

Abstract
This review provides insight into the intraneuronal transport of the Amyloid-β Precursor Protein (APP), the prototype of an extensively posttranslationally modified and proteolytically cleaved transmembrane protein. Uncovering the intricacies of APP transport proves to be a challenging endeavor of cell biology research, deserving increased priority, since APP is at the core of the pathogenic process in Alzheimer’s disease. After being synthesized in the endoplasmic reticulum in the neuronal soma, APP enters the intracellular transport along the secretory, endocytic, and recycling routes. Along these routes, APP undergoes cleavage into defined sets of fragments, which themselves are transported – mostly independently – to distinct sites in neurons, where they exert their functions. We review the currently known routes and mechanisms of transport of full-length APP, and of APP fragments, commenting largely on the experimental challenges posed by studying transport of extensively cleaved proteins. The review emphasizes the interrelationships between the proteolytic and posttranslational modifications, the intracellular transport, and the functions of the APP species. A goal remaining to be addressed in the future is the incorporation of the various views on APP transport into a coherent picture. In this review, the disease context is only marginally addressed; the focus is on the basic biology of APP transport in normal conditions. As shown, the studies of APP transport uncovered numerous mechanisms of transport, some of them conventional, and others, novel, awaiting exploration.
Keywords: Amyloid-β Precursor Protein, intracellular transport, microtubule motors, kinesin-1, phosphorylation, secretase cleavage
Introduction, or why should we care about APP transport
It is not uncommon for today’s biomedical research that the function of a protein at the center of a major disease is poorly understood. Amyloid-β Precursor Protein (APP), the protein viewed at the core of the pathogenesis of Alzheimer’s disease (AD) makes one good example. APP was discovered more than 25 years ago [1], and became one of the most studied proteins as soon as it was associated with AD. Multiple functions have been proposed for APP [2], but none of them is clearly demonstrated (Sam Sisodia of the University of Chicago once referred to APP as the All Purpose Protein – seminar delivered ~10 years ago to an audience at Case Western Reserve University). There are several explanations for this situation: (1) In mammals, APP is one of three related proteins, which may have overlapping functions; (2) Mice deficient in the APP gene, show a plethora of phenotypic changes – none essential for survival – that remain mechanistically unexplained [2]; (3) APP has complex biology, and is the precursor protein for Amyloid-β (Aβ), and several other polypeptides, which are generated from APP by successive cleavages operated by numerous proteases [3]. These polypeptides could have their own functions, independent of the parent protein, thus increasing the complexity of APP functions. Moreover, APP is extensively posttranslationally modified by glycosylation – both in the ecto- and endo-domain – and by phosphorylation at several residues within its short, cytoplasmic domain [4]. This domain interacts with multiple cytoplasmic proteins, including molecular motors that carry APP to different destinations [5]. In neurons, APP is transported along secretory, endocytic, and recycling routes that are currently being elucidated [6]. The cleavage of APP into fragments occurs along these routes, certainly at more than one cellular location.
APP is subjected to successive proteolytic cleavages by two of three endoproteases, called secretases, which operate along two mutually exclusive pathways [3]. Depending on its intracellular location, APP is cleaved by either α- and γ– secretase (non-amyloidogenic pathway) or β- and γ–secretase (amyloidogenic pathway) (Fig. 1A). Although the first and second cleavages may occur in the same subcellular compartment, they usually are temporarily and spatially separated. These two proteolytic pathways produce mostly distinct, but topologically similar, sets of protein fragments (Fig. 1A). It is the amyloidogenic pathway, which generates the potentially toxic Aβ peptide that is most relevant to AD. Other proteases, including caspases, also cleave APP, but these proteolytic pathways are less investigated. Obviously, the transport and cleavage of APP are intimately related processes, essential for both the physiology and pathology of the neuron, and cannot be dissociated from each other.
Figure 1.
