tumor growth and development. Thus, targeting by either downregulation of procancerous HSPs or upregulation of anticancerous HSPs could be a novel approach to control tumor growth and progression [53, 54].
Therefore, mitochondria dysfunction either because of metabolic defects or in response to deregulated HSPs signaling suggests that restoration of mitochondrial OXPHOS and inhibition of glycolytic pathway has potential therapeutic benefit for patients with cancer. Indeed, the seminal discovery by Michelakis and colleagues suggests that restoration of mitochondrial respiration by DCA exerts strong anticancer activities [22], which was further validated by various groups in multiple cancer types [80]. Despite significant recent interest in DCA, there are various challenges in establishing it as potent anticancer agent. For example, higher doses of DCA are required to achieve its anticancer activities, which are associated with toxicity to normal tissues. Identification of DCA transporters has provided the opportunity to accumulate DCA at a desired level in cancer cells and to exert its anticancer effects. The introduction of DCA transporters in cancer cells will reduce not only the amount of DCA required, but also the toxicity to patients with cancer during DCA-based therapy. Multiple strategies might help solve some of these problems, such as the development of DCA analogs that selectively target tumor cells. Of interest, multiple analogs have been synthesized and have shown slightly better efficacy compared with parent DCA. These analogs have been mostly synthesized as a complex of multiple DCA molecules with or without the inclusion of anticancer agents [80, 119–121]. Interesting, a recently developed DCA analog, Mito-DCA, specifically accumulates at mitochondria and has shown higher potency against prostate cancer cells at a micromolar concentration [21]. Therefore, future preclinical and clinical research focusing on DCA analogs either alone or as nanoparticle encapsulations or in combination with other anticancer agents could lead to the development of more efficacious anticancer agents.
Highlights.
Mitochondria dysfunction is a key reason for therapy resistance in cancer
Defective oxidative phosphorylation contributes to mitochondria dysfunction
Heat shock proteins and dichloroacetate restore mitochondria function
Restoration of mitochondria function induces apoptosis in cancer cells
Dichloroacetate exerts anticancer effects via multiple mechanisms of action
Acknowledgments
This work was supported in part by the National Cancer Institute of the National Institutes of Health under Award Number R01CA160685, and the American Cancer Society Research Scholar Grant RSG-12-214-01 – CCG to D.C.; and the National Cancer Institute Center Support Grant P30 CA016056 to the Roswell Park Cancer Institute. We apologize to those colleagues whose publications inadvertently could not be cited or are cited incorrectly.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Altman BJ, Rathmell JC. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb. Perspect. Biol. 2012;4:a008763. doi: 10.1101/cshperspect.a008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Spurgers KB, et al. Molecular mediators of cell death in multistep carcinogenesis: a path to targeted therapy. Cell Death Differ. 2006;13:1360–1370. doi: 10.1038/sj.cdd.4401986. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Mashima T, Tsuruo T. Defects of the apoptotic pathway as therapeutic target against cancer. Drug Resist. Updat. 2005;8:339–343. doi: 10.1016/j.drup.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Zhivotovsky B, Orrenius S. Defects in the apoptotic machinery of cancer cells: role in drug resistance. Semin. Cancer Biol. 2003;13:125–134. doi: 10.1016/s1044-579x(02)00130-x. [DOI] [PubMed] [Google Scholar]
- 7.Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010;123:3209–3214. doi: 10.1242/jcs.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandey PR, et al. Anti-cancer drugs targeting fatty acid synthase (FAS) Recent Pat. Anticancer Drug Discov. 2012;7:185–197. doi: 10.2174/157489212799972891. [DOI] [PubMed] [Google Scholar]
