

Abstract
Visual function in vertebrates is dependent on the membrane-bound retinoid isomerase, RPE65, an essential component of the retinoid cycle pathway that regenerates 11-cis-retinal for rod and cone opsins. The mechanism by which RPE65 catalyzes stereoselective retinoid isomerization has remained elusive due to uncertainty about how retinoids bind to its active site. Here we present crystal structures of RPE65 in complex with retinoid-mimetic compounds, one of which is in clinical trials for treatment of age-related macular degeneration. The structures reveal the active site retinoid-binding cavity located near the membrane-interacting surface of the enzyme as well as an Fe-bound palmitate ligand positioned in an adjacent pocket. With the geometry of the RPE65-substrate complex clarified we delineate a mechanism of catalysis that reconciles the extensive biochemical and structural research on this enzyme. These data provide molecular foundations for understanding a key process in vision and pharmacological inhibition of RPE65 with small molecules.
Keywords: RPE65, retinoid isomerase, non-heme iron enzyme, hydrolase, monotopic membrane protein
INTRODUCTION
Isomerization of retinoids lies at the heart of vertebrate vision 1. Photoisomerization of the 11-cis-retinylidene chromophore of rod and cone opsins initiates light perception, but it is the continuous regeneration of visual chromophore by a metabolic pathway known as the retinoid cycle that allows sustained visual function 2,3 (Fig. 1a). RPE65, a non-heme Fe-dependent monotopic membrane protein, is the essential retinoid isomerase of this pathway 4–6. Whereas loss-of-function mutations in RPE65 lead to devastating childhood blinding disorders such as Leber Congenital Amaurosis 7 partial blockade of RPE65 activity through the use of pharmacological inhibitors has been proposed as a therapeutic strategy for the treatment of “dry” age-related macular degeneration (AMD), a common, debilitating disease for which there are currently no FDA-approved medications 8. Two of these inhibitors, all-trans-retinylamine (Ret-NH2) and emixustat, were rationally designed and the latter has entered clinical trials for treatment of AMD 9,10.
Figure 1. Role of RPE65 in the RPE-based visual cycle.
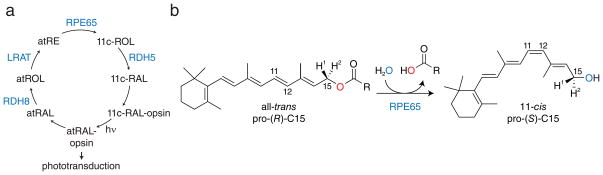
a, Schematic of the visual cycle pathway. Absorption of light (hν) by visual pigments (11c-RAL-opsin) results in their conversion to an active state capable of initiating phototransduction (atRAL-opsin). After a period of time activated pigments lose their retinoid chromophore by hydrolysis which generates free all-trans-retinal (atRAL). This molecule is reduced by retinol dehydrogenases (RDH), mainly RDH8, to form all-trans-retinol, which is then esterified by lecithin:retinol acyltransferase (LRAT) to produce all-trans-retinyl esters (atRE). RPE65 is the essential retinoid isomerase of this pathway that catalyzes the conversion of atRE into 11-cis-retinol (11c-ROL). 11c-ROL is oxidized, primarily by RDH5, to form 11-cis-retinal (11c-RAL), which recombines with opsins to complete the cycle. b, Ester cleavage/isomerization reaction catalyzed by RPE65. Unlike typical biological ester hydrolysis reactions isotope labeling experiments have demonstrated that ester dissociation occurs by O-alkyl cleavage rather than acyl cleavage. Notably, the prochirality of C15 inverts during the reaction.
The role of RPE65 in the visual cycle is to convert the fatty acid all-trans-retinyl esters (predominantly palmitate) produced by lecithin:retinol acyltransferase (LRAT) into 11-cis-retinol, a reaction that requires ester cleavage, geometric isomerization and addition of water (Fig. 1b) 11,12. This reaction, which to our knowledge is unique in biology, and the enzyme that catalyzes it exhibit several remarkable characteristics. 18O-labeling experiments have demonstrated that retinyl ester cleavage does not occur via a typical acyl tetrahedral intermediate but rather through O-alkyl bond cleavage (Fig. 1b) 13,14. Support for this mode of cleavage was also obtained from experiments showing that C15 undergoes an inversion of configuration during the isomerization reaction, a result that implies breakage of the C15-O bond 15,16. Mechanistically, this unusual mode of ester cleavage is thought to be critical for the subsequent isomerization reaction because it gives rise to a retinyl cation with altered molecular orbital structure that allows for isomerization at the C11-C12 bond to readily occur at biological temperatures 13. Additionally, ester cleavage provides energy required to drive the thermodynamically uphill trans-cis isomerization 11. Following ester cleavage and isomerization a water molecule attacks the C15 retinyl cation to produce 11-cis-retinol 17. The RPE65 crystal structure 17 and mutagenesis studies identifying the active site residues Thr147, Phe103, Tyr338 and Phe526 as key determinants of polyene isomerization stereoselectivity 14,18,19 have begun to shed light on the enzymology of this enzyme. However, hypothetical models of the RPE65-retinyl ester complex have placed the retinoid moiety in a region of the active site pocket that is distant from these residues 14,19. Crystallographic and X-ray absorption spectroscopy data indicate the presence of an Fe(II)-bound palmitate ligand in the “as-isolated” state of RPE65, suggesting that the Fe center serves the role of a Lewis acid to facilitate retinyl ester O-alkyl cleavage 20. But the weak and poorly connected electron density for the putative palmitate ligand precluded its definitive assignment. Further progress in understanding catalysis by RPE65 has been hampered by the lack of conclusive experimental data about how retinyl esters bind to its active site. No structures of RPE65 in complex with retinoids or retinoid derivatives have been reported to date. Indeed, no genuine ligand-bound structure has been determined for any member of the carotenoid cleavage oxygenase (CCO) family to which RPE65 belongs leaving a great deal of uncertainty about how these enzymes function catalytically 21–23.
Here, we identified emixustat and a β-ionone-substituted derivative of this compound called MB-001 (1) as potent RPE65 inhibitors suitable for structural studies. Crystal structures of bovine RPE65 in complex with these retinoid-mimetic agents revealed the location of the retinoid-binding site within a negatively charged region of the active site cavity proximal to the membrane-binding surface of the enzyme. An Fe-bound palmitate ligand was found in an adjacent pocket of the active site cavity. The simultaneous binding of these retinoid-mimetic compounds and palmitate thus established the orientation of the retinyl ester in the RPE65-substrate complex. The retinoid-binding site contained a constriction centered over the putative binding site of the retinoid 11-12 double bond formed by residues previously shown to control isomerization stereoselectivity. This structural feature likely stabilizes the retinyl cation intermediate of the retinoid isomerization reaction at carbon 11 to enable preferential production of 11-cis-retinol by RPE65.
RESULTS
Characterization of retinoid mimetics
The poor aqueous solubility of the isoprenoid substrates and products of CCOs appears to be a major factor that has prevented the formation of high occupancy complexes for crystallographic studies. To overcome this inherent limitation we sought to identify retinoid-like molecules with improved solubility characteristics. In analyzing the chemical structure of emixustat it was apparent that this molecule displays good structural correspondence to the retinoid carbon backbone (Fig. 2a). The positively charged amine nitrogen of emixustat aligns with the retinoid C15 atom suggesting that this compound could mimic the C15 retinyl cation transition state. Emixustat is therefore effectively one methylene group shorter than 11-cis-retinol, which could eliminate steric and electrostatic repulsion with the palmitate product of the retinoid isomerization reaction and allow simultaneous binding of both ligands. Importantly, the amino and hydroxyl groups of emixustat confer higher aqueous solubility as compared to retinoids (cLogP values: 3.2 and 6.4 for emixustat and retinol). This structural similarity was enhanced by substitution of a β-ionone ring for the cyclohexyl moiety of emixustat to afford a hybrid molecule referred to as MB-001 (Fig. 2a, Supplementary Results, Supplementary Fig. 1).
