

Abstract
IspG is the penultimate enzyme in non-mevalonate biosynthesis of the universal terpene building blocks isopentenyl diphosphate and dimethylallyl diphosphate. Its mechanism of action has been the subject of numerous studies but remained unresolved due to difficulties in identifying distinct reaction intermediates. Using a moderate reducing agent as well as an epoxide substrate analogue, we were now able to trap and crystallographically characterize various stages in the IspG catalyzed conversion of 2-C-methyl-D-erythritol-2,4-cyclo-diphosphate (MEcPP) to (E)-1-hydroxy-2-methylbut-2-enyl-4-diphosphate (HMBPP). In addition, the enzyme's structure was determined in complex with several inhibitors. These results, combined with recent electron paramagnetic resonance data, allowed us to deduce a detailed and complete IspG catalytic mechanism which describes all stages from initial ring opening to formation of HMBPP via discrete radical and carbanion intermediates. The data presented in this article provide a guide for the design of selective drugs against many pro- and eukaryotic pathogens to which the non-mevalonate pathway is essential for survival and virulence.
Keywords: terpene biosynthesis, methylerythritol-phosphate pathway, iron-sulfur cluster catalysis, reaction mechanisms
Introduction
The non-mevalonate pathway, also called the methylerythritol phosphate or MEP pathway [1, 2], is essential for isoprenoid biosynthesis in many human pathogens such as Plasmodium falciparum (the causative agent of malaria) and Mycobacterium tuberculosis (the causative agent of tuberculosis) [3, 4]. The pathway is, however, absent in humans and as such its enzymes are potential targets for the development of novel therapeutic compounds which are urgently required in light of the emerging threat of drug resistance [5]. The [Fe4S4] cluster containing protein IspG catalyzes the penultimate step in the MEP pathway, converting 2-C-methyl-D-erythritol-2,4-cyclo-diphosphate (MEcPP, 1) into (E)-1-hydroxy-2-methylbut-2-enyl-4-diphosphate (HMBPP, 2) by reductive dehydroxylation (Figure 1A). The efforts to develop IspG inhibitors by rational design have generated considerable interest in the unique mechanism of action of the IspG-catalyzed reaction.
Figure 1. The reaction and structure of IspG.

(a) IspG converts 2-C-methyl-D-erythritol-2,4-cyclo-diphosphate (MEcPP, 1) to (E)-1-hydroxy-2-methylbut-2-enyl-4-diphosphate (HMBPP, 2) under consumption of two electrons and two protons. (b) The enzyme forms head to tail homodimers which can alternate between an open and closed conformation through a 64° rotation of the C-terminal domain.
Previous work describes the crystal structures of both holo and substrate-bound IspG from Aquifex aeolicus and Thermus thermophilus (strain HB27) (PDB ID codes: 3NOY, 2Y0F and 4G9P), which provide crucial insights into the enzyme's structure and function [6-8]. Each subunit of the homodimer is known to fold into two domains: an N-terminal (βα)8 TIM-barrel, and a C-terminal domain that hosts a [Fe4S4] cluster. The active site is located at the interface of the N-terminal domain of one subunit with the C-terminal domain of the adjacent subunit, forming a catalytically active “head-to-tail” dimer. Catalysis proceeds in the “closed” active site cavities of the dimer. Substrate loading and product release require a transition to an open conformation, which occurs via a hinge movement of the two domains (Figure 1B).
Numerous reaction intermediates have been proposed to be involved in the IspG catalyzed conversion of MEcPP to HMBPP, including epoxide, ferraoxetane, radical, carbocation and carbanion structures [9-13]. Despite the availability of holo and substrate-bound crystal structures [6-8] as well as extensive spectroscopic data from electron paramagnetic resonance (EPR), and isotope labelling experiments [4, 14], the identity and lifetime of the IspG reaction intermediates has been controversial.
In this work, we have been able to trap and structurally characterize several putative reaction intermediates as well as several inhibitors bound to the IspG active site. In combination with data from previous EPR and isotope labeling experiments, these results help solve a long-standing mechanistic mystery and prepare the ground for rational inhibitor design against many important pathogens.