- 9.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 11.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochim. Biophys. Acta. 2011;1807:620–625. doi: 10.1016/j.bbabio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yadav N, Chandra D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim. Biophys. Acta. 2013;1836:336–344. doi: 10.1016/j.bbcan.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011;71:4585–4597. doi: 10.1158/0008-5472.CAN-11-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, et al. A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol. Cancer Ther. 2008;7:263–270. doi: 10.1158/1535-7163.MCT-07-0445. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, et al. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carew JS, Huang P. Mitochondrial defects in cancer. Mol. Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dey R, Moraes CT. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. J. Biol. Chem. 2000;275:7087–7094. doi: 10.1074/jbc.275.10.7087. [DOI] [PubMed] [Google Scholar]
- 19.Tomiyama A, et al. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J. Natl. Cancer Inst. 2006;98:1462–1473. doi: 10.1093/jnci/djj395. [DOI] [PubMed] [Google Scholar]
- 20.Fulda S, et al. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010;9:447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 21.Pathak RK, et al. Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem. Biol. 2014;9:1178–1187. doi: 10.1021/cb400944y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonnet S, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 23.Stacpoole PW. The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine: both or neither? Environ. Health Perspect. 2011;119:155–158. doi: 10.1289/ehp.1002554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vander Heiden MG, et al. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang M, et al. Cancer cell metabolism: implications for therapeutic targets. Exp. Mol. Med. 2013;45:e45. doi: 10.1038/emm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koppenol WH, et al. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 27.Yadava N, et al. Impaired mitochondrial metabolism and mammary carcinogenesis. J. Mammary Gland Biol. Neoplasia. 2013;18:75–87. doi: 10.1007/s10911-012-9271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JW, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Lum JJ, et al. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schindler A, Foley E. Hexokinase 1 blocks apoptotic signals at the mitochondria. Cell Signal. 2013;25:2685–2692. doi: 10.1016/j.cellsig.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 33.Cheung EC, et al. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. U. S. A. 2012;109:20491–20496. doi: 10.1073/pnas.1206530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goel A, et al. Glucose metabolism in cancer. Evidence that demethylation events play a role in activating type II hexokinase gene expression. J. Biol. Chem. 2003;278:15333–15340. doi: 10.1074/jbc.M300608200. [DOI] [PubMed] [Google Scholar]
- 35.Kim JW, et al. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007;27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suh DH, et al. Association of overexpression of hexokinase II with chemoresistance in epithelial ovarian cancer. Clin. Exp. Med. 2014;14:345–353. doi: 10.1007/s10238-013-0250-9. [DOI] [PubMed] [Google Scholar]
- 37.Sato-Tadano A, et al. Hexokinase II in breast carcinoma: a potent prognostic factor associated with hypoxia-inducible factor-1alpha and Ki-67. Cancer Sci. 2013;104:1380–1388. doi: 10.1111/cas.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McFate T, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulze A, Downward J. Flicking the Warburg switch-tyrosine phosphorylation of pyruvate dehydrogenase kinase regulates mitochondrial activity in cancer cells. Mol. Cell. 2011;44:846–848. doi: 10.1016/j.molcel.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Lu CW, et al. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008;283:28106–28114. doi: 10.1074/jbc.M803508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Madhok BM, et al. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br. J. Cancer. 2010;102:1746–1752. doi: 10.1038/sj.bjc.6605701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Z, Ristow M. Mitochondria and metabolic homeostasis. Antioxid. Redox Signal. 2013;19:240–242. doi: 10.1089/ars.2013.5255. [DOI] [PubMed] [Google Scholar]
- 43.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 44.Wallace DC. Mitochondria and cancer. Nat. Rev. Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plas DR, Thompson CB. Cell metabolism in the regulation of programmed cell death. Trends Endocrinol. Metab. 2002;13:75–78. doi: 10.1016/s1043-2760(01)00528-8. [DOI] [PubMed] [Google Scholar]
