

Abstract
Guanine-rich sequences can, under appropriate conditions, adopt a distinctive, four-stranded, helical fold known as a G-quadruplex. Interest in quadruplex folds has grown in recent years as evidence of their biological relevance has accumulated from both sequence analysis and function-specific assays. The folds are unusually stable and their formation appears to require close management to maintain cell health; regulatory failure correlates with genomic instability and a number of cancer phenotypes. Biologically relevant quadruplex folds are anticipated to form transiently in mRNA and in single-stranded, unwound DNA. To elucidate factors, including bound solvent, that contribute to the stability of RNA quadruplexes, we examine, by X-ray crystallography and small-angle X-ray scattering, the structure of a previously reported tetramolecular quadruplex, UGGGGU stabilized by Sr2+ ions. Crystal forms of the octameric assembly formed by this sequence exhibit unusually strong diffraction and anomalous signal enabling the construction of reliable models to a resolution of 0.88 Å. The solvent structure confirms hydration patterns reported for other nucleic acid helical conformations and provides support for the greater stability of RNA quadruplexes relative to DNA. Novel features detected in the octameric RNA assembly include a new crystal form, evidence of multiple conformations and structural variations in the 3′ U tetrad, including one that leads to the formation of a hydrated internal cavity.
Keywords: RNA quadruplex, high-resolution RNA structure
Introduction
Nucleic acid sequences in which guanines are both abundant and distributed in accordance with a distinct but permissive pattern have a propensity to fold into a G-quadruplex motif [1] composed of stacks of planar G-quartets, as outlined in Fig. 1. Once formed, such structures are stabilized by a complement of electrostatic, stacking and hydrogen-bonding forces that render them unusually stable relative to other nucleic acid conformations. Though many examples of in vitro folded structures obtained from G-rich sequences are available [2,3], the extent to which such structures occur in vivo and their physiological roles remain open to question.
Fig. 1.
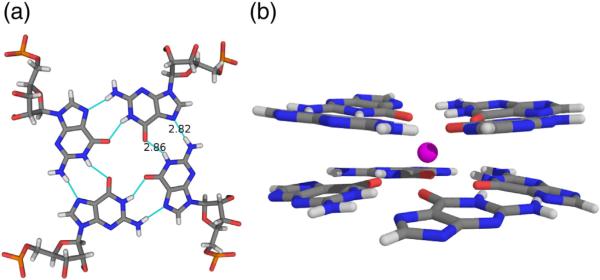
Guanine quartet secondary structure and quartet stacking, coordinates are from data set P1B.
Although our current understanding is limited, the following generalizations appear broadly applicable: a growing inventory of structures confirms the stability and conformational diversity of G-quadruplex folds; sequences with the potential to adopt a quadruplex fold are abundant, though globally counter-selected, and exhibit a markedly non-uniform genomic distribution suggestive of selective pressure for and against their formation in particular genomic regions; function-specific assays associate quadruplex folds with a range of cellular functions, notably regulation of genes associated with growth and development; and the formation and unwinding of quadruplex folds appear to require close management to maintain cell health—regulatory failure correlates with a number of cancer phenotypes [4–6].
Here we examine, by X-ray crystallography and small-angle X-ray scattering (SAXS), an RNA hexamer, UGGGGU, whose conformation adopts a guanine quadruplex fold. The crystallographic data analyzed in this work, three data sets whose resolution ranges from 0.88 to 1.01 Å, display unusually high resolution relative to current PDB depositions, particularly for RNAs. Thus, independently of insights into quadruplex structure, these data provide an opportunity for reliable quantification of RNA structure and hydration parameters, and this analysis is therefore of more general interest.
A TGGGGT repeat is synthesized by Tetrahymena telomerase and was the focus of the initial elucidation of telomerase activity in chromosome maintenance [7]. Its four-stranded, parallel quadruplex conformation has been the subject of numerous structural studies [8,9]. The RNA counterpart examined here has been the subject of previous NMR [10] and X-ray studies [11]. The DNA and RNA forms share a similar arrangement over the central stack of guanine quartets but differ in the conformation of capping nucleotides.
Biologically relevant quadruplex folds are anticipated to form transiently in mRNA and in single-stranded, unwound DNA [4]. In both cases, formation of stable structures in the substrate may interfere with recognition or processivity by polymerases. This effect has been documented in translational elongation and ribosomal frameshifting [12] and stalled transcription [13]. Understanding effects that contribute to the stability of quadruplex structures can help assess the roles they play. Two features of the present structure bear on the general issue of quadruplex stability: variability in the U tetrad formed by the capping nucleotides and evidence for helix stabilization by ribose O2′ atoms.
Capping the regularly spaced column of stacked G4 quartets poses an interesting structural challenge for quadruplex nucleic acid conformation [14]. A thymine tetrad at the 5′ terminus of a parallel-stranded TGGGGT quadruplex was reported by Cáceres et al. [15]. Recently, a thymine tetrad was also found in the locked-nucleic-acid form of the TGGGGT repeat [16]. However, as discussed by Cáceres et al., tetrad formation by terminal thymines is only one among several competing arrangements as evident in PDB entries 352D, 244D, 1S45 and 1S47. In contrast, the RNA form of this sequence appears to invariably favor U tetrads as a capping conformation. Formation of U tetrads and the unusual stability of the resulting structure were first reported by Cheong and Moore [10]. More recently, formation of U tetrads was shown to contribute to stabilization in human telomeric RNA repeats, UAGGGU [17,18]. Overall, U tetrads appear to provide a more stable solution for capping the quadruplex complex than the splayed-out thymines evident in the DNA structures. However, as reported in Structural variation and disorder, there is considerable variation in how U tetrads stack over guanines quartets.
Thermodynamic studies indicate that the RNA form of quadruplex structures is often more stable than DNA [19,20]. In Hydration: Distribution of water contacts, we show that, in guanine residues, the ribose O2′ hydroxyl is heavily involved in networks of water-mediated hydrogen bonds that link quadruplex chains. The hydrogen bonds form both inter-helix and intra-helix contacts that stabilize the right-handed helical fold formed from the core guanine residues. Taken together, these two effects likely to contribute to the unusual stability exhibited by this structure and provide insight into related RNA sequences.
Results and Discussion
Oligo assemblies
Crystallographic data reduction and refinement statistics for the three data sets examined are shown in Table 1. Two of the data sets share the tetragonal, P4212, space group of the previously reported structure (PDB entry 1J8G) [11], whereas the third exhibits a novel crystal form in an orthorhombic space group, C2221.
Table 1.