Results and discussion
IspG in complex with its reaction intermediates
Using the HB8 strain of T. thermophilus, we cloned and purified an IspG homologue differing in four positions of its primary sequence to the structures reported by Ermler, Jomaa and coworkers [6, 7]. After discovering alternative crystallization conditions, the IspG structure was determined by using the deposited coordinates (PDB ID code: 4G9P) as a starting model for molecular replacement [7]. With only one molecule in the asymmetric unit and a highly compact cell, structures were solved to a maximum resolution of 1.30 Å. Furthermore, in the presence of the reducing agent sodium dithionite, we were able to slow substrate conversion in the crystal sufficiently to trap distinct reaction intermediates (Figure 2).
Figure 2. X-ray structures of IspG reaction intermediates.

Close-up view of T. thermophilus IspG structures containing (a) MEcPP (1), (b) the radical/carbanion (I•), and (c) the carbanion (I-) intermediates as well as (d) HMBPP (2) (PDB ID codes 4S38, 4S3A, 4S3B and 4S39 respectively). The 2FoFc electron density is shown at a contour level of 1.0 sigma. The Glu232-H2O-Glu350 motif may serve as a proton shuttle during catalysis. Notably, the radical state (I•) may be interpreted as an average between the cyclic substrate and the carbanion (I-). As, however, all catalytic activity in the crystal is reduced dramatically, requiring more than 24 hours for a single reaction to conclude, a significant contribution of the anionic state seems unlikely.
Prior to substrate entry, IspG is thought to alternate between open and closed conformations, analogous to a “swinging door” [7]. When MEcPP enters the enzyme's catalytic centre, its functional groups are recognized and held in place by a variety of hydrogen bond forming active side residues (Figure 2A). The diphosphate moiety in particular interacts with a positively charged cavity formed by the side chains of Arg56, Arg110, Arg141, Lys204, and Arg260 [7]. IspG thus adopts a closed state in which the active site is protected from bulk solvent and catalysis can commence (Figure 3, steps a-f). As the substrate binds to the [Fe4S4] 2+ cluster it replaces the carboxylate side chain of Glu350. At the same time, the domain movement described above brings Glu232 into proximity of the MEcPP C3 hydroxyl group. This amino acid is then likely to accept a proton released on formation of the alkoxide complex between enzyme and substrate (steps a and b).
Figure 3. Proposed reaction mechanism for the IspG catalyzed conversion of MEcPP to HMBPP.

Structures observed crystallographically are labelled with black-framed characters. Grey labels for the short-lived reaction intermediates c1/c2 (in brackets) indicate availability of structural data from a different substrate, i.e. the epoxide analogue 5. Proposed oxidation and EPR spin states of the [Fe4S4] cluster are shown where available. EPR results from freeze-quenched solutions indicate that the ferraoxetane structure d3 is in equilibrium with d1/d2. IET = internal electron transfer.
Substrate binding is succeeded by the formation of a series of reaction intermediates (Figure 3, steps c-f). Liu and co-workers showed that (oxidized) IspG catalyzes positional isotopic exchange of the MEcPP's external diphosphate oxygen atoms [14] suggesting that reversible C2-O bond cleavage and ring opening occur in the closed state (steps c1/c2). Thus, the C2 atom may shuttle between a carbocation and a radical state at this point, mediated by an electron transfer via the [Fe4S4]2+/3+ cluster.
The subsequent reaction stages (steps d-f) could be analyzed by soaking crystals of the IspG-MEcPP complex with sodium dithionite for periods of 1, 24 or 72 hours, prior to cryoprotection and vitrification. In solution (and in the absence of an electron mediator such as a viologen) dithionite-promoted catalysis is comparatively slow. In the crystalline solid-state, diffusion and conformational restraints may reduce the reaction rates even further. This deceleration enabled us to trap and characterize three intermediates “en route” from substrate to product.Structures obtained from these crystals thus represent averaged snapshots of the individual reaction stages (Figure 2B-D).