- 46.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 47.Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion. 2013;13:577–591. doi: 10.1016/j.mito.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guha M, et al. Mitochondrial retrograde signaling induces epithelial–mesenchymal transition and generates breast cancer stem cells. Oncogene. 2013 doi: 10.1038/onc.2013.467. XX, YY–ZZ[LM4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chacinska A, et al. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jovaisaite V, et al. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014;217:137–143. doi: 10.1242/jeb.090738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20:214–225. doi: 10.1016/j.cmet.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011;76:91–99. doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- 53.Zhao YH, et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28:3689–3701. doi: 10.1038/onc.2009.229. [DOI] [PubMed] [Google Scholar]
- 54.Huang L, et al. HSP40 interacts with pyruvate kinase M2 and regulates glycolysis and cell proliferation in tumor cells. PLoS ONE. 2014;9:e92949. doi: 10.1371/journal.pone.0092949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williamson CL, et al. Mitochondria protection from hypoxia/reoxygenation injury with mitochondria heat shock protein 70 overexpression. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H249–H256. doi: 10.1152/ajpheart.00775.2007. [DOI] [PubMed] [Google Scholar]
- 56.Runkel ED, et al. Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 2013;9:e1003346. doi: 10.1371/journal.pgen.1003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Houtkooper RH, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baker BM, et al. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorska M, et al. Geldanamycin-induced osteosarcoma cell death is associated with hyperacetylation and loss of mitochondrial pool of heat shock protein 60 (hsp60) PLoS ONE. 2013;8:e71135. doi: 10.1371/journal.pone.0071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gogvadze V. Targeting mitochondria in fighting cancer. Curr. Pharm. Des. 2011;17:4034–4046. doi: 10.2174/138161211798764933. [DOI] [PubMed] [Google Scholar]
- 61.Shamas-Din A, et al. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013;5:a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013;19:546–558. doi: 10.1089/ars.2012.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moldoveanu T, et al. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014;39:101–111. doi: 10.1016/j.tibs.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Czabotar PE, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 65.Billard C. BH3 mimetics: status of the field and new developments. Mol. Cancer Ther. 2013;12:1691–1700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 66.Park D, et al. Novel small-molecule inhibitors of Bcl-XL to treat lung cancer. Cancer Res. 2013;73(17):5485–5496. doi: 10.1158/0008-5472.CAN-12-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sarosiek KA, et al. Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol. 2013;23:612–619. doi: 10.1016/j.tcb.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh KK, et al. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene. 1999;18:6641–6646. doi: 10.1038/sj.onc.1203056. [DOI] [PubMed] [Google Scholar]
- 69.Lawrence JW, et al. Delayed cytotoxicity and cleavage of mitochondrial DNA in ciprofloxacin-treated mammalian cells. Mol. Pharmacol. 1996;50:1178–1188. [PubMed] [Google Scholar]
- 70.Okamaoto M, et al. Ditercalinium chloride, a pro-anticancer drug, intimately associates with mammalian mitochondrial DNA and inhibits its replication. Curr. Genet. 2003;43:364–370. doi: 10.1007/s00294-003-0393-4. [DOI] [PubMed] [Google Scholar]
- 71.Prabhu V, et al. Resveratrol depletes mitochondrial DNA and inhibition of autophagy enhances resveratrol-induced caspase activation. Mitochondrion. 2013;13:493–499. doi: 10.1016/j.mito.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Z, et al. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin. Cancer Res. 2006;12:5817–5825. doi: 10.1158/1078-0432.CCR-06-1037. [DOI] [PubMed] [Google Scholar]