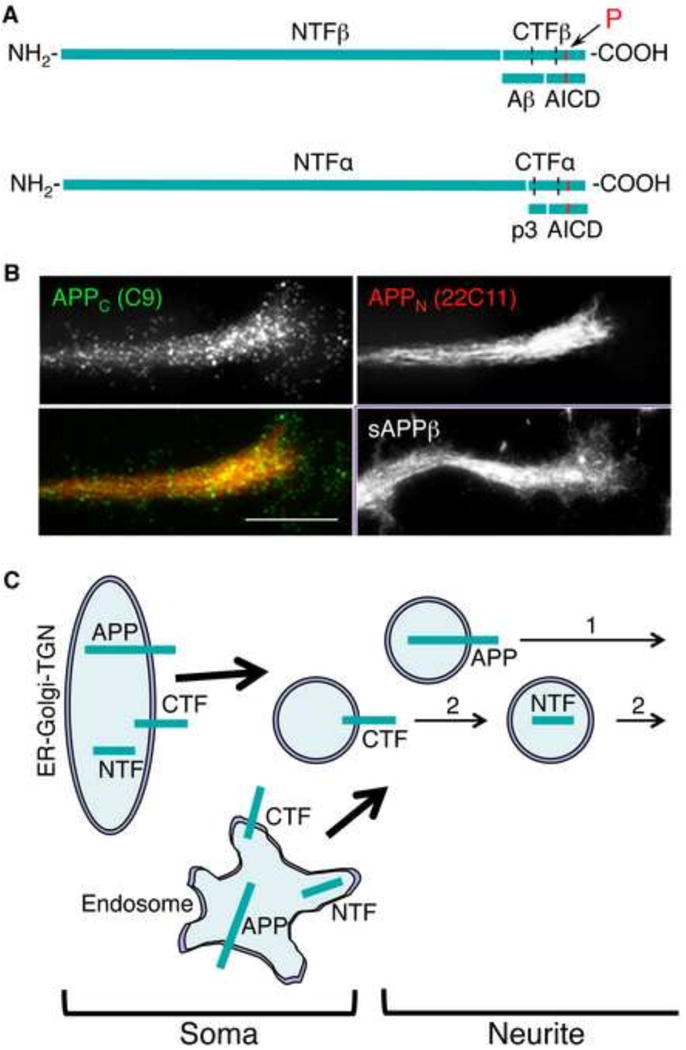
Segregated transport of N- and C-terminal fragments of APP. (A) Processing of APP via the amyloidogenic (by β/γ-secretase; upper) and non-amyloidogenic (also called anti-amyloidogenic; by α/γ-secretase; lower) pathways. The transmembrane domain of APP (dotted lines), and the APP-derived fragments are shown. The arrow indicates the location of phosphorylatable Thr668 (red bar). Cleavage via the amyloidogenic pathway produces NTFβ (known as sAPPβ) and CTFβ. The Aβ (or p3) and the APP intracellular domain (AICD) are generated from CTFβ (or CTFα) by cleavage by γ-secretase.
(B) Punctate, discrete versus filamentary distribution of APP C- and N-terminal epitopes (APPC and APPN) in extending neurites. Immunocytochemistry was carried out in CAD cells with antibodies C9 (kindly provided by Dr. Denis Selkoe) and 22C11 (Millipore). Note that APPN accumulates in the center of the growth cone. The antibodies recognizing the cleaved C-terminal end of sAPPβ show a labeling similar to antibodies recognizing APPN. Bar = 20 μm.
(C) Two possible trafficking pathways of APP. In pathway (2), the cleaved C- and N-terminal fragments of APP are segregated in different vesicles, and transported independently into neurites. Cleavage and sorting into distinct transport carriers could occur in ER-Glogi-TGN or endosomes, in the soma.
This short review provides a glimpse into the transport of APP in relation to its processing; due to space limitations, it is not a comprehensive list of all identified APP transport routes, and their regulation. We aim to reveal the complexity of APP transport, which comprises the transport of the full-length APP, and of its derived fragments; we will also discuss the experimental challenges encountered when trying to track down what is, in fact, transported: full-length APP, or APP fragments. We will limit our analysis to a few examples, focusing on neurons. For the most part, we will avoid the beaten path, and stress on novelty.
Where does APP localize in neurons? Short answer: Almost everywhere
APP, a type I transmembrane protein, is synthesized at the endoplasmic reticulum, and enters the intracellular transport along the secretory, endocytic, and recycling routes, in the soma and neuronal processes. With immunocytochemistry at light or electron microscopy level, APP was detected at the ER, the ER-Golgi Intermediate Compartment (ERGIC), in all subcompartments of the Golgi apparatus, the trans-Golgi network (TGN), post-Golgi secretory vesicles, the plasma membrane, in the early, sorting/recycling, and late endosomes, lysosomes, and autophagosomes. APP is transported to both axons and dendrites, but the fates of axonally- and dendritically-localized APP likely differ [7]. APP – full length and/or fragments – was also found in unexpected places, such as mitochondria [8], the nucleus [9], the ciliary rootlet of the retinal photoreceptor cells [10], and even – apparently free – in the cytoplasm [11]. In some of these locations, cleaved fragments were predominant. Still, some anomalies are obvious: the detection of Aβ epitope in the cytoplasm is not explainable, unless intracellular degradation of membrane-bounded compartments, retrotranslocation from the membrane-bounded organelles, or some other – yet to be explained – phenomenon, reverses the topology of the polypeptide. Also, the extent of APP localization to mitochondria needs to be carefully assessed, because ER-localized APP is enriched at ER-mitochondria contact sites [12]. The localization to the nucleus of APP represents the nuclear targeting of the cytoplasmic fragment of APP (the APP Intracellular Domain, AICD), presumed to enter the nucleus alone, or in association with the APP binding protein, Fe65 [9].