With the first electron transfer from the external reducing agent, conversion of MEcPP becomes irreversible. As a single electron is shuttled through the [Fe4S4] 2+/3+ cluster and into the substrate, C2 is reduced and the ring opens permanently (step d1/d2 and Figure 2B). This electron uptake leads to generation of a radical-(I•)-[Fe4S4] 2+ or carbanion-(I-)-[Fe4S4]3+ intermediate. Again, both forms may be interconvertible via internal electron transfer. After incubation with sodium dithionite for 1 hour, this intermediate is observed crystallographically. Although the phosphoester link is still discernible in the electron density map, C2-O bond breakage is clearly apparent. As a result, C2 shows neither an entirely planar nor a fully sp3 hybridized geometry (Figure 2B) and the structure is more likely to represent an averaged state between steps d1 and d2. EPR data further suggest the presence of a third transition state (step d3). The observed spin of S=1/2 together with large hyperfine couplings are consistent with an [Fe4S4]3+‐ferraoxetane intermediate whose structure was deduced from the results of extensive spectroscopic and computational work [13]. This state has not been observed crystallographically but is likely to be in equilibrium with the radical/carbanion species in solution.
Uptake of a second electron by either the carbon radical (d1) or the oxidized cluster of the carbocation (d2) leads to formation of a proposed mono-carbanionic (I-) species bound to an [Fe4S4]2+ cluster (step e). This stage of the reaction was trapped and characterized after dithionite incubation for 24 hours (Figure 2C). The structural data provide evidence for pyramidalization at C2 (sum of angles = 343°), suggesting carbanion formation, with the Fe-O(C3) bond intact (Figure 2C). Subsequent elimination of the Fe-bound oxygen from the substrate may be facilitated twofold: while (i) Glu232 could donate a proton to the resulting oxyanion (O-O distance is reduced to 2.9 Å), (ii) the syn-orientation of the anion's free electron pair would allow compensation of the positive partial charge developing on the C3 carbon. This would finally result in the formation of a double bond to give the HMBPP product 2 (step f in Figure 3). Glu232 links the active site pocket to a proton channel, which may deliver a second proton to the oxygen and thus generate a water molecule as a leaving group. Correspondingly, at 72 hours of dithionite incubation, the Fe-O3 bond is no longer apparent in the electron density and there is a well-defined H2O/OH- attached the unique 4th Fe in the cluster. The ligand has planar C2 and C3 centers (C-C distance 1.4 Å, Figure 2D). Structural rearrangements required to return IspG to its open conformation eventually trigger the release of HMBPP product and water from the active site, leaving the enzyme ready for a new catalytic cycle. Constraints imposed by molecular packing prevent this last stage from occurring in the crystalline state.
IspG in complex with an epoxide substrate analogue
Earlier IspG mechanistic proposals included formation of the epoxide 5 (Figure 4a) as one possible intermediate of the reaction trajectory [11]. Specifically, 5 was shown to be converted to MEcPP under oxidizing conditions [15] and, when reduced, reacts to give the HMBPP product at catalytic turnover rates comparable to the natural substrate [16]. However, the present study confirms a different function of this molecule and its downstream reaction intermediate. Under reducing conditions, it forms the same EPR detectable signal (Figure 3, d3, Figure 4) found with the native substrate, 1.
Figure 4. Crystallographic and spectroscopic analysis of IspG with the epoxide substrate 5.
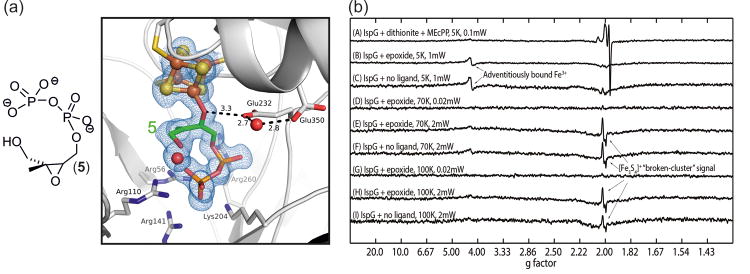
(a) The structure (PDB ID code: 4S3C) was obtained after incubation of oxidized IspG with 5 for 24 hours. It reveals an open epoxide ring and an overall geometry resembling Figure 2B (contour level of the 2FoFc electron density = 1.0 sigma). (b) The 9 GHz EPR spectra of IspG + 5. The top spectrum was obtained in the presence of dithionite and corresponds to the proposed ferraoxetane intermediate and the HMBPP product. The other spectra were obtained in the absence of any reducing agent and do not indicate the formation of any radical species.