- 73.Choi YW, Lim IK. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014;346:300–308. doi: 10.1016/j.canlet.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 74.Palorini R, et al. Mitochondrial complex I inhibitors and forced oxidative phosphorylation synergize in inducing cancer cell death. Int. J. Cell Biol. 2013;2013:243876. doi: 10.1155/2013/243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen YC, et al. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer. 2013;108:72–81. doi: 10.1038/bjc.2012.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jose C, et al. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim. Biophys. Acta. 2011;1807:707–718. doi: 10.1016/j.bbabio.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 77.Lim SH, et al. Rosamines targeting the cancer oxidative phosphorylation pathway. PLoS ONE. 2014;9:e82934. doi: 10.1371/journal.pone.0082934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen LB. Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- 79.Sun L, et al. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol. Cell Biol. 2008;28:1007–1017. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta. 2014 doi: 10.1016/j.bbcan.2014.08.005. XX,YYY–ZZZ[LM5] [DOI] [PubMed] [Google Scholar]
- 81.Vella S, et al. Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int. J. Cancer. 2012;130:1484–1493. doi: 10.1002/ijc.26173. [DOI] [PubMed] [Google Scholar]
- 82.Kumar A, et al. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem. Biol. Interact. 2012;199:29–37. doi: 10.1016/j.cbi.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 83.Sutendra G, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2013;32:1638–1650. doi: 10.1038/onc.2012.198. [DOI] [PubMed] [Google Scholar]
- 84.Cao W, et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008;68:1223–1231. doi: 10.1002/pros.20788. [DOI] [PubMed] [Google Scholar]
- 85.Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994–1002. doi: 10.1038/sj.cdd.4401908. [DOI] [PubMed] [Google Scholar]
- 86.Liu JW, et al. Induction of prosurvival molecules by apoptotic stimuli: involvement of FOXO3a and ROS. Oncogene. 2005;24:2020–2031. doi: 10.1038/sj.onc.1208385. [DOI] [PubMed] [Google Scholar]
- 87.Lin G, et al. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br. J. Cancer. 2014;111:375–385. doi: 10.1038/bjc.2014.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gong F, et al. Dichloroacetate induces protective autophagy in LoVo cells: involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 2013;4:e913. doi: 10.1038/cddis.2013.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Delaney LM, et al. Dichloroacetate affects proliferation but not survival of human colorectal cancer cells. Apoptosis. 2015;20:63–74. doi: 10.1007/s10495-014-1046-4. [DOI] [PubMed] [Google Scholar]
- 90.Olszewski U, et al. In vitro cytotoxicity of combinations of dichloroacetate with anticancer platinum compounds. Clin. Pharmacol. 2010;2:177–183. doi: 10.2147/CPAA.S11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie J, et al. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int. J. Oncol. 2011;38:409–417. doi: 10.3892/ijo.2010.851. [DOI] [PubMed] [Google Scholar]
- 92.Ishiguro T, et al. Co-treatment of dichloroacetate, omeprazole and tamoxifen exhibited synergistically antiproliferative effect on malignant tumors: in vivo experiments and a case report. Hepatogastroenterology. 2012;59:994–996. doi: 10.5754/hge10507. [DOI] [PubMed] [Google Scholar]
- 93.Dai Y, et al. Dichloroacetate enhances adriamycin-induced hepatoma cell toxicity in vitro and in vivo by increasing reactive oxygen species levels. PLoS ONE. 2014;9:e92962. doi: 10.1371/journal.pone.0092962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sanchez WY, et al. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br. J. Cancer. 2013;108:1624–1633. doi: 10.1038/bjc.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xie Q, et al. Combination of Taxol(R) and dichloroacetate results in synergistically inhibitory effects on Taxol-resistant oral cancer cells under hypoxia. Mol. Med. Rep. 2015;11:2935–2940. doi: 10.3892/mmr.2014.3080. [DOI] [PubMed] [Google Scholar]
- 96.Zheng MF, et al. DCA increases the antitumor effects of capecitabine in a mouse B16 melanoma allograft and a human non-small cell lung cancer A549 xenograft. Cancer Chemother. Pharmacol. 2013;72:1031–1041. doi: 10.1007/s00280-013-2281-z. [DOI] [PubMed] [Google Scholar]