Figure 2. Structural analysis and inhibitory effects of retinoid-mimetic visual cycle modulators.

a, Structures and structure relationships between retinoids and non-retinoid visual cycle modulators. By virtue of its rotatable single bonds, MB-001 can adopt a conformation whereby it overlaps with the backbone atoms of the 11-cis-retinyl cation with its amino moiety corresponding to C15 of the retinoid. Selected carbon atoms referenced in the text are numbered in each of the compounds. b, Inhibitory effects of visual cycle modulators on retinoid isomerase activity in vitro. Data points are shown as mean ± s.d. calculated from five replicates. c, Inhibitory effects of visual cycle modulators on visual chromophore regeneration in vivo. Data points are shown as mean ± s.d.; n = 5.
Although emixustat was proposed to directly bind and inhibit the activity of RPE65 there currently are no published data to support this mode of action. Thus, we first measured the inhibitory effects of emixustat and MB-001 in an in vitro retinoid isomerase activity assay using bovine RPE microsomes as the enzyme source. The compounds strongly inhibited 11-cis-retinol production with IC50 values of 232 ± 3 nM and 396 ± 58 nM for emixustat and MB-001, respectively (Fig. 2b). Both compounds were more potent than the extensively studied RPE65 inhibitor, Ret-NH2, which had an IC50 value of 903 ± 41 nM. The effect of these compounds on visual cycle function was also examined in an in vivo rhodopsin regeneration assay (Fig. 2c). Mice were administered the indicated compounds by oral gavage and then subjected to intense light that bleached a large fraction of rhodopsin. Following a 6 h dark incubation period in which rhodopsin regeneration could occur, ocular retinoids were extracted and analyzed by HPLC. Similar to the in vitro results emixustat and MB-001 both strongly suppressed visual cycle function in vivo (Fig. 2c). Interestingly, when RPE65 was exposed to MB-001 during its purification from RPE microsomes the purified protein sample lost its typical red-brown color (Supplementary Fig. 2a) 17. HPLC analysis demonstrated the absence of retinyl esters in MB-001-treated samples suggesting competition for binding sites within the sample including the RPE65 active site (Supplementary Fig. 2b).
RPE65 in complex with emixustat, MB-001 and palmitate
With the inhibitory activity of these compounds confirmed we crystallized RPE65 in the presence of both emixustat and MB-001 and determined the respective crystal structures using diffraction data extending to 1.8 Å and 2.3 Å resolution. (Supplementary Table 1 and Supplementary Fig. 1). The bound inhibitors were unambiguously identified from the initial electron density maps within a V-shaped region of the RPE65 active site cavity proximal to the membrane-embedded substrate-entry port (Fig. 3, a and b and Supplementary Fig. 3a and Supplementary Movie 1). Additional residual electron density in an adjacent hydrophobic pocket within the active site cavity could clearly be assigned to a bound palmitate molecule in both structures (Fig. 3, a and b and Supplementary Fig. 3a and Supplementary Movie 1). The binding site and conformation of the 3-amino-1-phenylpropan-1-ol moiety common to both inhibitors was highly similar between the two structures (Supplementary Fig. 3b). The hydroxyl group of the inhibitors participated in a hydrogen bonding interaction with the hydroxyl moiety of Thr147 whereas their positively charged amino groups formed ionic interactions with the carboxylate moieties of Glu148 and the bound palmitate molecule (Fig. 3c and Supplementary Fig. 3c). A distance of ~5.7 Å separated the inhibitor amine nitrogen from the catalytic Fe. The inhibitor C-O and C-N bonds were roughly parallel, which resulted in an intramolecular hydrogen bonding interaction between the hydroxyl and amine groups. Phe61 and Tyr338 engaged in non-polar interactions with the side chain propyl backbones of both inhibitors. Despite use of racemic emixustat for the crystallization experiments the electron density surrounding the chiral center was consistent with exclusive binding of the (R)-isomer and very similar to that observed for MB-001, which was synthesized as the enantiomerically pure (R)-isomer (Fig. 3b and Supplementary Fig. 3a). The central phenyl rings of the molecules made favorable non-polar and π–π contacts with side chain atoms of Phe103, Tyr275, Thr129, Thr149, and Glu148 (Fig. 3c and Supplementary Fig. 3c). The most extensive contact involved a tilted edge-to-face π–π interaction between the Cδ atom of Phe103 and the carbon atoms of the phenyl ring with contact distances in the range of 3.7 to 3.8 Å. Opposite of this interaction was a ~3.4 Å contact between the inhibitor phenyl ring and the hydroxyl group of Thr147. The phenyl rings of both inhibitors were in relatively close contact (~3.2–3.4 Å) with two water molecules located at the base of the V-shaped cavity. The terminal aliphatic rings of both inhibitors were the most sparsely stabilized portions of the molecules as evidenced by their elevated atomic displacement parameters. The locations of the rings were similar between emixustat and MB-001 but a 180° difference in rotation about the phenoxy bond resulted in mirror-like correspondence between their carbon backbones (Supplementary Fig. 3b). The cyclohexyl ring of emixustat engaged in van der Waals interactions with side chain atoms of Ile259, Phe264, Phe196, Thr149 and Asn194, main chain atoms of Glu148 and Asn175 and two nearby water molecules (Fig. 3c). The gem-dimethyl group of the MB-001 β–ionone ring was oriented towards a hydrophobic surface formed by the side chains of Ile259, Phe279, Tyr239 whereas the monomethyl group formed non-polar contacts with Thr149 and Val128 (Supplementary Fig. 3, a and c). Phe264 and Phe196 formed additional long-range contacts with the β–ionone ring. We speculate that the MB-001 and retinoid β–ionone rings share a common mode of binding. The V-shaped cavity promoted binding of the inhibitors in a bent conformation with the C2-C3-C4-C5 dihedral at a ~50°, cis-like angle consistent with these compounds mimicking the 11-cis retinoid configuration. A cis-like conformation promoted by intramolecular hydrogen bonding was also observed for the N1-C1-C2-C3 dihedral. (Fig. 3, b and c and Supplementary Fig. 3, a and c). In comparison to the “as-isolated” RPE65 crystal structure 17 only subtle structural differences in active site residues close in proximity to the bound inhibitors were observed. By contrast, residues Phe196 and Phe264 located at the mouth of the substrate entry port adopted substantially different conformations similar to those observed in a lipid-embedded RPE65 crystal structure, which likely represents the active conformation of the protein (Supplementary Fig. 4, a and b) 20. Long-range van der Waals interactions between these residues and the aliphatic rings of the inhibitors could contribute to their mutual conformational stabilization.
Figure 3. Crystal structure of RPE65 in complex with emixustat at 1.8 Å resolution.

a, Cut-through view of the RPE65 active site cavity showing the locations of the emixustat and palmitate-binding sites. The asterisk indicates the substrate entry port of the enzyme. The view shown here is rotated ~180° around the vertical axis relative to the view show in ref 22 for a bacterial member of the CCO family. b, Unbiased 2Fo-Fc electron density map contoured at 1σ shown within 2 Å of the emixustat and palmitate ligands. The map was calculated prior to inclusion of the ligands in the structural model. Residues within 4.5 Å of the bound ligands are shown as grey sticks. c, Two-dimensional interaction diagram for the emixustat and palmitate ligands. Polar interactions are shown with dashed lines. Residues participating in non-dipolar interactions are shown as spiked arcs.