Crystallographic data reduction and refinement statistics [33]
| Space group |
P4212 |
C2221 |
|
|---|---|---|---|
| Data set | P1A | P1B | P2C |
| Cell dimensions (Å) | |||
| a | 36.65 | 36.62 | 37.66 |
| b | — | — | 54.20 |
| c | 37.08 | 37.38 | 95.68 |
| Resolution range (Å) | 36.68–0.88 (0.93–0.88) | 18.31–0.92 (0.97–0.92) | 95.68–1.01 (1.06–1.01) |
| R meas | 0.082 (0.249) | 0.153 (0.631) | 0.103 (0.946) |
| R pim | 0.024 (0.157) | 0.033 (0.205) | 0.030 (0.364) |
| Number of observations | 139,429 (2889) | 391,879 (15,631) | 624,238 (46,394) |
| Number of unique observations | 19,031 (1684) | 17,321 (1759) | 51,294 (7088) |
| Mean [(I)/σ(I)] | 18.500 (2.400) | 21.900 (5.300) | 18.000 (2.600) |
| CC(1/2) | 0.998 (0.945) | 0.989 (0.824) | 0.997 (0.748) |
| Completeness (%) | 93.50 (58.80) | 95.00 (69.30) | 99.20 (94.90) |
| Multiplicity | 7.30 (1.70) | 22.60 (8.90) | 12.20 (6.50) |
| Refinement | |||
| R work | 0.093 | 0.091 | 0.102 |
| R free | 0.098 | 0.109 | 0.119 |
| Bond length RMSD (Å) | 0.013 | 0.011 | 0.012 |
| Bond angle RMSD (°) | 1.744 | 1.568 | 1.670 |
| Chiral volume RMSD (Å3) | 0.106 | 0.096 | 0.100 |
Entries in parentheses apply to the outermost shell.
In all unit cell arrangements observed, the overall crystal packing is arranged in infinite columns composed of coaxially stacked, oblong, octamers. The columns are parallel with the cell c and a axes in tetragonal and orthorhombic space groups, respectively. Each octamer consists of two stacked and intercalated tetramers. Four UGGGGU oligos with parallel polarity wrapped about a central axis with a right-handed helical twist make up a tetramer, as shown in Fig. 2a and b. The two tetramers that form the octameric unit are arranged with opposing polarity. Thus, the center of the octamer is formed by the intercalated 5′ termini of two tetramers whereas the 3′ termini form the ends of the octamer.
Fig. 2.

Components of UGGGGU octamer assembly. (a) An isolated RNA hexamer, the 3′ uridine that participates in a U tetrad, is shown in blue, and the 5′ uridine involved in tetramer intercalation is shown in cyan. Two central Sr2+ cations involved in G-quartet stabilization are shown in magenta. (b) The tetramer component produced by rotation of an individual strand about a 4-fold axis. (c) Two adjacent, stacked, strands related by 2-fold symmetry and (d) the resulting octamer. Coordinates correspond to data set P1B.
Within each oligo, the four guanine residues and the 3′ uridine point toward the central axis and the orbit resulting from rotation about 4-fold yields the stacked G-quartets characteristic of a G4 fold as shown in Fig. 2b. The 5′ and 3′ terminal uridines adopt very different conformations. The vector corresponding to the C1′–N1 bond of the 3′ uridine points toward the central axis but diagonally away from it in the 5′ uridine, though in both cases, the χ angle is in the anti range. The G-quartets formed by four hydrogen-bonded guanine bases are approximately planar and all nucleotides adopt an anti conformation about the glycosidic bond with χ values around −145°, near the center of the anti range.
Cations that stabilize the stacked G-quartets are positioned along the 4-fold axis. In these structures, Sr2 + cations are invariably positioned between two G-quartets, as shown in magenta in Fig. 2b and d, and Na+ cations coordinate the 3′ U tetrads.
Column arrangement and crystal packing
The columns formed by stacked octamers are arranged differently in the two space groups observed as shown in Fig. 3. In the tetragonal space group , each column is in contact with four neighbors. The cations at the center of the neighboring columns form a square and a relatively large volume of bulk solvent fills the space near the center of this square. In the ortho-rhombic space group, each column is in contact with six neighbors. The column centers form a hexagon and nearly all the solvent volume is taken up by interstitial waters that form networks of hydrogen bonds around each column.
Fig. 3.

Arrangement of quadruplex columns in (a) tetragonal (P4212) and (b) orthorhombic (C2221) space groups. Stacked octamers are shown with the central cation axis directed toward the reader. The ASU is shown in lighter colors.
In both cases, the overall symmetry of the crystal results from application of multiple symmetry operations to the hexamer chains. However, the division between crystallographic and non-crystallographic operations differs between space groups. In the tetragonal case, two strands, related by a noncrystallographic symmetry (NCS) 2-fold, occur in the asymmetric unit (ASU). Octamers result from rotation of these strands about the space group's 4-fold axis.
Orthorhombic space groups include no crystallo-graphic 4-fold operator. Here eight RNA chains occur in the ASU. Of these, four form a complete tetramer whose chains are related by 4-fold NCS. The partner tetramer is formed by a crystallographic 2-fold. The remaining four chains in the ASU, which are related by two NCS 2-fold operations, form an adjacent octamer by rotation around the space group's 2-fold axis.
Though data sets P1A and P1B share the P4212 space group of structure 1J8G, the unit cell is only approximately half as large. Whereas the 1J8G unit cell spans two octamers along the c axis, here it only spans a single octamer.
Small-angle X-ray scattering
Solution small-angle scattering data were collected for UGGGGU samples over a range of concentrations and exposure times as described in the methods section. Scattering curves indicated good contrast and a mono-disperse solution with little aggregation or radiation damage yielding data amenable to more detailed analysis. The Kratky curves indicate a folded conformation. Intensity and Kratky plots for a representative sample are shown in Fig. 4a and b and derived parameters are given in Table 2.
Fig. 4.

SAXS analysis. (a) Intensity of buffer-adjusted scattering for UGGGGU samples at concentrations of 0.4 mg/ml (red), 2 mg/ml (green) and 4 mg/ml (blue). (b) The corresponding Kratky plots. (c) The ab initio model derived by DAMMIF from merged scattering data displayed as a black mesh and fit to the symmetry-expanded octamer from structure P1B. (d) Comparison of the experimental scattering curve, merged over all concentrations and shown in blue, relative to predicted scattering from octameric and tetrameric models derived from structure P1B, shown in red and green.
Table 2.