Upon binding to IspG, 5 immediately releases the ring strain of the epoxide moiety and undergoes heterolytic ring-opening prior to any [Fe4S4] cluster mediated reduction, which is a clear distinction to the observed reactivity of the natural substrate (Figure 3, steps c1-c3). In this state, the ligand now corresponds to the carbocation intermediate (step c1) of MEcPP conversion. Small differences in the binding mode of epoxide vs. natural substrate explain why, instead of collapsing into the energetically more favorable MEcPP, the carbocation intermediate can be observed for the epoxide substrate analog. When the free diphosphate tail of 5 is bound in the IspG active site cavity, it is accompanied by a water molecule of its former hydration sphere. This water, which is absent in the IspG:MEcPP complex structures, is sandwiched between C2 and the terminal phosphate. Thus bound, it contributes in two ways to stabilization of the intermediate's dipolar state: i) In contrast to MEcPP, the epoxide analogue's terminal phosphate group is thought to be protonated at physiological pH as well as under crystallization conditions (pH 6.5). As a result, two rather than three negative charges surround the diphosphate, resulting in a reduced driving force for ring closure. ii) In the event of occasional ring closure, the protonated water (H3O+) can immediately act as a Brønsted acid, catalyze heterolytic cleavage of the phosphoester bond and return the ligand to its open conformation. Analogous to MEcPP, the carbocation is eventually reduced, forming the intermediates described above, before being released from the active site as HMBPP product (Figure 3). As such, IspG complexed with 5 represents a high-resolution crystallographic snap-shot of an otherwise short-lived intermediate.
The question then arises as to whether there is any evidence for the presence of the alternate, C2 radical-[Fe4S4]3+ species that could form on internal electron transfer from a carbocation-[Fe4S4]2+ cluster. Such a species could exist as a closed shell singlet (S=0), an open shell singlet (S=0), or as a triplet (S=1). We obtained 9 GHz EPR spectra (of oxidized IspG + 5) over a broad range of temperature, power level and sweep width conditions (Figure 4B). No evidence for any radical-type signals was found, favouring the presence of a cationic [Fe4S4]2+ species.
Inhibition of IspG
IspG has previously been proposed to be an attractive drug target against a range of infectious diseases. In response, a variety of diphosphate-containing inhibitors have been designed and screened against the enzyme [4, 13, 17-19]. Several of them were shown to exhibit activity against both IspG as well as IspH, the downstream catalyst and final component of the MEP pathway [18, 20]. In addition to the identification of the reaction intermediates described above, minimal requirements for ligand binding are essential pieces of information for the design and improvement of such inhibitors. The complex structures of IspG with two alkyne inhibitors (7 and 8; both of which have also been structurally analyzed with IspH [21]), as well as diphosphate (6, see Figure 5), reveal a clear relationship between the compounds' structure and their potency.
Figure 5. Crystallographic analysis of IspG inhibition.
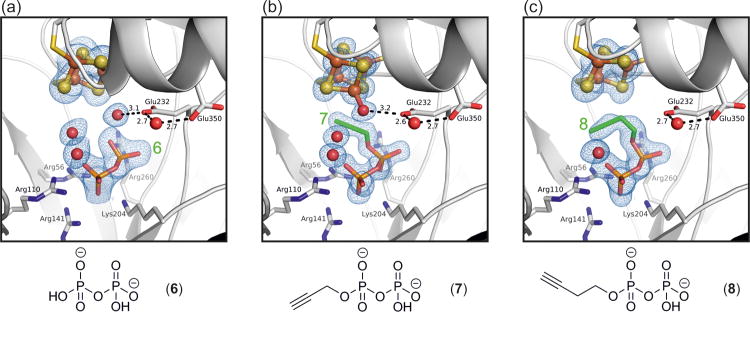
Structures of IspG are shown (a) in the presence of a diphosphate fragment (6) as well as in complex with the propargyl diphosphate inhibitors (b) 7 and (c) 8 (PDB ID codes 4S3D, 4S3E and 4S3F respectively). The 2FoFc electron density maps are contoured at 1.0 sigma.