Located deeper within the active site cavity was a curved hydrophobic pocket that contained the bound palmitate ligand (Fig. 3a). The palmitoyl-acyl chain was of ideal length to completely fill the lipophilic cavity. The carboxylate moiety coordinated Fe in a monodentate fashion with an Fe-O bond length of ~ 2.0 Å (Fig. 3, b and c). The Fe center was thus 5-coordinate with a distorted trigonal bipyramidal geometry (Supplementary Table 2), which is consistent with its description based on X-ray absorption spectroscopy 20. In previously reported RPE65 structures the acyl chain of a putative palmitate ligand appeared to occupy the region presently identified as the inhibitor-binding site 17,20. It is conceivable that this represents an alternative, off-pathway mode of binding for palmitate in the absence of retinoids and inhibitors, but ambiguity in the electron density maps from these structures prohibits a definitive conclusion. Because the purification procedure for RPE65 destined for crystallization trials did not involve extensive washing of the column-bound protein with emixustat or MB-001-containing buffer it is possible that the observed fatty acid was derived from residual co-purified retinyl palmitate that was cleaved during the crystallization trial with the palmitate product subsequently trapped by inhibitor binding. Because retinyl palmitate is the most common native retinyl ester in RPE microsomes, it is likely that the RPE65-emixustat-palmitate complex is at least one relevant form of the inhibited RPE65 molecule in vivo. Taken together these structures predict a mode of binding for the retinyl ester substrate wherein the retinoid and acyl moieties occupy proximal and distal regions of the active site cavity, defined with respect to the substrate entry port, with the ester functional group centered over the Fe cofactor.
Retinoid docking experiments
To independently assess the chemical suitability of the proposed retinoid-binding site we conducted retinoid ligand docking experiments using Autodock Vina 24. The program placed MB-001 and emixustat into their known binding site with RMSDs of < 2 Å, which validated the parameterization of the docking calculations. In multiple docking runs the top binding pose for all-trans-retinol and 11-cis-retinol was always found to directly overlap with the MB-001-binding site with a more favorable free energy of binding for 11-cis-retinol (Supplementary Table 3). Consistent with the two-dimensional structure alignment (Fig. 2a) the C3-C4 bond of MB-001 and the C11-C12 double bond of the retinoids occupied common positions within the active site cavity (Supplementary Table 3 and Supplementary Movie 2). Additionally, the conformation of the retinoid β–ionone ring frequently overlapped with that of MB-001. Docking of all-trans-retinyl palmitate into the active site cavity supported our proposed orientation of the enzyme-substrate complex (Supplementary Table 3). Interestingly, when all-trans-retinol was docked with the C11-C15 bonds allowed to rotate the 11-12 bond was invariably found in a cis-like state in the top scoring pose indicating a chemical and geometric preference for active site binding of the 11-cis configuration (Supplementary Table 3). The excellent geometric overlap between MB-001 and the docked 11-cis-retinol molecule, as well as the structural equivalence of the inhibitor primary amine group and retinol C15 atom together support the notion that MB-001 acts as a structural mimetic of an 11-cis-like retinyl cation intermediate.
Structural basis of stereoselective isomerase activity
Analysis of the active site electrostatics revealed a negative potential surrounding the retinoid-binding cavity that terminated abruptly at the Fe cofactor (Fig. 4a). By contrast, the palmitate-binding site was relatively neutral with the exception of a slight positive region surrounding the Fe cofactor. The inhibitor-binding site thus appeared well suited to stabilize the retinyl cation intermediate. Although a resonance-stabilized retinyl cation contains lower average polyene bond order compared with its retinoid precursor, the fully delocalized charge actually rigidifies the polyene making it less susceptible to bond rotation 25. Full delocalization of the cation throughout the polyene backbone is also inconsistent with the 11-cis stereospecificity of RPE65. The protein therefore must be able to transiently stabilize the cation at the C11 position to enable selective 11-12 bond rotation and proper positioning of C15 for subsequent nucleophilic attack by solvent. The retinoid-binding pocket contained a slight constriction formed by the aromatic side chain of Phe103 and the hydroxyl group of Thr147 that could serve this purpose (Fig. 4b and Supplementary Movie 2). The line connecting the Cδ atom of Phe103 with the Oγ atom of Thr147, where the constriction is centered, precisely intersected with the predicted binding position of the retinoid C11 atom. In support of this proposal, Phe103, Thr147 and two other residues in close proximity, Tyr338 and Phe526, are known determinants of RPE65 isomerization specificity (Supplementary Fig. 5) 14,18,19. All of these residues are strictly conserved from zebrafish to man. The Phe103 and Thr147 side chains were properly positioned to stabilize the cationic intermediate through aromatic-cation 26 and dipole interactions, respectively. Similar modes of carbocation stabilization have been proposed for other isoprenoid-metabolizing enzymes, e.g. squalene cyclase 27 and pentalenene synthase 28. Diverse mutations in these two residues results in preferential production of 13-cis-retinol by RPE65 likely due to disruption of carbocation stabilization at C11 14. The only examined substitution in which 11-cis isomerization stereospecificity is maintained and indeed enhanced is a Thr to Ser substitution at position 147 14. The side chain of Ser contains a hydroxyl group that can adopt a conformation similar to that of the corresponding wild-type Thr side chain. These data are thus consistent with critical roles for the Phe103 aromatic side chain and the hydroxyl moiety of Thr147 in RPE65 isomerization selectivity.
Figure 4. Structural determinants of RPE65 ester cleavage and isomerization specificity.

a, Electrostatic potential mapped to the molecular surface of the RPE65 active site cavity shown at the + 5 kT/e (blue) and − 5 kT/e (red) levels. b, Constriction in the retinoid-binding pocket of RPE65 imposed by Phe103 and Thr147. Emixustat is shown as orange sticks. Atomic spheres are scaled to their appropriate van der Waals radii and the molecular surface is represented as mesh. c, Location of the candidate hydrolytic water molecule. The water molecule (light-red sphere circumscribed in orange) is located in a relatively narrow cavity that is partially formed by the catalytic Fe. The dotted arrow delineates a path the solvent must travel to reach the scissile O-alkyl bond. Glu148, which engages in an electrostatic interaction with this water, may serve as a proton acceptor during cleavage. The molecular surface was computed using a probe radius of 1.2 Å.
The specialized structure of the retinoid-binding site also explained why unwanted side products of retinyl cation formation are not produced by RPE65. Retinyl esters in acidic solutions readily form retinyl cations that undergo elimination to form anhydroretinol 13,29, a compound that also is enzymatically produced by a protein known as retinol dehydratase. The proposed catalytic mechanism of retinol dehydratase has features in common with that of RPE65, including O-alkyl bond cleavage and a retinyl cation intermediate 30. In retinol dehydratase, the β–ionone ring of retinol is snugly bound in an interior pocket of the enzyme where deprotonation at C4 occurs 30. This contrasts sharply with the loose binding site for the β–ionone ring in RPE65, whereby no selective stabilization is possible. This paucity of electrostatic stabilization of carbon atoms other than C11 through C15 is consistent with the fact that only 11-cis-retinol and under some in vitro conditions, 13-cis-retinol, are produced by RPE65 13,14.
Besides the retinyl cation mechanism two other mechanisms of retinoid isomerization have been proposed. The first of these posits a key role for an unspecified active site nucleophile in the retinoid isomerization reaction 11. However, the retinoid-binding site identified in this study does not reveal any good nucleophilic candidates in close proximity to the polyene chain that could fulfill this role. The second mechanism involves a retinyl radical cation intermediate that is generated prior to retinyl ester cleavage by an unspecified electron acceptor 19. The retinoid-binding site environment does not feature any obvious candidate redox-active moieties that could achieve one-electron oxidation of the retinoid. Although iron in a +3 state might be able to serve as a suitable cofactor for this type of mechanism, RPE65 requires Fe(II) for catalytic activity 6 and contains a bound Fe(II) atom in its as isolated state 20, which is not able to be further reduced under biological conditions. Moreover, the predicted polyene-binding region is not in close proximity to the iron center so electron transfer between these sites likely would be inefficient.