Derived parameters from SAXS data collection
| Guinier-based I(0) (cm–1), Rg (Å) | 4707.3 ± 6.05, 14.77 ± 0.0.02 |
| P(R)-based I(0) (cm–1), Rg (Å) | 4654 ± 1.3, 14.63 ± 0.0.003 |
| D max | 45.82 |
| Porod volume | 16,711 |
The estimates of Rg and Dmax are in good agreement with the dimensions of the octamer augmented by a hydration layer. The molecular mass estimate of 14.3 kDa, obtained via the volume-of-correlation method [21], is in reasonable agreement with the value of 17.2 kDa expected for an octamer and, along with the Rg and Dmax estimates, excludes larger multimeric assemblies in solution.
The fit of the molecular envelope obtained from an ab initio model to the crystallographic structure is shown in Fig. 4c. A comparison of the experimental scattering curve, merged over all concentrations, with predicted scattering from octameric and tetrameric models derived from structure P1B is shown in Fig. 4d.
Overall, these scattering data suggest that the preferred conformation in solution is an octameric assembly with dimensions in good agreement with the crystal form. This result differs from the tetramer conformation observed via NMR analysis by Cheong and Moore [10], a difference likely due to the identity of the central coordinating cation, K+ versus Sr2+. The backbone geometry of the NMR structure, as reported in their Table IV, shows only rough agreement with the torsion angles [46] of structures P1A, P1B and P2C (Supplemental Table 3). It is noteworthy that angles for which they report the widest variation among the three NMR models for nucleotides G5 and U6, G5-ζ and U6-α, are the same angles that show pronounced variation in conformations 3′U-A, 3′U-B and 3′U-C of the crystal structures as discussed in Structural variation and disorder.
At higher scattering angles, discrepancy in intensity between that predicted for the octamer derived from the crystallographic model and the experimental solution data is apparent. Conformations of the octameric assemblies in solution could differ from the crystallographic model, for example, by populating multiple U tetrad conformations.
Structural variation and disorder
Guanine tetraplex structures are distinguished among nucleic acid conformations by remarkable stability. As outlined in Fig. 1, three distinct bonding effects contribute to this stability: eight hydrogen bonds that link the Hoogsteen face of each guanine to the Watson–Crick face of its neighbor occur in each quartet; a universally present, dehydrated, central cation forms electrostatic links to the O6 carbonyl oxygen atoms of four or eight surrounding guanines; and the planes of adjacent quartets are offset at angles that favor π–π stacking interactions [3,14].
The stability of stacked G-quartets is evident in the distribution of B-factors shown in Fig. 5. The base moiety of the four pairs of G-quartets coordinated by a central Sr2+ cation form the most stable elements in the structure. Conversely, the more disordered parts of the octamer assembly are the outward-directed 5′ uridines and the 3′ U tetrads at either end of the octamer. Among atoms with the highest B-factors, there is clear evidence of structural variation; three instances are prominent.
Fig. 5.

Isotropic B-factor distribution of octamer assemblies for data sets (a) P1A and (b) P1B.
The first involves alternate arrangements of 3′ U tetrad. Superposition over the first five residues in 5′–3′ order of 16 oligo chains from four structures, P1A, P1B, P2C and 1J8G with THESEUS [22], is shown in Fig. 6a. There is very good structural alignment over three guanine residues and minor variation in the 3′ guanine and 5′ uridine. The average, pairwise least-square RMSD over these residues is 1.687 Å.
Fig. 6.

Structural variation in UGGGGU conformation. (a) Superposition of 16 RNA strands from four structures over residues 1–5 in 5′–3′ order [22] identifies three principal conformations for the 3′ uridine: 3′U-A (red), 3′U-B (green) and 3′ U-C (magenta). (b) ζ and α torsion angles for the three conformations fall into distinct clusters. (c) Alternate conformation of a phosphate group in UGGGGU structure P1A. Isocontours of a σA-weighted 2Fo − Fc density map contoured at 2.0σ are shown in blue; conformation A is shown in magenta and conformation B is shown in cyan.
However, the 3′ uridine can adopt three distinct conformations, labeled 3′U-A, 3′U-B and 3′U-C and colored red, green and magenta. The conformations are not equally populated, with 3′U-A appearing most frequently. 3′U-A and 3′U-B are related by mirror symmetry and result from a nearly 180 difference in ζ and α angles flanking the phosphodiester linkage between the last two residues. Average values for 3′ U-A residue 5-ζ and residue 6-α are 58.4 and 159.0 relative to 284.6 and 298.2 for 3′U-B.
Conformation 3′U-C results from a significantly smaller α angle coupled with a different sugar pucker. This oligo chain is unique in adopting a c3′ endo pucker at residue 6 with phase angle of 21.6.
Octamers can form from different combinations of these conformations, utilizing either two inward-pointing U tetrads, as occurs in structures P1A and 1J8G, or with one inward U tetrad and one outward, as occurs in structure P1B.
Crystal packing requires the stacking of octamers into columns. At the junction between two stacked octamers, a void forms in one of two ways. If both 3′ U tetrads point toward the center of the respective octamer in a concave arrangement, the void forms between two octamers. In contrast, a convex arrangement at one end of the octamer results when the octamer includes chains with both inward-pointing and outward-pointing 3′ U tetrads. In this case, an internal void forms between the outward-pointing U tetrad and its neighboring G tetrad, within the same octamer. In Fig. 5, structures P1A and P1B exhibit, respectively, the concave and convex conformations. We did not observe a crystal form exhibiting a convex arrangement at both ends of the octamer, though the ab initio model derived from SAXS data does not seem to preclude this possibility.
The RNA structure surrounding the void between octamers is the least well-ordered region of the octamer as evident in Fig. 5. The occurrence of different conformations for the 3′ uridine shown in Fig. 6a suggests an ensemble of distinct arrangements. However, within a given crystal, a single conformation occurs predominantly and there is insufficient density to model alternate conformations of the 3′ U tetrad. Selection of “concave” or “convex” octamer conformations appears fixed for a given crystal packing. Though some water molecules are apparent in the void, they are not clearly resolved and significant residual density remains unmodeled in this region.
A second instance of structural variation occurs in the phosphate group that connects uridine 1006 to guanine 1005 in chain B of structure P1A as shown in Fig. 6b. A similar alternate conformation was detected in the TGGGGT structure deposited as PDB entry 352D [8] but not in a previously published UGGGGU structure, 1J8G [11]. The 5′ uracils are the least ordered bases in the octamer. However, there is little evidence to support extension of the alternate conformation beyond the phosphate group. Density for the corresponding O1P and O2P oxygens only becomes visible at about the 1.5σ level and displacement of the O3′ and O5′ oxygens appears minimal.