- 97.Tong J, et al. Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J. Biomed. Biotechnol. 2011;2011:740564. doi: 10.1155/2011/740564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saha S, et al. A potent tumoricidal co-drug ‘Bet-CA’ – an ester derivative of betulinic acid and dichloroacetate selectively and synergistically kills cancer cells. Sci. Rep. 2015;5:7762. doi: 10.1038/srep07762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haugrud AB, et al. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast Cancer Res. Treat. 2014;147:539–550. doi: 10.1007/s10549-014-3128-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen J, et al. Dominant-negative hypoxia-inducible factor-1 alpha reduces tumorigenicity of pancreatic cancer cells through the suppression of glucose metabolism. Am. J. Pathol. 2003;162:1283–1291. doi: 10.1016/s0002-9440(10)63924-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Agrawal A, et al. Normoxic stabilization of HIF-1alpha drives glycolytic metabolism and regulates aggrecan gene expression in nucleus pulposus cells of the rat intervertebral disk. Am. J. Physiol. Cell Physiol. 2007;293:C621–C631. doi: 10.1152/ajpcell.00538.2006. [DOI] [PubMed] [Google Scholar]
- 102.Dang DT, et al. Hypoxia-inducible factor-1alpha promotes nonhypoxia-mediated proliferation in colon cancer cells and xenografts. Cancer Res. 2006;66:1684–1936. doi: 10.1158/0008-5472.CAN-05-2887. [DOI] [PubMed] [Google Scholar]
- 103.Papandreou I, et al. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 104.Fukuda R, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 105.Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 106.Bhat TA, Singh RP. Tumor angiogenesis: a potential target in cancer chemoprevention. Food Chem. Toxicol. 2008;46:1334–1345. doi: 10.1016/j.fct.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 107.Michelakis ED, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 108.Kumar K, et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. 2013;91:749–758. doi: 10.1007/s00109-013-0996-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kluza J, et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012;72:5035–5047. doi: 10.1158/0008-5472.CAN-12-0979. [DOI] [PubMed] [Google Scholar]
- 110.Campisi J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liao EC, et al. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis. 2014;5:e1255. doi: 10.1038/cddis.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dunbar EM, et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest. New Drugs. 2014;32:452–464. doi: 10.1007/s10637-013-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Strum SB, et al. Case report: sodium dichloroacetate (DCA) inhibition of the ‘Warburg Effect’ in a human cancer patient: complete response in non-Hodgkin’s lymphoma after disease progression with rituximab-CHOP. J. Bioenerg. Biomembr. 2013;45:307–315. doi: 10.1007/s10863-012-9496-2. [DOI] [PubMed] [Google Scholar]
- 114.Garon EB, et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2014;140:443–452. doi: 10.1007/s00432-014-1583-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Babu E, et al. Role of SLC5A8, a plasma membrane transporter and a tumor suppressor, in the antitumor activity of dichloroacetate. Oncogene. 2011;30:4026–4037. doi: 10.1038/onc.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013;5:a008722. doi: 10.1101/cshperspect.a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Altieri DC. Hsp90 regulation of mitochondrial protein folding: from organelle integrity to cellular homeostasis. Cell Mol. Life Sci. 2013;70:2463–2472. doi: 10.1007/s00018-012-1177-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Haynes CM, Ron D. The mitochondrial UPR: protecting organelle protein homeostasis. J. Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 119.Dhar S, Lippard SJ. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. U. S. A. 2009;106:22199–22204. doi: 10.1073/pnas.0912276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu W, et al. Two mixed-NH3/amine platinum (II) anticancer complexes featuring a dichloroacetate moiety in the leaving group. Sci. Rep. 2013;3:2464. doi: 10.1038/srep02464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang N, Palmer AF. Development of a dichloroacetic acid-hemoglobin conjugate as a potential targeted anti-cancer therapeutic. Biotechnol. Bioeng. 2011;108:1413–1420. doi: 10.1002/bit.23071. [DOI] [PubMed] [Google Scholar]