Examination of the high resolution RPE65-emixustat crystal structure revealed a single plausible water molecule within the RPE65 active site that could quench the C15 retinyl cation (Fig. 4c and Supplementary Movie 2). It was located in a narrow cavity adjacent to the ester-binding region of the active site where it formed hydrogen bonding and ion-dipole interactions with Ala179, His241 and Glu148. This cavity was also in direct contact with the Fe center. A linear distance of 5.3 Å separated the solvent oxygen atom from the emixustat nitrogen atom, indicating that solvent and the C15 cation mimicked by the amine nitrogen must more closely approach one another to react. Different possibilities can be envisioned to explain how solvent traverses from its binding site to approach C15. Despite the narrowness of the cavity a strong interaction with the Glu148 side chain carboxylate could allow movement of the solvent molecule through the cavity towards C15 31. The solvent could be deprotonated by the Glu148 side chain carboxylate either before or after nucleophilic attack on C15. An alternative mechanism involves a transient coordination of the deprotonated solvent molecule to the Fe center and its subsequent direct attack on C15. As previously noted, the side chain methyl group of Val134 limits the ability of the open Fe coordination site in its vicinity to accept ligands as a result of steric hindrance 17. However, the short (~1.9 Å) bond length associated with Fe-hydroxo complexes might allow such coordination to occur. As expected, no solvent molecules were observed in close proximity to the carbonyl carbon of the palmitate carboxylate group. Additionally, the faces of the palmitate carbonyl carbon were in close contact with carbon atoms of His 241 and Phe526 which shield it from solvent nucleophiles (Fig. 3b). The observed interaction between the inhibitor primary amine and the palmitate carboxylate implies that, following ester cleavage, C15 and palmitate remain associated as an ion pair. Such ion pairing would maintain the orientation of C15 and shield the leaving group side from solvent attack. This finding could explain the inversion of stereochemical configuration that occurs at C15 during the isomerization reaction 16.
DISCUSSION
The preceding results allow us to propose a detailed mechanism of catalysis by RPE65 that reconciles prior biochemical and structural findings. The reaction follows an AAL1, SN1 mechanism wherein the carbocation intermediate undergoes geometric trans-cis isomerization (Fig. 5, a and b) 32. Though possibly unique in biological systems this mechanism has a strong precedent in organic chemistry 33. all-trans-Retinyl esters are taken up from the membrane with the acyl chain entering the active site first. Favorable van der Waals and hydrophobic interactions associated with acyl binding likely provide a strong driving force for substrate uptake (Fig. 5c, i). During substrate binding the curved retinoid-binding cavity likely partially twists the 11-12 double bond of the retinoid moiety such that it is poised to undergo isomerization. Binding of the substrate positions the ester group in close proximity to the Fe center resulting in an electrostatic interaction between Fe and the ester carbonyl oxygen. Acting as a Lewis acid, Fe polarizes the ester group which results in development of a partial positive charge on C15 of the retinoid that is stabilized by polyene delocalization as well as electrostatic interactions with the carboxylate group of Glu148. The C15-O bond then breaks to yield a retinyl cation-palmitate ion pair (Fig. 5c, ii). The carbocation is partially stabilized on C11 due to favorable interactions with Phe103 and Thr147 which allows rotation of the C11-C12 bond from trans to a cis-like configuration. Repositioning of C15 strengthens electrostatic stabilization of the carbocation at this position by the Glu148 and palmitate carboxylate groups so that it is poised for attack by the solvent molecule located opposite to the dissociated carboxylate group. Nucleophilic attack of the solvent molecule on C15 is accompanied by proton transfer to Glu148. The nascent 11-cis-retinol dissociates back into the membrane first followed by palmitic acid after a proton transfer event (Fig. 5c, iii). Protonation of the Fe-bound carboxylate group likely facilitates its dissociation to generate the resting state of the enzyme containing a Fe(II) center with a coordinated solvent molecule, as deduced from the resting-state structure of the RPE65 homolog apocarotenoid oxygenase (ACO) (Fig. 5d, iv) 23. 11-cis-Retinal, formed from the 11-cis-retinol product of RPE65, is transported to photoreceptor outer segments where it combines with opsins to regenerate their light-sensitive, ground-states 34 thus completing the visual cycle (Fig. 1a).
Figure 5. Proposed mechanism of retinoid isomerization catalyzed by RPE65.

a, General AAL1, SN1 reaction scheme. b, Variant of the AAL1 scheme with isomerization of the carbocation intermediate. c, Proposed catalytic mechanism for RPE65.
The inhibitor-bound structures reported here provide a critical link between the extensive biochemical research that has been performed over the past 3 decades regarding the visual cycle retinoid isomerization mechanism and recent structural studies on RPE65. Moreover, they also represent the first genuine examples of CCO-ligand complexes. We expect that this approach of using substrate/product derivatives or transition state analogs with elevated aqueous solubility that exploit critical active site polar pockets will be applicable to structural studies of other CCOs as well as to other enzymes that metabolize lipophilic substrates. This study also provides a molecular foundation for understanding therapeutic inhibition of RPE65 with primary amine compounds and will enable structure-guided development of next-generation visual cycle modulators for the treatment of AMD.
ONLINE METHODS
Synthetic Chemistry
all-trans-Retinylamine (Ret-NH2) was synthesized as previously described 9. Racemic emixustat (>95% pure as assessed by NMR and HPLC) was synthesized as previously described 35. 1H NMR values matched those previously reported 35. 13C NMR (100 MHz, CDCl3) δ 159.50, 146.54, 129.37, 117.80, 113.19, 111.85, 74.40, 73.50, 39.78, 38.67, 37.85, 30.04, 26.66, 25.94. Stereoselective synthesis of MB-001 was carried out by the following procedure, which is based partially on reported methods 36–38.
3-hydroxy-3-(3-hydroxyphenyl)propanenitrile (8)
An oven-dried 1 liter, two-necked, round-bottomed flask was sealed under argon. The flask was charged with anhydrous THF (150 mL) and anhydrous acetonitrile (6.4 mL, 114.2 mmol), cooled to −42 °C and then treated dropwise with a solution of potassium tert-butoxide (28.4 g, 253.2 mmol) in THF (300 mL). After stirring for 45 min, 3-hydroxy-benzaldehyde (13.78 g, 112.8 mmol) in THF (60 mL) was added dropwise. The reaction was allowed to warm to −15 °C over a 4 h period, and was quenched by the slow addition of saturated aq. (~25%) NH4Cl (200 mL). The layers were partitioned, and the aqueous phase was extracted with ethyl acetate (EtOAc) (2x). The combined organics were washed with H2O (2x), brine, dried over MgSO4, and evaporated under reduced pressure. Purification by silica gel column chromatography (EtOAc/hexanes 50:50) afforded 14.7 g (80%) of compound 8 as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.26 (t, 1H, J= 8 Hz), 6.95-6.91 (m, 2H), 6.82 (ddd, 1H, J = 1.2, 2.4, 8.0 Hz), 5.01 (t, 1H, J= 6.4 Hz), 2.77 (d, 2H, J= 6 Hz). 1H NMR values matched reported literature values 36. 13C NMR (100 MHz, CDCl3) δ 156.5, 143.0, 130.4, 117.7, 117.4, 116.0, 112.5, 68.1, 25.7.