A third instance of structural variation occurs in structure P2C in chains “A” and “F”. Once again, the 3′ uridines are involved; however, here the alternate conformations extend over the entire terminal residue rather than being limited to the phosphodiester linkage. In chain “A”, the alternate conformation begins at O3′ of the N2 guanine, residue 104. A key distinguishing characteristic of the two conformations are different values of the ζ torsion angle at the linkage between residues 104 and 105: −82.2 for conformation A and −64.4 for conformation B. In chain “F”, the alternate conformation begins at C1′ of the N1 guanine, residue 605, and a distinguishing feature of the two conformations is the difference in [notdef] values, −148.5 versus −160.7. In both cases, the net effect of the two conformations is a lateral shift of the 3′ residue that maintains the angle of the base relative to the central cation axis.
It is noteworthy that the alternate conformations in structure P2C appear to be near the limit of what can be observed by diffraction in this relatively disordered part of the structure. Crystal packing in the orthorhombic space group enables observation of multiple chains within the same tetramer component: chains “A”–“D” belong to the same tetramer assembly, chains “E” and “F” belong to another and chains “G” and “H” belong to a third; furthermore, the two tetramers in chains “E”–“H” belong to the same octamer assembly. Though the abundance of independent chains within the ASU allows observation of multiple constituents of the capping 3′ U tetrads, alternate conformations could only be modeled for one chain within each tetramer.
Cation coordination and quartet stacking
The close packing of bases characteristic of quartet structures places the carbonyl oxygens, that is, the O6 atoms in G-quartets, as well as the O4 atoms in U-quartets, within unusually close proximity. The repulsion between the partial negative charges residing on neighboring oxygen atoms is screened by inner-sphere coordination of dehydrated cations, such as Sr2+, in the central channel formed by the stacked quartets. Coordination observed in the UGGGGU structure exhibits three distinct patterns, one defined by the four central guanine quartets and two others defined by quartets formed by the 3′ uridines that cap the ends of the octameric unit. As described previously, the 5′ uridines rotate away from the central channel and thus do not participate in quartets or cation coordination.
The coordination geometry of the guanine quartets is invariant across all structures examined in this work and is summarized in Fig. 7a. The coordinating cation is always Sr2+, centrally positioned between the eight O6 carbonyl oxygens of two adjacent G-quartets in a bipyramidal antiprism geometry. The Sr–O6 distance, at 2.60 Å, is shorter than O6–O6 distances to atoms in either the same or the adjacent quartets, 3.16 Å and 3.54 Å, respectively. The guanines in each quartet are tilted away from a common plane and toward the Sr2+ cation; a cation occurs only between every other quartet. This alternating distribution, which allows a separation of 6.43 Å between adjacent cations, appears to be unique to Sr2+ and accommodates their electrostatic repulsion [23]. Though Sr2+ has no direct biological relevance, this cation is known to confer high stability to the resulting quadruplex structure [24], thus enabling more detailed examination of the fold's geometry and of the capping U tetrads.
Fig. 7.

Cation coordination by guanine quartets. (a) Coordination of a Sr2+ cation, shown in green, between the eight guanine O6 atoms of two G-quartets, shown in magenta. (b and c) Views of a UGGGGU octamer emphasizing the regular arrangement of O6 atoms about the central cation induced by the structure's helical twist. Coordinates are from the refinement of data set P1B.
Within each octamer, 32 O6 carbonyl oxygens are wrapped about the four central Sr2+ cations in a helical arrangement as shown in Fig. 7b and c. As evident from the distribution of B-factors summarized in Fig. 5, cation coordination by G-quartets yields the most stable, well-ordered, part of the overall structure.
Coordination of uridine O4 atoms follows a similar but less well-defined pattern. Here a Na+ cation with split-occupancy centered on the central axis serves to screen accumulated charge. The U tetrad's four O4 carbonyl atoms define a square of approximately 3.27 Å. Two adjacent sites near the center of the square are alternately occupied by a Na+ cation. The most frequently occupied site is coplanar with the oxygens whereas the site with minor occupancy lies 2.2–2.3 Å out of the plane, away from the octamer. As discussed in Structural variation and disorder, the two 3′ U tetrads that occur at each octamer–octamer boundary can adopt either a concave or a convex disposition. In the concave arrangement, the two U tetrads tilt away from one another toward the center of their respective octamer. In this case, the coordination pattern by the central Na+ cation is similar for the two tetrads. However, in the convex arrangement, both U tetrads tilt toward the same octamer. In this case, the two U tetrads are brought close to one another and the two alternate position of a single Na+ cation appear to coordinate both U tetrads.
Geometric outliers
Maintaining the regularity of stacked guanine and uridine tetrads in the octameric unit imposes constraints that tax the flexibility of the RNA backbone. Accordingly, the structures exhibit some features that, though well-supported by the data, are unusual with respect to nucleotide geometry: not all nucleotide conformations match the clusters of torsion angles used in the definition of rotamers for the RNA backbone [25], there is a marked deviation from planarity between the guanine and sugar moieties and some bonds and angles fall outside the center point of distributions derived from reference structures.
The RNA backbone has been shown to be rotameric [25]. Nevertheless, for certain UGGGGU residues, rotamer assignment as implemented in molprobity version 4.1 and suitename version 0.3.070628 [25] does not identify a known cluster. For example, for structure P1B, no rotamer assignment is reported for residue 1004 in chain “A” and residue 2004 in chain B. For both, the δ−1δγ classification is “23p”. Inspection indicates that the observed ∈−1 torsion angles of the two residues, with values of −170° and −172°, respectively, do not match any peak among currently defined “23p” rotamers.
The strong electrostatic interaction between O6 and Sr2+ atoms distorts both the planar arrangement of atoms within individual guanine residues and the planar arrangement of residues in a G4 quartet. The deviation from a plane is most pronounced at the ribose C1′. For example, in structure P1A, the connecting ribose C1′ of residues 1002 and 1003 are markedly below and above the plane defined by the guanine atoms: seven and eight times the plane's RMSD, respectively.
An instance where the orderly arrangement of stacked quartets strains the RNA backbone occurs in structure P1A. The “concave” U tetrad conformation of chain “A” requires the terminal uridine to stack above the adjacent 3′ guanine in a direction opposed to the strand's helical twist. This geometry places a tight constraint on the connecting phosphodiester bond. Two conformations of the C3′-O3′-P-O5′-C5′ linkage are evident. Though both are well-supported by 2Fo − Fc density as shown in Fig. 6c, the C2′-C3-O3 angle and the C5′-O5′ bond length for one of the conformations fall over five standard deviations outside the centroid of the reference distribution. A similar situation occurs at the 3′ guanine–uridine linkage of the tetramer formed by the structure's “B” chain.