Shared between all inhibitors is the binding mode of the diphosphate moiety in the positively charged pocket and the presence of a ‘conserved’ water molecule as observed for the epoxide intermediate. Isolated diphosphate (half maximal inhibitory concentration IC50 = 20 μM, Figure S2), is the structurally simplest inhibitor. The molecule occupies the positively charged binding pocket and, in that way, competes directly with the natural MEcPP substrate in solution (Figure 5A). In contrast to the larger inhibitor structures, diphosphate does not fill the active site cavity and interactions with amino acid side chains are limited. Nevertheless, it is the smallest species that is able to maintain IspG in a locked conformation, albeit only while its diffusion is restricted by the closed state of the crystal structure. Surprisingly, an [Fe3S4] configuration is observed in which the apical iron is lost. This is, however, likely to be an artifact of the crystalline state in which the cluster is left without coordination by either Glu350 (as seen in the holo structure) or a stabilizing ligand.
The propargyl inhibitor 7 has an IC50 value of 770 nM and does not displace the apical iron (Figure 5B). In fact, its binding mode is reminiscent of the natural HMBPP product (Figure 2D) and inhibition is more likely the result of competitive binding to the enzyme's active site. Neither IspG nor IspH catalyze any conversion of this molecule (Figure S2) [21]. Compound 8, the most potent inhibitor, has an IC50 value of 580 nM. It is likely that a steric clash between the alkyne group and the enzyme's iron-sulfur cluster, as a consequence of which the apical iron is lost in the complex, is an important contribution to its inhibitory effect (Figure 5C). In the active site of IspH, 8 reacts with a water bound to the apical iron of the [Fe4S4] cluster to form an enolate which is then converted into the corresponding aldehyde in solution [21]. This hydration is not catalyzed by IspG as the acetylene group of 8 points in opposite directions when bound to IspH and IspG, providing a ready explanation for the lack of reactivity with the latter (Figure S3).
Despite inhibiting both enzymes, the binding modes observed for 7 and 8 vary between IspG and IspH. This observation holds true as a general feature for all structurally analyzed ligands, and also applies to natural substrates, intermediates and products [22]. With the exception of [Fe4S4] cluster coordination, the respective active site cavities also differ accordingly. For both enzymes, however, compounds with an increased carbon chain length and/or sterically more demanding substituents have been found to be less potent inhibitors [17] and could not be visualized as complex structures in our X-ray crystallographic studies.
Conclusions
By identifying each of the IspG reaction intermediates we are now able to reconstruct the enzyme's complete catalytic reaction. The conversion of MEcPP to HMBPP is now visualized on a molecular level, from initial ring opening to product release via formation of distinct radical and anionic intermediates at the C2 carbon atom. The present study on mechanism and ligand/inhibitor binding provides highly valuable information for future research into drug design targeting the MEP pathway. It may enable novel, rational design strategies in the global battle against bacterial infections and diseases caused by eukaryotic pathogens such as Plasmodium spp.. The final two enzymes of this pathway, IspG and IspH, are of particular interest since they can be targeted simultaneously. It remains to be determined if the mechanistic findings outlined here also apply to closely related enzymes, such as the three-domain IspG homologues in plants [17]. If so, they may be of future use for even further applications such as herbicide development.
Materials and Methods
Cloning
The ispG gene from T. thermophilus HB8 (NCBI Gene ID: 3168579) was amplified by polymerase chain reaction (PCR) using the oligonucleotides 5′-ACACACGGATCCATGGAGGGGATGAGGCGAC-3′ and 5′-ACACACCTGCAGTCAGGCCTTGGGGGCGAAG-3′ as primers and genomic DNA as template. The amplification product was digested with BamHI-HF and PstI-HF (New England Biolabs, Ipswich, MA) and subsequently ligated into a modified pET-28b(+) vector (Invitrogen, Darmstadt, Germany) comprising a SUMO protein coding region (Smt3p from Saccharomyces cerevisiae, amino acids 1–98). The Escherichia coli strain BL21(DE3) (Novagen, Darmstadt, Germany) was transformed by the resulting plasmid pET28bSUMOispGTherm.th. together with the plasmid pACYC184iscSfdx [23] containing the isc operon for iron-sulfur cluster assembly.
Bacterial culture
The recombinant E. coli strain was grown in shaking flasks containing 3 L of lysogeny broth supplemented with kanamycin (50 mg/L), chloramphenicol (25 mg/L), ferric ammonium citrate (16 mg/L), and cysteine (121 mg/L). The cultures were incubated at 37 °C under agitation to an OD600 of 0.7. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM and incubation was continued under agitation over night at 20 °C. Cells were harvested by centrifugation, washed with 0.9% (w/v) NaCl and stored at -20 °C.