3-(3-amino-1-hydroxypropyl) phenol (7)
An oven-dried 500 mL, two-necked, round-bottomed flask was sealed under argon. The flask was charged with anhydrous THF (100 mL) and compound 8 (10 g, 61.28 mmol). To the stirred reaction mixture at room temperature, borane methyl sulfide complex 5 M in ether (36.8 mL, 184 mmol) was slowly added. After complete addition, the reaction mixture was heated under reflux (2–4 h) and cooled to 0 °C. Subsequent treatment with methanol was utilized to decompose the unreacted borane complex. The organic solvent was evaporated under reduced pressure, and the residue was purified by silica gel column chromatography (NH4OH/MeOH/dichloromethane (CH2Cl2) 6:30:70) to afford 9.2 g (90%) of compound 7 as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.07 (t, 1H, J = 7.6Hz),6.74-6.72 (m, 1H), 6.70 (d, 1H, J = 7.6 Hz), 6.58 (ddd, 1H, J = 0.8, 2.4, 8.0 Hz), 4.55 (dd, 1H, J= 7.2, 7.6 Hz), 2.65-2.57 (m, 2H), 1.65-1.57 (m, 2H). 1H NMR values matched those previously reported 39. 13C NMR (100 MHz, DMSO-d6) δ 157.2, 148.2, 128.9, 116.4, 113.5, 112.6, 71.3, 48.7, 42.3.
tert-butyl 3-hydroxy-3-(3-hydroxyphenyl)propylcarbamate (6)
An oven-dried 2 liter, two-necked, round-bottomed flask was charged with compound 7 (20.0 g, 109.9 mmol), 1,4-dioxane (1 L) and K2CO3 (15.3 g, 110.64 mmol). To the stirred reaction mixture at room temperature, di-tert-butyl dicarbonate (Boc2O) (25 mL, 110 mmol) was slowly added. The mixture was stirred at room temperature for a subsequent 2 h. It was then quenched by addition of water and extracted with EtOAc. The organic layer was washed with water, brine, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. Purification by silica gel column chromatography (EtOAc/hexanes 20:80) afforded 18.5 g (63%) of compound 6 as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.10 (t, 1H, J = 7.6 Hz), 6.82-6.78 (m,1H), 6.75 (d, 1H, J = 7.6 Hz), 6.71 (ddd, 1H, J = 0.8, 2.4, 8.0 Hz), 4.57 (t, 1H, J =6.8 Hz), 3.30-3.12 (m, 1H), 3.12-3.10 (m, 1H), 1.82-1.73 (m, 2H), 1.42 (s, 9H). 1H NMR values matched those previously reported 39. 13C NMR (100 MHz, CDCl3) δ 157.1, 156.5, 145.7, 129.7, 117.6, 114.8, 112.8, 80.0, 71.9, 39.1, 37.7, 28.5.
tert-butyl 3-(3-hydroxyphenyl)-3-oxopropylcarbamate (5)
An oven-dried 1 liter, two-necked, round-bottomed flask was charged with CH2Cl2 (200 mL), celite (22.6 g) and pyridinium chlorochromate (PCC) (22.4 g, 104 mmol). The mixture was cooled to 0 °C. Compound 6 (18.5 g, 69 mmol) in CH2Cl2 (100 mL) was slowly added to the reaction mixture over a period of 15 min. The reaction mixture was allowed to stir at room temperature for 2 h. The mixture was then filtered through a pad of celite and the filter bed was washed with CH2Cl2. Concentration of the filtrate under reduced pressure gave a black residue. Purification by silica gel column chromatography (EtOAc/hexanes 40:60) afforded 12.7 g (46%) of compound 5 as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.50-7.43 (m, 2H), 7.31 (t, 1H, J = 8 Hz), 7.09 (dd, 1H, J= 2.4, 8 Hz), 5.23 (brs, 1H), 3.53 (t, 2H, J = 5.6 Hz), 3.16 (t, 2H, J = 5.6 Hz), 1.43 (s, 9H). 1H NMR values matched those previously reported 39. 13C NMR (100 MHz, CDCl3) δ 199.5, 156.8, 156.5, 138.0, 130.1, 121.0, 120.3, 114.8, 79.9, 38.9, 35.7, 28.5.
3-(3-hydroxyphenyl)-3-oxopropan-1-aminium 2,2,2-trifluoroacetate
A pre-weighed, oven-dried 250 mL, one-necked, round-bottomed flask was charged with anhydrous CH2Cl2 (55 mL) and trifluoroacetic acid (55 mL) and cooled to 0 °C. Then compound 5 (13.38 g, 50.4 mmol) was slowly added to the reaction mixture. The resulting reaction was allowed to stir at room temperature for 2 h. Upon completion of the reaction, the solvent was removed under reduced pressure and the residue was triturated with toluene. Toluene was discarded and residual toluene was removed in vacuo (0.5 torr). The crude amine was directly utilized for the next transformation without purification 39.
2-(3-(3-hydroxyphenyl)-3-oxopropyl)isoindoline-1,3-dione (4)
To a solution of crude amine (14.05 g, 48.2 mmol) in a mixture of acetonitrile/toluene (1:3; 168 mL) at 0 °C was added N,N-diisopropylethylamine (DIPEA) (20.9 mL, 120 mmol). The resulting mixture was stirred at room temperature for 10 min. Then phthalic anhydride (7.14 g, 48.2 mmol) was added and the reaction mixture was refluxed for 2 h using a Dean-Stark assembly. After completion of the reaction the solvent was removed under reduced pressure and the reaction mass was treated with CH2Cl2. The organic layer was washed with water, saturated NH4Cl, saturated NaHCO3, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. Purification by silica gel column chromatography (EtOAc/hexanes 75:25) afforded 13.0 g (80%) of compound 4 as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 7.88-7.82 (m, 2H), 7.76-7.68 (m, 2H), 7.50-7.43 (m, 2H), 7.30 (t, 1H, J = 8 Hz), 7.06 (d, 1H, J = 0.8, 2.8, 8.0 Hz), 6.11 (bs, 1H), 4.13 (t, J = 7.6 Hz, 2H), 3.39 (t, 2H, J = 7.2 Hz). 1H NMR values matched those previously reported 39. 13C NMR (100 MHz, CDCl3) δ 197.5, 168.5, 156.4, 137.8, 134.2, 132.1, 130.1, 123.5, 120.9, 120.7, 114.6, 37.1, 33.7.
(R)-2-(3-hydroxy-3-(3-hydroxyphenyl)propyl)isoindoline-1,3-dione (3a)
An oven-dried 100 mL, two-necked, round-bottomed flask was sealed under argon and charged with anhydrous THF (20 mL) and compound 4 (3 g, 10.1 mmol). A solution of (+)-diisopinocampheylchloroborane ((+)-Ipc2B-Cl) in hexane (1.5 M, 26.9 mL, 40.4 mmol) was added under argon to the reaction mixture at room temperature. On completion of the reaction (2.5 to 3.5 h at 24 °C), the reaction mixture was partitioned between 25% NH4Cl and THF. The aqueous layer was additionally twice extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous MgSO4 and concentrated under reduced pressure. Purification by silica gel column chromatography (EtOAc/hexanes 60:40) afforded 2.78 g (92%) of compound 3a as a white solid. An enantiomeric excess (ee) of 88% was determined by HPLC [Daicel Chiralcel OD column, n-hexane/i-PrOH) 85:15, 1 mL/min, 272 nm; tR) 21.8 min (major) and 19.2 min]. Percent ee was enriched to 98% by two crystallizations in acetone.1H NMR (400 MHz, DMSO-d6) δ 9.27(s, 1H), 7.86-7.77 (m, 4H), 7.06 (t, 1H, J = 7.6 Hz), 6.77-6.74 (m, 1H), 6.73 (d, 1H, J = 7.6Hz),6.58 (ddd, 1H, J = 1.2, 2.4, 8.0Hz), 5.26 (d, 1H, J = 4.4 Hz), 4.52 (t, 1H, J = 6.4 Hz), 3.72-3.57 (m, 2H), 1.93-1.83 (m, 2H). 1H NMR values matched those previously reported 39. 13C NMR (100 MHz, DMSO-d6) δ 168.0, 157.2, 147.2, 134.4, 131.8, 129.1, 123.0, 116.4, 113.8, 112.6, 70.4, 37.4, 35.1.