Solvent ions
Ordered Sr2+ and Ca2+ ions are evident in the solvent channels between adjacent octamers. They are distributed at regular intervals and appear to serve two distinct functions: stabilizing the geometry of 3′ and 5′ uridines or stabilizing octamer assembly and crystal packing via contacts that link strands within an octamer and between adjacent octamers.
Examples of the first instance are residues 9004 and 2106 in structure P1B. Residue 9004 forms bifurcated contacts with the ribose O2′ and O3′ oxygens of the chain's 3′ uridine, residue 1006. A contact to water 9111 places the cation near the center of a triangle defined by the three oxygens. Five water molecules, 9110, 9108, 9102, 9112 and 9101, are evenly distributed in a plane perpendicular to this triangle. Each forms contacts with both the cation and either the ribose O2′ oxygen or the O3′ oxygen. The location of the metal and the regularly arranged cluster of water molecules suggest a role in stabilizing the unusual backbone geometry that enables formation of the terminal U tetrad. Residue 2106 appears to serve a similar function near the 5′ terminal uridine. A short, 2.48 Å, contact links the Ca2+ cation with the uridine O4 and both atoms share similar, low, B-factors of 7.88 and 8.11, respectively.
The locations of other metals suggest roles in stabilizing both the helical twist of neighboring chains forming the octameric assembly and the packing of adjacent columns enabling crystal formation. Representative examples are Ca2+ residues 2104 and 2105 in structure P1B. Residue 2104 forms near-equidistant contacts, of 2.40 and 2.47 Å with the OP1 oxygens of guanine residues from octamers in adjacent columns. The two oxygen atoms are separated by a distance of only 2.80 Å. Contacts to four adjoining waters, 2204, 2209, 2214 and 2225, form a network that further stabilizes cross-column contact. Similarly, residue 2105 forms contacts, of distances 2.39, 2.34 and 2.29 Å with the phosphate oxygens of residues in three distinct chains; two are OP2 oxygens from residues 1002 and 2002 in different chains of the same octamer, and the third is the OP1 oxygen from residue 2005 in a neighboring column.
Interestingly, in a number of cases, solvent positions that support inter-octamer and intra-octamer linkages are alternately occupied by a metal cation or by a water molecule separated by an angstrom or less. In most instances, the X-ray anomalous signal unambiguously distinguishes the identity of the two alternately occupied scattering peaks and corroborates metal assignment for one of the two peaks.
Hydration: Distribution of water contacts
Hydration of the octameric assembly largely agrees with expectations based on similar nucleic acid structures. No crystallographically detectable waters are observed in the central channel within each octamer, whereas water contacts occur throughout the external surface and at the cavity formed between stacked octamers. Closer examination of the distribution of water contacts indicates that they are not uniformly distributed over the external surface.
To examine this point, we constructed a list of potential contacts by considering the neighborhood of solvent atoms in each of the UGGGGU structures. As described in Materials and Methods, each contact record includes the source solvent atom, a target atom that lies within 3.6 Å and attributes of the two atoms. The list of 1852 possible water contacts was used to define a simple hydration index for each atom type: the ratio of the number of contacts to the number of occurrences. The results are summarized in Supplemental Table 1.
In Fig. 8a, the RNA atoms from structure P1A have been colored by value of the atom type's hydration index with thin, blue, lines indicating a value less than 0.25. In Fig. 8b, the full symmetry-expanded octamer and its surface are colored by hydration value and modeled waters are included.
Fig. 8.
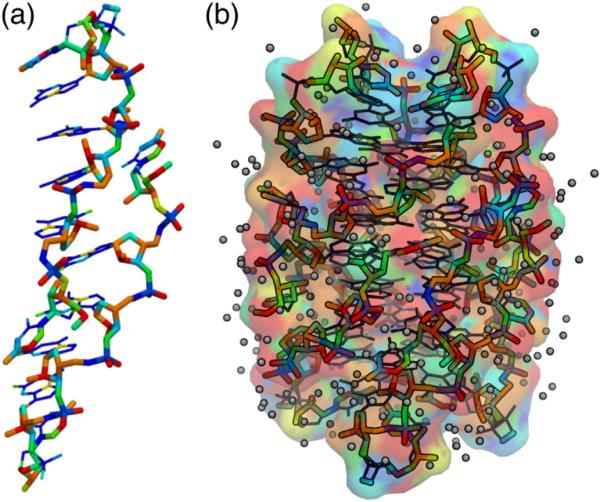
Hydration by atom type. (a) The two strands from structure P1A are shown with atom types colored by hydration index value (Supplemental Table 1). Atoms with a hydration score below 0.25 are shown by blue thinner lines. (b) The hydration-based coloring scheme extended to the symmetry-expanded P1A octamer with waters included; the molecular surface is also colored by hydration value.
Distinctly high values of the hydration index occur at uridine C5′ atoms. Inspection suggests that the network of water molecules near C5′ stabilize the terminal uridines. At the 3′ end, hydration of C5′ serves to maintain orientation of the capping U tetrads, whereas at the 5′ end, C5′ hydration helps maintain the strand intercalation that stabilizes octamer assembly.
As expected, phosphate oxygens, which protrude into solvent away from the octamer, exhibit high hydration scores. Significantly, the hydration index of ribose O2′ guanine atoms falls close to this peak, underscoring a stabilizing structural feature accessible to RNA over DNA G-quadruplexes.
Hydration: Role of the ribose O2′ hydroxyl group
Residency times for bound waters are fleeting relative to those of macromolecular atoms [26]. Thus, repeated exchange of waters at a given site that occurs frequently enough to permit observation of the site by diffraction implies a structural role for a water molecule at that location. Inspection of water linkages in the UGGGGU data sets confirms three well-established roles for occupied sites: helix stabilization, crystal packing and charge shielding of phosphates along the RNA backbone.
Structural features mediated through water-based hydrogen bonding can resist definitive classification because of promiscuity and plasticity. Nevertheless, even an approximate and coarse-grained classification is sufficient to allow some general patterns to emerge. To this end, we analyzed the distribution of waters across the three data structures examined, focusing on hydration of ribose O2′ oxygens. As documented above, the ribose O2′ hydroxyl is a well-hydrated group in the UGGGGU structures: on average, each instance is the target of contacts from two water molecules. This finding is consistent with previous analyses of RNA hydration [27,28]. Hydrogen bonding associated with the 2′-OH group has been suggested to account for greater rigidity of RNA A-duplexes [27]. Experimental data indicate that RNA G-quadruplexes are more stable than corresponding DNA structures [29]. We hypothesized that hydration of O2′ contributes to this effect and analyzed the list of potential water contacts to determine whether the data support this hypothesis.