Protein purification
All purification steps were carried out under anoxic conditions (pO2 < 1 ppm) in an anaerobic chamber (Coy Labs, Grass Lake, MI). Frozen bacterial cells were thawed in 100 mM Tris hydrochloride, pH 7.5, containing 300 mM NaCl and 20 mM imidazole hydrochloride (buffer A). The cells were lysed using a cell lysis device (Basic Z Model, Constant Systems, Daventry, England). The resulting suspension was centrifuged at 17,000 g (9,600 rpm) for 60 min at 4 °C. The supernatant was applied to a column of nickel-chelating Sepharose FF (GE Healthcare Life Science, Uppsala, Sweden; column volume, 12.5 mL), which had been equilibrated with buffer A (flow rate, 3 mL/min). The column was washed with buffer A (125 mL) and subsequently developed with a linear gradient of 20–500 mM imidazole in buffer A (total volume, 125 mL). Fractions were combined and His6-SUMO protease (Ulp1 from S. cerevisiae, 0.5 mg) was added. The solution was dialyzed overnight at 4 °C against 100 mM Tris hydrochloride, pH 7.5, containing 300 mM NaCl, and applied to a nickel-chelating column of Sepharose FF (column volume, 12.5 mL) that was washed with 75 mL of buffer A. The percolate was dialyzed overnight at 4 °C against 30 mM Tris hydrochloride, pH 7.5, containing 150 mM NaCl. The solution was concentrated to approximately 15 – 26 mg/mL using a 30 kDa Amicon Ultra Centrifugal Filter Device (Merck Millipore, Billerica, MA) and was stored at -80 °C.
IspG Crystallization and X-Ray analysis
Crystals of IspG grew at 20 °C within one week using the sitting drop vapor diffusion method. All complex structures were obtained by cocrystallization of IspG with 5 mM of the respective ligand. Drops comprised equal volumes of protein and reservoir solutions (100 mM Bis-Tris propane/HCl, pH 6.5, 20 % PEG 3350, and 0.2 mM Na2SO4). Complex structures IspG:I•, IspG:I- and IspG:2 were obtained by incubating IspG:1 crystals with 50 mM sodium dithionite for 1 h (IspG:I•), 24 h h (IspG:I-) or 72 h (IspG:2) and 72 h (IspG:HMBPP). Crystals were soaked for 1 min in a mixture of mother liquor and 50 % glycerol (1:1, v/v) prior to vitrification in liquid nitrogen. Datasets were recorded using synchrotron radiation at the beamline X06SA, SLS (Villigen, Switzerland). X-ray intensities were evaluated with XDS [24]. The space group of IspG structures was C2 with unit cell dimensions of approximately a = 111 Å, b = 63 Å, and c = 86 Å. Phases were obtained by molecular replacement using PHASER [25] and the coordinates of IspG from T. thermophilus strain HB27 in complex with MEcPP (PDB ID code: 4G9P) [7] as starting model. The model was completed with COOT [26] and rigid body, TLS and positional refinements with REFMAC5 [27] (Table S1). The quality of the stereochemistry was confirmed by the Ramachandran plot determined with the program PROCHECK [28]. Graphical illustrations were drawn with the program PyMOL [29].
T. thermophilus IspG Assays
T. thermophilus IspG was purified as described previously [13], degassed by bubbling nitrogen through the solution vigorously, and then transferred into an anaerobic chamber (Coy Labs, Grass Lake, MI). The as-purified IspG was reconstituted by incubating with 0.5 mM Fe(NH4)2(SO4)2, 2.5 mM L-cysteine, 5 mM DTT, and ∼0.1 μM purified IscS (expressed from a plasmid which was the kind gift of Professor James A. Imlay) until the A410/A280 ratio reached ∼0.4. The protein was desalted prior to use. Protein concentration was determined using a Bio-Rad (Hercules, CA) Protein Assay kit. Inhibition assays were performed anaerobically at room temperature as described previously [13].
Continuous-Wave EPR Spectroscopy
All EPR samples were prepared as described previously [13]. CW-EPR experiments were performed at X-band using a Varian E-122 spectrometer together with an Air Products (Allentown, PA) helium cryostat. Various temperatures and power levels were used as indicated in Fig. 3.