2,6,6-trimethylcyclohex-1-ene-1-methanol (3b)
An oven-dried 25 mL, two-necked, round-bottomed flask was sealed under argon and charged with sodium borohydride (735 mg, 19.5 mmol), ethanol (2.5 mL) and propane-2-ol (2.5 mL). The mixture was cooled to 0 °C. Then β–cyclocitral (Sigma-Aldrich, purity 94.9%) (2 g, 13 mmol) in propan-2-ol (3.5 mL) was added dropwise to the stirred reaction mixture. The mixture was allowed to warm to ambient temperature by removing the ice bath while stirring was maintained for 1 h. The reaction mixture then was poured into water (10 mL) and subsequently saturated with sodium chloride and extracted with ether (3×5 mL). The organic extracts were combined, washed with water (10 mL), brine (10 mL), dried over MgSO4 and concentrated under reduced pressure. Purification by silica gel chromatography (EtOAc/petroleum ether 20:80) afforded 1.7 g (85%) of compound 3b as colorless waxy solid 37. 1H NMR (400 MHz, CDCl3) δ 4.1 (s, 2H), 1.95(t, 2H, J = 6.4 Hz), 1.72(s, 3H), 1.61-1.53(m, 2H), 1.45-1.39(m, 2H), 1.01(m, 6H). 13C NMR (100 MHz, CDCl3) δ 137.7, 133.7, 58.9, 39.4, 34.1, 32.8, 28.6, 19.7, 19.4. 1H NMR and 13C values matched those previously reported 38.
(R)-2-(3-hydroxy-3-(3-((2,6,6-trimethylcyclohex-1-en-1-yl)methoxy)phenyl)propyl)isoindoline-1,3-dione (2)
An oven-dried 10 mL, two-necked, round-bottomed flask was charged with compound 3b (100 mg, 0.648 mmol), compound 3a (202.2 mg, 0.680 mmol), triphenylphosphine (341.0 mg, 1.3 mmol) and 5 mL anhydrous tetrahydrofuran under an atmosphere of argon. The mixture was cooled to 0 °C, then 1,1′-(azodicarbonyl)dipiperidine (ADDP) (328 mg, 1.3 mmol) was added in one portion. The resulting bright-orange solution was stirred overnight. The yellow suspension was concentrated under reduced pressure, and purified by silica gel column chromatography (EtOAc/hexanes 30:70) to afford 60 mg (21%) of compound 2 as a colorless oil. The ee was 82 % [Daicel Chiralcel OD column, n-hexane/i-PrOH) 90:10, 1 mL/min, 272 nm; tR) 9.6 min (major) and 8.3 min (minor)].1H NMR (400 MHz, CDCl3) δ 7.86-7.81(m, 2H), 7.74-7.68 (m, 2H), 7.19 (t, 1H, J = 8.0 Hz), 6.97 (t, 1H, J = 1.6 Hz), 6.9 (d, 1H, J = 7.6 Hz), 6.8 (ddd, 1H, J = 0.8, 2.8, 8.4 Hz), 4.67 (dd, 1H, J = 4.8, 5.2 Hz), 4.04 (s, 2H), 3.91 (t, 2H, J = 6.4 Hz), 2.92 (bs, 1H), 2.14-2.06 (m, 2H), 2.06-2.00 (m, 2H), 1.69 (S, 3H), 1.68-1.59 (m, 2H), 1.51-1.47 (m, 2H), 1.03 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 168.9, 159.7, 145.3, 135.8, 134.1, 132.1, 129.5, 123.4, 117.9, 114.1, 111.8, 71.4, 64.5, 39.4, 37.7, 35.1, 34.2, 33.0, 28.5, 19.9, 19.4.
(R)-3-amino-1-(3-((2,6,6-trimethylcyclohex-1-en-1-yl)methoxy)phenyl)propan-1-ol (1, MB-001)
An oven-dried 10 mL, one-necked, round-bottomed flask was charged with compound 2 (60 mg, 0.138 mmol), 3 mL EtOH and N2H4.H2O (33.9 μl, 34.9 mg, 0.697 mmol). The reaction mixture was stirred at room temperature for 26 h and then concentrated under reduced pressure. Purification by silica gel column chromatography NH4OH/MeOH/CH2Cl2 4:30:70) afforded 31 mg (75%) of compound 1 (MB-001) as a colorless oil. 1H NMR (400 MHz, CD3OD) δ 7.24 (t, J = 8 Hz, 1H), 6.98 (t, 1H, J = 2.0 Hz), 6.93 (d, 1H, J = 7.6 Hz), 6.8 (ddd, 1H, J = 0.8, 2.8, 8.4 Hz), 4.71 (dd, 1H, J = 7.6, 8Hz), 4.46 (s, 2H), 3.82-2.68 (m, 2H), 2.08-2.02 (m, 2H), 1.95-1.80 (m, 2H), 1.69 (S, 3H), 1.70-1.63 (m, 2H), 1.54-1.48 (m, 2H), 1.05 (s, 6H) ppm; 13C NMR (100 MHz, CD3OD) δ 160.8, 148.0, 136.4, 134.5, 130.3, 119.1, 114.4, 113.0, 73.4, 65.4, 42.4, 40.6, 39.6, 35.1, 33.8, 28.9, 20.4, 20.0 ppm. HRMS (ESI): m/z calculated for C19H30NO2 [M+H]+ 304.22711, found 304.22714.
In vitro retinoid isomerization assay
Retinoid isomerization assays were performed as previously described using bovine RPE microsomes as the enzyme source 13,40. Test compounds ((R,S)-emixustat, MB-001 and Ret-NH2) were delivered to the assay mixtures in DMF. 11-cis-Retinol production was analyzed by normal phase HPLC using pure 11-cis-retinol (a gift from Dr. R. Crouch, Medical University of South Carolina) as a standard for quantification.
In vivo mouse experiments
Mice handling and drug administration
Mice were housed in the animal facility at the Case Western Reserve University School of Medicine where they were maintained under either complete darkness or a 12 h light (~300 lux) and 12 h dark cycle. Test compounds were suspended in 100 μL of soybean oil with less than 10% (v/v) DMSO and were administered by oral gavage with a 22-gauge feeding needle. Experimental manipulations in the dark were done under dim red light transmitted through a Kodak No. 1 safelight filter (transmittance >560 nm). All animal procedures and experiments were approved by the Case Western Reserve University Animal Care Committees and conformed to recommendations of the American Veterinary Medical Association Panel on Euthanasia and the Association of Research for Vision and Ophthalmology
Visual chromophore recovery assay
Four-week-old wild-type C57BL6J mice were administrated 50 μg of the primary amine inhibitors by oral gavage and then kept in darkness for 24 h. After exposure to bright light that resulted in photoactivation of ≥90% of rhodopsin, mice were placed back into total darkness for 6 h to allow visual chromophore recovery. The animals then were euthanized and their eyes were enucleated. Whole eyes were homogenized in 10 mM sodium phosphate buffer, pH 7.4 containing 50% methanol (v/v) and 50 mM hydroxylamine. Retinoids were extracted from the resulting mixture by addition 4 mL of hexanes followed by vigorous shaking. The organic extracts were dried under a vacuum and then reconstituted in 300 μL of hexanes. One-hundred μL of the extract was analyzed by normal-phase HPLC on a silica column with 10% (v/v) EtOAc in hexanes used as the mobile phase.