For analysis, we aggregated the 1852 possible contacts from 238 waters in two ways. The first considered whether a given water molecule makes contacts exclusively with atoms of a single octamer or whether its contacts span two or more adjacent octamers, typically symmetry-related copies. For example, for structure P1A, the octamer formed by the two strands in the ASU is linked by water contacts to five adjacent octamers. The distinction between the two groups of water molecules is illustrated in Fig. 9a. The octamer formed by the ASU strands is shown in yellow thick lines and adjacent octamers linked by water contacts are shown in thin blue. Water molecules limited to intra-octamer contacts are shown in magenta and waters with possible inter-octamer contacts are shown in teal. Most, but not all, intra-octamer waters make contact with the ASU-defined octamer. This classification assigns a given water to one of three disjoint subsets depending on whether its contacts are inter-octamer, intra-octamer or part of a secondary hydration shell that includes no contacts to RNA.
Fig. 9.

Waters involved in intra-octamer and inter-octamer contacts. (a) The octamer formed from the ASU strands of structure P1A is shown in yellow thick lines and adjacent octamers linked by water contacts are shown in blue thin lines. Waters whose potential contacts are limited to a single octamer are colored magenta; waters that form possible cross-octamer contacts are colored teal. (b) A network of hydrogen bonds involving two waters and four O2′ residues stabilizes cross-strand linkage in structure P1B.
Within each subset, we considered whether the target of a potential water-based contact is a phosphate oxygen or a ribose O2′ oxygen. These atom types are among the most frequent targets found as hydration contacts, though the overall distribution of target atom types is quite broad (Supplemental Table 2). The secondary classification does not partition waters into mutually exclusively subsets: among the 238 waters combined from three data sets, 143 include contacts to phosphate oxygens, 96 include contacts to O2′ oxygens, 44 include contacts to both types of targets and 43 include contacts to neither.
The properties of potential water contacts classified according to these two levels of aggregation are summarized in Table 3.
Table 3.
Contact properties
| Count | Contacts | Distance | Solvent B-factor | Target B-factor | |
|---|---|---|---|---|---|
| All | 238 | 1852 | 3.07 (0.33) | 17.15 (8.91) | 12.83 (7.58) |
| Phosphate | 143 | 200 | 2.94 (0.29) | 17.51 (8.40) | 10.84 (2.70) |
| Ribose O2′ | 96 | 126 | 2.98 (0.30) | 14.95 (7.80) | 11.23 (7.43) |
| Secondary shell | 6 | 16 | 2.79 (0.33) | 33.86 (16.42) | 20.96 (10.09) |
| Inter-octamer | 79 | 648 | 3.08 (0.33) | 18.73 (10.27) | 14.14 (8.58) |
| Phosphate | 54 | 92 | 2.98 (0.31) | 17.25 (9.28) | 10.67 (2.78) |
| Ribose O2′ | 15 | 16 | 2.98 (0.37) | 18.49 (10.14) | 15.23 (14.77) |
| Intra-octamer | 153 | 1188 | 3.07 (0.33) | 16.06 (7.52) | 12.00 (6.77) |
| Phosphate | 89 | 108 | 2.90 (0.26) | 17.73 (7.62) | 10.99 (2.64) |
| Ribose O2′ | 81 | 110 | 2.98 (0.29) | 14.44 (7.31) | 10.65 (5.53) |
Properties of overall water contacts in comparison to three disjoint subsets: waters in contact with phosphate group oxygens, with the ribose O2′ hydroxyl group and with waters in a secondary hydration shell. For each group, the number of contacts found among all three structures with the mean and standard deviation of the contact distance, target B-factor and solvent B-factor are listed.
As expected, contacts among waters in the secondary hydration layer exhibit higher B-factors at both source and target though the mean contact distance remains unaffected. More interestingly, the analysis reveals differences between waters bound to O2′ oxygens relative to those bound to the phosphate group. Two differences arise: contacts to O2′ are more frequent among waters involved in intra-octamer binding and the locations of those waters are more tightly bound as evidenced by lower B-factors. Whereas only 15 (18%) of 79 inter-octamer waters include O2′ contacts, 81 (53%) of 153 intra-octamer waters do. Though the B-factors of contact targets among phosphate oxygens and O2′ hydroxyl groups are very similar, the B-factors of the waters are significantly lower for contacts to the O2′ group. This difference, though minor, is significant at 0.005 level among all waters and at 0.001 level among intra-octamer waters.
Overall, these data indicate that the displacement of solvent positions near the O2′ group, most of which form intra-octamer contacts, is more tightly constrained than displacement of positions involved in charge shielding along the RNA backbone. An example of the strand-linking stabilization provided by water contacts to the O2′ hydroxyl groups in structure P1B is illustrated in Fig. 9b. The O2′ groups of residues 1001 and 1002 from chain “A” and residues 2001 and 2002 from chain “B” form a network of hydrogen bonds with waters 9505 and 9521.
Conclusions
The findings reported here contribute to the understanding of quadruplex folds and RNA structure. Evidence that mammalian telomeric repeats, TTAGGG, are selectively transcribed [30] has increased interest in RNA quadruplexes. The r(UAGGGU) repeat was shown to adopt a parallel-stranded G-quadruplex conformation capped and stabilized by a U tetrad [17,18]. Here we show that the terminal U tetrads in the previously reported UGGGGU crystal structure [11] exhibit considerable conformational variability.
Detailed analysis of hydration patterns in the structures reported confirms that the O2′ hydroxyl group plays a major role in stabilization of inter-strand and intra-strand contacts that support the quadruplex helical fold. Comparisons of the stability of similar DNA and RNA quadruplex-forming sequences have shown that the RNA form often exhibits greater stability [19,20]. Our analysis supports the hypothesis that networks of water-mediated contacts within the helical grooves of RNA quadruplexes contribute to enhanced stability. However, our investigation is limited to the structures presented. A more extensive comparison of hydration across diverse DNA and RNA quadruplexes is needed to assess the importance of this effect.
The high resolution is unusual for RNA and contributes to the understanding of RNA geometry. In particular, some features of these structures, including bond lengths, angles and tuples of torsion angle values used for rotamer assignment that are flagged as outliers by current structure validation software, are shown to be legitimate expressions of RNA conformation.