Supplementary Material
Acknowledgments
This work was supported by the Hans-Fischer Gesellschaft, the Deutsche Forschungsgemeinschaft (DFG Grant 1861/5-1), the United States Public Health Service (National Institutes of Health Grant GM065307), a Harriet A. Harlin Professorship (E.O.), the University of Illinois/Oldfield Research Fund, and the King Abdullah University of Science and Technology (FIC/2010/07 and KAUST-TUM joint degree program). We are grateful to Ms. Katrin Gärtner for invaluable protein purification and crystallization work. We also thank the staff of the X06SA-beamline at the Paul Scherrer Institute, Swiss Light Source, Villigen, Switzerland for help during data collection.
Abbreviations
- MEP
methylerithrol phosphate
- MEcPP
2-C-methyl-D-erythritol-2,4-cyclo-diphosphate
- HMBPP
(E)-1-hydroxy-2-methylbut-2-enyl-4-diphosphate
- IET
internal electron transfer
Footnotes
Accession numbers: The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB ID codes: 4S38, 4S3A, 4S3B, 4S39, 4S3C, 4S3D, 4S3E and 4S3F).
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Rohmer M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants[dagger] Natural Product Reports. 1999;16:565–74. doi: 10.1039/a709175c. [DOI] [PubMed] [Google Scholar]
- 2.Campos N, Rodríguez-Concepción M, Seemann M, Rohmer M, Boronat A. Identification of gcpE as a novel gene of the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis in Escherichia coli. FEBS Letters. 2001;488:170–3. doi: 10.1016/s0014-5793(00)02420-0. [DOI] [PubMed] [Google Scholar]
- 3.Eisenreich W, Bacher A, Arigoni D, Rohdich F. Biosynthesis of isoprenoids via the non-mevalonate pathway. CMLS, Cell Mol Life Sci. 2004;61:1401–26. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Oldfield E. Bioorganometallic Chemistry with IspG and IspH: Structure, Function, and Inhibition of the [Fe4S4] Proteins Involved in Isoprenoid Biosynthesis. Angewandte Chemie International Edition. 2014;53:4294–310. doi: 10.1002/anie.201306712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rohdich F, Bacher A, Eisenreich W. Isoprenoid biosynthetic pathways as anti-infective drug targets. Biochemical Society transactions. 2005;33:785–91. doi: 10.1042/BST0330785. [DOI] [PubMed] [Google Scholar]
- 6.Rekittke I, Nonaka T, Wiesner J, Demmer U, Warkentin E, Jomaa H, et al. Structure of the E-1-hydroxy-2-methyl-but-2-enyl-4-diphosphate synthase (GcpE) from Thermus thermophilus. FEBS Letters. 2011;585:447–51. doi: 10.1016/j.febslet.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Rekittke I, Jomaa H, Ermler U. Structure of the GcpE (IspG)–MEcPP complex from Thermus thermophilus. FEBS Letters. 2012;586:3452–7. doi: 10.1016/j.febslet.2012.07.070. [DOI] [PubMed] [Google Scholar]
- 8.Lee M, Grawert T, Quitterer F, Rohdich F, Eppinger J, Eisenreich W, et al. Biosynthesis of isoprenoids: crystal structure of the [4Fe-4S] cluster protein IspG. Journal of molecular biology. 2010;404:600–10. doi: 10.1016/j.jmb.2010.09.050. [DOI] [PubMed] [Google Scholar]
- 9.Kollas AK, Duin EC, Eberl M, Altincicek B, Hintz M, Reichenberg A, et al. Functional characterization of GcpE, an essential enzyme of the non-mevalonate pathway of isoprenoid biosynthesis. FEBS Letters. 2002;532:432–6. doi: 10.1016/s0014-5793(02)03725-0. [DOI] [PubMed] [Google Scholar]
- 10.Seemann M, Bui BTS, Wolff M, Tritsch D, Campos N, Boronat A, et al. Isoprenoid Biosynthesis through the Methylerythritol Phosphate Pathway: The (E)-4-Hydroxy-3-methylbut-2-enyl Diphosphate Synthase (GcpE) is a [4Fe–4S] Protein. Angewandte Chemie International Edition. 2002;41:4337–9. doi: 10.1002/1521-3773(20021115)41:22<4337::AID-ANIE4337>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 11.Rohdich F, Zepeck F, Adam P, Hecht S, Kaiser J, Laupitz R, et al. The deoxyxylulose phosphate pathway of isoprenoid biosynthesis: Studies on the mechanisms of the reactions catalyzed by IspG and IspH protein. Proceedings of the National Academy of Sciences. 2003;100:1586–91. doi: 10.1073/pnas.0337742100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brandt W, Dessoy MA, Fulhorst M, Gao W, Zenk MH, Wessjohann LA. A Proposed Mechanism for the Reductive Ring Opening of the Cyclodiphosphate MEcPP, a Crucial Transformation in the New DXP/MEP Pathway to Isoprenoids Based on Modeling Studies and Feeding Experiments. ChemBioChem. 2004;5:311–23. doi: 10.1002/cbic.200300743. [DOI] [PubMed] [Google Scholar]