RPE65 purification
Bovine RPE microsomes were isolated as previously described 17. Proteins were extracted from the microsomes with hexaethylene octyl ether (C8E6, anagrade, Anatrace) and RPE65 was purified from the sample by ion-exchange chromatography 17. (R,S)-emixustat or MB-001, delivered in DMF, was added to the microsomal suspension at final concentrations of 1 mM during the detergent extraction portion of the purification procedure. RPE65-containing fractions from the ion exchange column were pooled and concentrated to 10–15 mg protein/mL. RPE65 purity was judged to be > 90% based on Coomassie-stain SDS-PAGE analysis (Supplementary Fig. 2a). Prior to crystallization trials additional emixustat or MB-001 was added to the concentrated samples to concentrations of 1 mM. The final concentration of inhibitors in the samples was thus > 1 mM, the exact concentration being dependent on an unknown amount of inhibitor carried over from the first addition.
Crystallization, structure determination and analysis
Crystallization trials were carried out by the hanging drop-vapor diffusion method. Crystals of the RPE65-emixustat complex were grown by mixing 1 μL of purified protein solution with 1 μL of crystallization solution consisting of 30% PEG 200, 200 mM (NH4)2PO4 and 100 mM Tris-HCl, pH 8.5 (condition #25 of the Wizard Cryo I screen, Rigaku) and incubating the drops over 500 μL of crystallization solution. Crystals of the RPE65-MB-001 complex were grown in the same manner except that 40% v/v PEG 300, 200 mM NaCl and 100 mM CHES-NaOH, pH 9.5 (condition #26 of the Wizard Cryo I screen, Rigaku) was used as the crystallization solution. Crystals with approximate sizes of 50 × 50 × 200 μm were harvested 2-4 weeks after initiation of the crystallization trial. Crystals were harvested and flash cooled in liquid nitrogen prior to X-ray exposure. No additional cryoprotective agents were required to suppress ice formation.
Diffraction data sets were collected at the X29 beamline of the National Synchrotron Light Source (NSLS) using 1.07500 Å wavelength X-rays. The crystal temperature was maintained with a nitrogen cryosteam during data collection. The data were indexed, integrated and scaled using the XDS package 41. The crystals were isomorphous to the previously reported P65 crystal form. Scaled intensities were converted to amplitudes by the French-Wilson procedure 42 as implemented in XDSCONV 41. Analysis of the diffraction amplitudes was performed with phenix.xtraige 43. The RPE65-MB-001 crystal was merohedrally twinned to a small degree (α ~15%) but this did not complicate structure refinement. Diffraction data statistics are shown in Supplementary Table 1.
Structures were solved by rigid body refinement in REFMAC with the deposited RPE65 atomic coordinates (PDB accession code: 3FSN) used as a starting model. The bound inhibitor and palmitate ligands were clearly visible in the initial electron density maps. Adjustments to the protein structure and addition of water molecules were performed using COOT 44. Restrained refinement was carried out using REFMAC 45. Following several cycles of manual model adjustments and computational refinement inhibitor and palmitate ligands were placed into the residual active site electron density. Atomic coordinates and geometric restraints were generated using the GRADE server (Global Phasing). As previously reported, segments 109-126, 197-201 and 266-271, which comprise part of the membrane-binding region of the protein, exhibited weak electron density; therefore, most of these residues were excluded from the final models. The structures were validated using MOLPROBITY 46, EDSTATS 47 from the CCP4 suite 48 and the wwPDB structure validation server 49. Real-space difference density Z score statistics computed using EDSTATS were used to determine the correct conformation of the bound MB-001 molecule 47. The final emixustat and MB-001 models had 97.3% and 97.2% of residues, respectively, in favored regions of the Ramachandran plot. No Ramachandran outliers were present in either model. In both structures electron density for the ligands in chain B was slightly stronger compared to chain A so the former was used for the structure analysis. Final crystallographic statistics are shown in Supplementary Table 1. Figures of the crystallographic models prepared using PyMOL (Schrödinger). Electrostatics calculations were carried out using APBS 50.
Computational ligand docking
Preparation of the receptor
The 1.8 Å resolution structure of RPE65 in complex with emixustat was used for the docking calculations. Ligands, water molecules and side chain alternative conformations were removed from the model. The resulting coordinate file was converted to pdbqt format using Autodock Tools. Polar hydrogens were included in the protein model. The charge on the Fe cofactor was varied from 0 to +2, but its value did not significantly affect the docking results.
Preparation of the ligand
Ligand coordinate and stereochemical restraint files were generated using either the GRADE or PRODRG servers. Adjustments to the polyene configuration and regularization of the ligand bond lengths and angles were performed using COOT. The ligand coordinate files then were processed with Autodock Tools to generate pdbqt files with polar hydrogens added to the models. For polyene compounds the alternating single bonds were allowed to freely rotate whereas double bonds were fixed in ideal trans or cis configurations except for those specified in Supplementary Table 3.
Docking calculations
Docking experiments were carried out with Autodock Vina version 1.1.2 24. Initial docking trials were conducted using the Fe atom as the center of a search area that included an entire RPE65 monomer. After observing that the retinoid and emixustat ligands were consistently placed into the active site cavity the search area was narrowed to this region to allow for a more exhaustive sampling of the ligand docking space.
Calculation of clogP values
The clogP values reported in the manuscript were computed using ChemBioDraw Ultra v 14.0.0.117.
Supplementary Material
Acknowledgments
We thank Dr. Leslie T. Webster Jr. for helpful comments on this manuscript. This work was supported by funding from the National Institutes of Health EY023948 (MG), EY009339 (KP), EY021126 (KP), CA157735 (GPT), the Department of Veterans Affairs IK2BX002683 (PDK) and National Science Foundation MCB-084480 (GPT). Data for this study were measured at beamline X29 of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources (P41RR012408), the National Institute of General Medical Sciences (P41GM103473) of the National Institutes of Health and the National Institute of Biomedical Imaging and Bioengineering (NIBIB) (P30-EB-09998). We thank the staff at the APS NECAT beamlines, supported by a grant from the National Institute of General Medical Sciences (P41 GM103403), for assistance with collection of preliminary diffraction data. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. K.P. is John H. Hord Professor of Pharmacology.
Footnotes
DISCLOSURE STATEMENTS
K.P. and M.G. are inventors of US Patent No. 8722669 - “Compounds and Methods of Treating Ocular Disorders” and US Patent No. 20080275134 - “Methods for Treatment of Retinal Degenerative Disease” issued to Case Western Reserve University (CWRU) whose values may be affected by this publication. CWRU may license this technology for commercial development. KP is a member of the scientific board of Vision Medicine, Inc. involved in developing visual cycle modulators whose values may be affected by this publication.
CONTRIBUTIONS
P.D.K., J.Z., G.P.T. and K.P. conceived and designed experiments. P.D.K. purified and crystallized RPE65, solved and refined the structures and performed structural analyses. J.Z. performed in vitro and in vivo retinoid isomerization assays. M.B., Q.L. and G.P.T. carried out chemical synthesis. W.S. collected diffraction data. X.S. assisted with crystallization and crystal harvesting. M.G. assisted with in vitro and in vivo retinoid isomerization assays. P.D.K. wrote the paper. K.P. coordinated and oversaw the research project. All authors discussed the results and commented on the manuscript.
Coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under accession codes 4RSC and 4RSE for the RPE65-emixustat and RPE65-MB-001 crystal structures.