The SAXS-derived solution structure for UGGGGU shows good agreement with the octameric assembly observed in the crystal structure and suggests monodisperse collections of the octamer readily form in solution.
Materials and Methods
RNA oligos were purchased from Dharmacon and deprotected as per manufacturer's instructions. Crystals were grown using previously reported conditions [11]. Usable crystals grew within 1–2 weeks. Variation in the neighborhood of these conditions did not appear to affect crystal quality consistently. However, high mosaicity and irregular growth yielding multiple lattices were a common problem and required screening multiple crystals to obtain high-quality data sets. Growth of irregular crystals was significantly improved by a simple microseeding protocol [31].
The 33 data sets used for analysis were collected on beamlines 7-2, 9-1, 9-2 and 11-1 at the Stanford Synchrotron Radiation Laboratory (SSRL) from 17 single crystals selected from a larger pool of candidates. For several crystals, multiple image batches were collected to optimize anomalous differences or improve data statistics via high- and low-resolution passes. Each of the final P1A, P1B and P2C data sets results from images collected from individual single crystals.
For several crystals, multiple data sets were collected, either to optimize anomalous differences or to improve data statistics via high- and low-resolution passes.
Data reduction used mosflm, scala, pointless and aimless from the Collaborative Computational Project Number 4 [32,33] suite, as well as programs from the PHENIX and labelit [34,35] suites.
Preliminary model fitting was based on iterative rigid-body refinement with phenix.refine [35] in the tetragonal space group and by molecular replacement with Phaser [36] in the orthorhombic space group.
Structure refinement relied on the refmac [37] and shelxl [38] programs. Occupancy refinement with shelxl was applied to all solvent, to alternate conformations and, in certain chains, to the terminal, less-ordered, uridine residues. B-Factors were refined individually and anisotropically other than in regions where weak density did not support the additional parameterization. Non-hydroxyl hydrogens were modeled and refined with shelxl except for residues modeled in weak density. Refinement solutions obtained from shelxl were input to refmac for bulk solvent modeling.
Anomalous difference maps were computed with the Collaborative Computational Project Number 4 fft program and with the Phaser log-likelihood gradient module and used for identification of solvent ions.
Waters were placed by means of a novel approach based on partitioning density difference maps into Morse-Smale complexes [39]. Briefly, a preliminary model was built and preliminary waters and ions were placed via the Coot [40] “findwaters” command. All waters were then removed and the resulting difference map was partitioned into basins bounded by zero flux surfaces (A. C. Fyfe, and W. G. Scott unpublished results). Basins enclosing an acceptable total density with a peak within a plausible distance of non-hydrogen atoms of the existing working model were selected as candidate waters. Those yielding improvements in model quality were retained for further rounds of refinement. The procedure was iterated outward from the working model to uncover waters in outer hydration shells.
A number of adjacent solvent sites were detected from their elongated profile in difference density maps and modeled as alternate split-occupancy locations. Partial occupancy of waters and solvent ions was modeled; however, occupancies that refined over 0.9 were assigned full occupancy and waters whose occupancies refined below 0.20 and that did not fall on special positions were discarded.
Solvent contact statistics were calculated in two steps. First, solvent positions were scanned to determine all atoms, including symmetry equivalents, lying within 3.6 Å [41]. For solvent positions with alternate occupancy, both positions were considered but atoms with alternate occupancy within the radial distance sphere were excluded. Thus, for each potential contact, either the source or the target but not both may occupy an alternate position. The resulting list of potential contacts was annotated with properties of the source and target atoms, including occupancy, B-factor and peak height in the associated 2Fo − Fc map and analyzed with R [42]. Aggregating potential contacts over structures P1A, P1B and P2C yielded a data set of 2364 contact records and 267 solvent entries. For most analyses, we focused on the subset of 1852 contacts from 238 water molecules.
SAXS data were collected on SSRL beamline 4-2 for an RNA concentration series in the range 0.4–4 mg/ml in a solution of 10 mM sodium cacodylate, 10 mm MgCl2, 40 mM SrCl2 and 20 mM CacCl2. Samples were allowed to equilibrate against buffer overnight in 50 μl dialysis buttons (Hampton Research). For each sample, scattering from ten 1-s exposures of sample and two exposures of buffer was recorded. Data analysis relied on the ATSAS suite of programs [43]. Ab initio molecular models were built from 15 iterations of DAMMIF, aligned with SUP-COMB, averaged with DAMAVER and filtered with DAMFILT. The symmetry-expanded octamer derived from structure P1B was fit to the resulting bead model with the Chimera “Fit-to-Map” routine [44]. Predicted scattering profiles for models derived from crystallographic coordinates were computed with FoXS [45].
Supplementary Material
Acknowledgments
Use of the Stanford Synchrotron Radiation Light-source, SLAC (Stanford Linear Accelerator Center) National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) . The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of National Institute of General Medical Sciences or National Institutes of Health.This work was supported by the National Institutes of Health grant R01GM087721 to WGS.
Abbreviations used
- SAXS
small-angle X-ray scattering
- NCS
non-crystal-lographic symmetry
- ASU
asymmetric unit
- SSRL
Stanford Synchrotron Radiation Laboratory
Footnotes
Accession numbers
Data sets P1A, P1B and P2C have been deposited in the PDB with codes 4RKV, 4RJ1 and 4RNE, respectively.
Dedication
We dedicate this paper to the memory of Muttaiya and Indrani Sundaralingam.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2015.03.022.