- 13.Wang W, Li J, Wang K, Huang C, Zhang Y, Oldfield E. Organometallic mechanism of action and inhibition of the 4Fe-4S isoprenoid biosynthesis protein GcpE (IspG) Proceedings of the National Academy of Sciences. 2010;107:11189–93. doi: 10.1073/pnas.1000264107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao Y, Rooker D, You Q, Freel Meyers CL, Liu P. IspG-Catalyzed Positional Isotopic Exchange in Methylerythritol Cyclodiphosphate of the Deoxyxylulose Phosphate Pathway: Mechanistic Implications. ChemBioChem. 2011;12:527–30. doi: 10.1002/cbic.201000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao Y, Nyland RL, 2nd, Meyers CL, Liu P. Methylerythritol cyclodiphosphate (MEcPP) in deoxyxylulose phosphate pathway: synthesis from an epoxide and mechanisms. Chemical communications (Cambridge, England) 2010;46:7220–2. doi: 10.1039/c0cc02594a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nyland Ii RL, Xiao Y, Liu P, Freel Meyers CL. IspG Converts an Epoxide Substrate Analogue to (E)-4-Hydroxy-3-methylbut-2-enyl Diphosphate: Implications for IspG Catalysis in Isoprenoid Biosynthesis. Journal of the American Chemical Society. 2009;131:17734–5. doi: 10.1021/ja907470n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu YL, Guerra F, Wang K, Wang W, Li J, Huang C, et al. Structure, function and inhibition of the two- and three-domain 4Fe-4S IspG proteins. Proceedings of the National Academy of Sciences. 2012;109:8558–63. doi: 10.1073/pnas.1121107109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerra F, Wang K, Li J, Wang W, Liu YL, Amin S, et al. Inhibition of the 4Fe-4S proteins IspG and IspH: an EPR, ENDOR and HYSCORE investigation. Chemical Science. 2014;5:1642–9. doi: 10.1039/C3SC53301H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakagawa K, Takada K, Imamura N. Probable novel MEP pathway inhibitor and its binding protein, IspG. Bioscience, biotechnology, and biochemistry. 2013;77:1449–54. doi: 10.1271/bbb.130094. [DOI] [PubMed] [Google Scholar]
- 20.Rohdich F, Hecht S, Bacher A, Eisenreich W. The deoxyxylulose phosphate pathway of isoprenoid biosynthesis. Discovery and function of the ispDEFGH genes and their cognate enzymes. Pure and Applied Chemistry. 2003:393. [Google Scholar]
- 21.Span I, Wang K, Wang W, Zhang Y, Bacher A, Eisenreich W, et al. Discovery of acetylene hydratase activity of the iron-sulphur protein IspH. Nature communications. 2012;3:1042. doi: 10.1038/ncomms2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gräwert T, Span I, Eisenreich W, Rohdich F, Eppinger J, Bacher A, et al. Probing the reaction mechanism of IspH protein by x-ray structure analysis. Proceedings of the National Academy of Sciences. 2010;107:1077–81. doi: 10.1073/pnas.0913045107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gräwert T, Kaiser J, Zepeck F, Laupitz R, Hecht S, Amslinger S, et al. IspH Protein of Escherichia coli: Studies on Iron– Sulfur Cluster Implementation and Catalysis. Journal of the American Chemical Society. 2004;126:12847–55. doi: 10.1021/ja0471727. [DOI] [PubMed] [Google Scholar]
- 24.Kabsch W. XDS. Acta Crystallographica Section D. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–74. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–91. [Google Scholar]
- 29.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.