References
- 1.Wald G. Molecular basis of visual excitation. Science. 1968;162:230–9. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- 2.Kiser PD, Golczak M, Palczewski K. Chemistry of the retinoid (visual) cycle. Chem Rev. 2014;114:194–232. doi: 10.1021/cr400107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saari JC. Vitamin A metabolism in rod and cone visual cycles. Annu Rev Nutr. 2012;32:125–45. doi: 10.1146/annurev-nutr-071811-150748. [DOI] [PubMed] [Google Scholar]
- 4.Golczak M, Kiser PD, Lodowski DT, Maeda A, Palczewski K. Importance of membrane structural integrity for RPE65 retinoid isomerization activity. J Biol Chem. 2010;285:9667–82. doi: 10.1074/jbc.M109.063941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redmond TM, et al. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci USA. 2005;102:13658–63. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moiseyev G, et al. RPE65 is an iron(II)-dependent isomerohydrolase in the retinoid visual cycle. J Biol Chem. 2006;281:2835–40. doi: 10.1074/jbc.M508903200. [DOI] [PubMed] [Google Scholar]
- 7.Marlhens F, et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet. 1997;17:139–41. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 8.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci USA. 2005;102:8162–7. doi: 10.1073/pnas.0503318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubota R, et al. Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina. 2014;34:603–9. doi: 10.1097/01.iae.0000434565.80060.f8. [DOI] [PubMed] [Google Scholar]
- 11.Deigner PS, Law WC, Canada FJ, Rando RR. Membranes as the energy source in the endergonic transformation of vitamin A to 11-cis-retinol. Science. 1989;244:968–71. doi: 10.1126/science.2727688. [DOI] [PubMed] [Google Scholar]
- 12.Moiseyev G, et al. Retinyl esters are the substrate for isomerohydrolase. Biochemistry. 2003;42:2229–38. doi: 10.1021/bi026911y. [DOI] [PubMed] [Google Scholar]
- 13.McBee JK, et al. Isomerization of all-trans-retinol to cis-retinols in bovine retinal pigment epithelial cells: dependence on the specificity of retinoid-binding proteins. Biochemistry. 2000;39:11370–80. doi: 10.1021/bi001061c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redmond TM, Poliakov E, Kuo S, Chander P, Gentleman S. RPE65, visual cycle retinol isomerase, is not inherently 11-cis-specific: support for a carbocation mechanism of retinol isomerization. J Biol Chem. 2010;285:1919–27. doi: 10.1074/jbc.M109.027458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Law WC, Rando RR. Stereochemical inversion at C-15 accompanies the enzymatic isomerization of all-trans- to 11-cis-retinoids. Biochemistry. 1988;27:4147–52. doi: 10.1021/bi00411a037. [DOI] [PubMed] [Google Scholar]
- 16.Jang GF, McBee JK, Alekseev AM, Haeseleer F, Palczewski K. Stereoisomeric specificity of the retinoid cycle in the vertebrate retina. J Biol Chem. 2000;275:28128–38. doi: 10.1074/jbc.M004488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiser PD, Golczak M, Lodowski DT, Chance MR, Palczewski K. Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc Natl Acad Sci USA. 2009;106:17325–30. doi: 10.1073/pnas.0906600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi Y, Moiseyev G, Nikolaeva O, Ma JX. Identification of the key residues determining the product specificity of isomerohydrolase. Biochemistry. 2012;51:4217–25. doi: 10.1021/bi300144n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chander P, Gentleman S, Poliakov E, Redmond TM. Aromatic residues in the substrate cleft of RPE65 protein govern retinol isomerization and modulate its progression. J Biol Chem. 2012;287:30552–9. doi: 10.1074/jbc.M112.364596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiser PD, et al. Structure of RPE65 isomerase in a lipidic matrix reveals roles for phospholipids and iron in catalysis. Proc Natl Acad Sci USA. 2012;109:E2747–56. doi: 10.1073/pnas.1212025109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messing SA, et al. Structural insights into maize viviparous14, a key enzyme in the biosynthesis of the phytohormone abscisic acid. Plant Cell. 2010;22:2970–80. doi: 10.1105/tpc.110.074815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kloer DP, Ruch S, Al-Babili S, Beyer P, Schulz GE. The structure of a retinal-forming carotenoid oxygenase. Science. 2005;308:267–9. doi: 10.1126/science.1108965. [DOI] [PubMed] [Google Scholar]
- 23.Sui X, et al. Analysis of carotenoid isomerase activity in a prototypical carotenoid cleavage enzyme, apocarotenoid oxygenase (ACO) J Biol Chem. 2014;289:12286–99. doi: 10.1074/jbc.M114.552836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blatz PE, Pippert DL. Fluorescence Spectrum of Retinylic Cation. Chem Commun. 1968:176-&. [Google Scholar]
- 26.Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–8. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 27.Wendt KU, Poralla K, Schulz GE. Structure and function of a squalene cyclase. Science. 1997;277:1811–5. doi: 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]
- 28.Lesburg CA, Zhai G, Cane DE, Christianson DW. Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science. 1997;277:1820–4. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 29.Blatz PE, Pippert DL. Carbonium Ion of All-Trans-Retinyl Acetate. Spectroscopic Detection and Identification of Absorbing Species. Effect of Environment on Spectral Properties. J Am Chem Soc. 1968;90:1296-&. [Google Scholar]
- 30.Pakhomova S, Kobayashi M, Buck J, Newcomer ME. A helical lid converts a sulfotransferase to a dehydratase. Nat Struct Biol. 2001;8:447–51. doi: 10.1038/87617. [DOI] [PubMed] [Google Scholar]
- 31.Bhat S, Purisima EO. Molecular surface generation using a variable-radius solvent probe. Proteins. 2006;62:244–261. doi: 10.1002/prot.20682. [DOI] [PubMed] [Google Scholar]
- 32.Smith M, March J. March’s advanced organic chemistry: reactions, mechanisms, and structure. xviii. Wiley; New York: 2001. March’s advanced organic chemistry: reactions, mechanisms, and structure; p. 2083. [Google Scholar]
- 33.Kirby AJ. Hydrolysis and Formation of Esters of Organic Acids. In: Bamford CH, Tipper CFH, editors. Ester Formation and Hydrolysis and Related Reactions. Vol. 10. Elsevier; New York: 1972. pp. 57–207. [Google Scholar]
- 34.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 35.Scott IL, et al. WO patent 2009045479. Alkoxy Compounds for Disease Treatment. 2009
- 36.Caspi DD, Ebner DC, Bagdanoff JT, Stoltz BM. The resolution of important pharmaceutical building blocks by palladium-catalyzed aerobic oxidation of secondary alcohols. Adv Synth Catal. 2004;346:185–189. [Google Scholar]
- 37.Crombie BS, Smith C, Varnavas CZ, Wallace TW. A conjugate addition-radical cyclisation approach to sesquiterpene-phenol natural products. J Chem Soc [Perkin 1] 2001:206–215. [Google Scholar]
- 38.Srikrishna A, Krishnan K. Synthesis of (+/−)-Thaps-7(15)-Ene and (+/−)-Thaps-6-Enes. J Chem Soc [Perkin 1] 1993:667–673. [Google Scholar]
- 39.Scott IL, Kuksa V, Hong F, Kubota R, Gage J. WO patent 2010048332. Compounds for treating ophthalmic diseases and disorders. 2010
- 40.Stecher H, Gelb MH, Saari JC, Palczewski K. Preferential release of 11-cis-retinol from retinal pigment epithelial cells in the presence of cellular retinaldehyde-binding protein. J Biol Chem. 1999;274:8577–85. doi: 10.1074/jbc.274.13.8577. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W. Xds. Acta Crystallogr D. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.French S, Wilson K. Treatment of Negative Intensity Observations. Acta Crystallogr A. 1978;34:517–525. [Google Scholar]
- 43.Zwart PH, Grosse-Kunstleve RW, Lebedev AA, Murshudov GN, Adams PD. Surprises and pitfalls arising from (pseudo)symmetry. Acta Crystallogr D. 2008;64:99–107. doi: 10.1107/S090744490705531X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D. 2011;67:355–67. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tickle IJ. Statistical quality indicators for electron-density maps. Acta Crystallogr D. 2012;68:454–67. doi: 10.1107/S0907444911035918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Read RJ, et al. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19:1395–412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.