References
- 1.Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–6. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 2.Phan AT, Kuryavyi V, Patel DJ. DNA architecture: from G to Z. Curr Opin Struct Biol. 2006;16:288–98. doi: 10.1016/j.sbi.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–15. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maizels N. Neidle S, Balasubramanian S, editors. Quadruplexes and the biology of G-rich genomic regions. Quad-ruplex Nucleic Acids. 2006 [Google Scholar]
- 5.Huppert JL. Hunting G-quadruplexes. Biochimie. 2008;90:1140–8. doi: 10.1016/j.biochi.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 6.Neidle S. Therapeutic applications of quadruplex nucleic acids. Academic Press; 2012. [Google Scholar]
- 7.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–7. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 8.Phillips K, Dauter Z, Murchie AI, Lilley DM, Luisi B. The crystal structure of a parallel-stranded guanine tetraplex at 0.95 Å resolution. J Mol Biol. 1997;273:171–82. doi: 10.1006/jmbi.1997.1292. [DOI] [PubMed] [Google Scholar]
- 9.Laughlan G, Murchie AI, Norman DG, Moore MH, Moody PC, Lilley DM. The high-resolution crystal structure of a parallel-stranded guanine tetraplex. Science. 1994;265:520–4. doi: 10.1126/science.8036494. [DOI] [PubMed] [Google Scholar]
- 10.Cheong C, Moore PB. Solution structure of an unusually stable RNA tetraplex containing G- and U-quartet structures. Biochemistry. 1992;31:8406–14. doi: 10.1021/bi00151a003. [DOI] [PubMed] [Google Scholar]
- 11.Deng J, Xiong Y, Sundaralingam M. X-ray analysis of an RNA tetraplex (UGGGGU)(4) with divalent Sr(2+) ions at subatomic resolution (0.61 Å). Proc Natl Acad Sci U S A. 2001;98:13665–70. doi: 10.1073/pnas.241374798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Endoh T, Sugimoto N. Unusual -1 ribosomal frameshift caused by stable RNA G-quadruplex in open reading frame. Anal Chem. 2013;85:11435–9. doi: 10.1021/ac402497x. [DOI] [PubMed] [Google Scholar]
- 13.Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, Hanawalt PC. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc Natl Acad Sci U S A. 2010;107:12816–21. doi: 10.1073/pnas.1007580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lech CJ, Heddi B, Phan AT. Guanine base stacking in G-quadruplex nucleic acids. Nucleic Acids Res. 2013;41:2034–46. doi: 10.1093/nar/gks1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cáceres C, Wright G, Gouyette C, Parkinson G, Subirana JA. A thymine tetrad in d(TGGGGT) quadruplexes stabilized with Tl+/Na+ ions. Nucleic Acids Res. 2004;32:1097–102. doi: 10.1093/nar/gkh269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russo Krauss I, Parkinson GN, Merlino A, Mattia CA, Randazzo A, Novellino E, et al. A regular thymine tetrad and a peculiar supramolecular assembly in the first crystal structure of an all-LNA G-quadruplex. Acta Crystallogr D Biol Crystallogr. 2014;70:362–70. doi: 10.1107/S1399004713028095. [DOI] [PubMed] [Google Scholar]
- 17.Kimura T, Xu Y, Komiyama M. Human telomeric RNA r(UAGGGU) sequence forms parallel tetraplex structure with U-quartet. Nucleic Acids Symp Ser (Oxf) 2009:239–40. doi: 10.1093/nass/nrp120. http://dx.doi.org/10.1093/nass/nrp120. [DOI] [PubMed]
- 18.Xu Y, Ishizuka T, Kimura T, Komiyama M. A U-tetrad stabilizes human telomeric RNA G-quadruplex structure. J Am Chem Soc. 2010;132:7231–3. doi: 10.1021/ja909708a. [DOI] [PubMed] [Google Scholar]
- 19.Mergny JL, Lacroix L. UV melting of G-quadruplexes. Curr Protoc Nucleic Acid Chem. 2009 doi: 10.1002/0471142700.nc1701s37. http://dx.doi.org/10.1002/0471142700.nc1701s37. [DOI] [PubMed]
- 20.Joachimi A, Benz A, Hartig JS. A comparison of DNA and RNA quadruplex structures and stabilities. Bioorg Med Chem. 2009;17:6811–5. doi: 10.1016/j.bmc.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 21.Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–81. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theobald DL, Wuttke DS. THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics. 2006;22:2171–2. doi: 10.1093/bioinformatics/btl332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plavec J. Metal ion coordination in G-quadruplexes. In: Hadjiliadis N, Sletten E, editors. Metal complexes: DNA interactions. Blackwell Publishing Ltd; 2009. [Google Scholar]
- 24.Tran P, De Cian A, Gros J, Moriyama R, Mergny J-L. Tetramolecular quadruplex stability and assembly. In: Chaires JB, Graves D, editors. Quadruplex Nucleic Acids. Vol. 330 of topics in current chemistryBerlin Heidelberg. Springer; 2013. pp. 243–73. [DOI] [PubMed] [Google Scholar]
- 25.Richardson JS, Schneider B, Murray LW, Kapral GJ, Immormino RM, Headd JJ, et al. RNA backbone: consensus all-angle conformers and modular string nomenclature (an RNA Ontology Consortium contribution). RNA. 2008;14:465–81. doi: 10.1261/rna.657708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levitt M, Park BH. Water: now you see it, now you don't. Structure. 1993;1:223–6. doi: 10.1016/0969-2126(93)90011-5. [DOI] [PubMed] [Google Scholar]
- 27.Berman HM, Schneider B. Nucleic acid hydration. In: Neidle S, editor. Oxford Handbook of Nucleic Acid Structure. Oxford University Press; 1999. [Google Scholar]
- 28.Sundaralingam M, Pan B. Hydrogen and hydration of DNA and RNA oligonucleotides. Biophys Chem. 2002;95:273–82. doi: 10.1016/s0301-4622(01)00262-9. [DOI] [PubMed] [Google Scholar]
- 29.Mergny J, Gros J, De Cian A, Bourdoncle A, Rosu F, Sacca B, et al. Neidle S, Balasubramanian S, editors. Energetics, kinetics and dynamics of quadruplex folding. Quad-ruplex Nucleic Acids. 2006 [Google Scholar]
- 30.Schoeftner S, Blasco M. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol. 2008;10:228–36. doi: 10.1038/ncb1685. [DOI] [PubMed] [Google Scholar]
- 31.Bergfors T. Succeeding with seeding: some practical advice. Evolving Methods for Macromolecular Crystallography. Vol. 245 of NATO Science Series. In: Read RJ, Sussman JL, editors. Springer; Netherlands: 2007. pp. 1–10. [Google Scholar]
- 32.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–3. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sauter NK, Grosse-Kunstleve RW, Adams PD. Robust indexing for automatic data collection. J Appl Crystallogr. 2004;37:399–409. doi: 10.1107/S0021889804005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 36.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–22. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 39.King H, Knudson K, Mramor N. Generating discrete Morse functions from point data. Exp Math. 2005;14:435–44. [Google Scholar]
- 40.Emsley P, Lohkamp B, Scott W, Cowtan K. Acta Cryst. Acta Crystallographica Section D-Biological Crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. D66, 486-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cowtan KD. The Clipper Project. Joint CCP4 and ESFEACBM Newsletter on Protein Crystallography. 2002 [Google Scholar]
- 42.R Core Team . R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. 3-900051-07-0. [Google Scholar]
- 43.Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr. 2012;45:342–50. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goddard T, Huang C, Ferrin T. Visualizing density maps with UCSF Chimera. J Struct Biol. 2007;157:281–7. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 45.Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J. 2013;105:962–74. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009;37:5917–29. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.