Exogenously expressed APP, usually C-terminally tagged with YFP [13], also localizes to many intraneuronal compartments. While the resolution of detection of the tag varies among reports, expressed, tagged APP usually shows fewer details, being often localized throughout the neuronal soma and processes, unless the expression levels are kept low [14]. Little justification is found in expressing APP-derived polypeptides with incorrect topology, such as the expression, or injection, of Aβ in the cytoplasm, without demonstrating that such situation really occurs under specific physiological or pathological conditions.
APP detection with antibodies in situ versus tagged APP
Since APP is extensively proteolytically processed, it is uncertain whether the immunostaining with antibodies reflects the full-length APP, or an APP fragment containing the cognate epitope(s). Therefore, the studies relying on only one antibody to detect endogenous APP provide results that are difficult to interpret. Dual immunolabeling of C- and epitopes of APP in cultured primary neurons and neuronal cells shows both colocalization, consistent with labeling of full-length APP, and non-overlapping staining, consistent with labeling of the cleaved fragments with distinct cellular targeting [14]. Nonoverlapping distributions of APP C- and N-terminal epitopes are clearly detected both in the neuronal soma and processes. However, the data of immunocytochemistry can be misleading, because of steric hindrance or epitope masking by proteins bound to APP. This particularly applies to epitopes from the APP cytoplasmic domain, which interacts with numerous proteins, including those that contain a phosphotyrosine binding (PTB) domain [15]. A solution to this problem is provided by the expression of dual tagged APP constructs, with distinct tags introduced into the C- and the N-terminal regions. Although largely used in the study of APP transport, the single tag reporters do not solve the problem, since they do not indicate whether the tag represents full-length, or a cleaved APP fragment. Dual tagged APP, where one fluorescent tag is attached to the extreme C-terminus of APP, and the other either next to the signal sequence or inserted into a more internal region of the ectodomain, have been generated recently [16–18]. The expressed, fluorescent, dual tagged APP appears to be correctly processed by the secretases, generating the expected, tagged C- and N-terminal fragments (CTFs and NTFs) [16, 18]. However, the immunoblot detection of the tagged APP fragments only indicates that the dual tagged APP can be cleaved, not that it is cleaved at the same rate, and at the same location as endogenous APP. Two-channel imaging of the two fluorescent tags revealed some separation of the tags, especially within the neurites, but mostly showed colocalization. Colocalization was interpreted as indicating the presence of full-length APP, although it could also indicate colocalization of cleaved NTFs and CTFs. A quantitative analysis of the published data clearly shows that the intensity ratio of the two fluorescent tags varies largely between the labeled structures in the neurites [16, 18]; this indicates that those compartments contain mixtures of full-length APP and APP fragments. Interestingly, the degree of colocalization of the tags increases with increasing the levels of expression of dual tagged APP, which indicates that at high levels of expression the cleavage rate drops artificially, or that the sorting and transport machinery of the fragments are saturated. It is likely that the voluminous, fluorescent tags attached to, or inserted into, APP hinder the interaction of APP with normal binding partners. For instance, the YFP molecule immediately following the C-terminus of APP – as it is in a widely used APP-YFP construct [13] – likely interferes with the binding of proteins that normally attach to the C-terminal region of APP. Thus, it is likely that the YFP-tagged APP species will be transported within neurons in a manner that differs from that of endogenous APP. The intraneuronal distribution of APP tagged with small tags at its N- and C-termini showed a better resemblance to the distribution of endogenous APP epitopes compared to the dual tagged APP containing large, fluorescent tags of the GFP type [18]. Presently, the solution to the problem of studying APP localization and transport in neurons could be found in optimizing the placement of the tags, and undertaking combined investigation of APP localization and transport using both immunocytochemistry of endogenous APP, and exogenous expression of tagged APP.
APP destinations within the neuronal soma, axons, and dendrites. How does APP reach these destinations?
After exiting the ER, APP reaches multiple intracellular compartments in neurons; some of these are only obligate landmarks along the itinerary, while others are the sites where APP is cleaved into fragments, sorted for targeting to specific intracellular sites, or represent the “final destination” where APP species reside to fulfill specific functions, before their decay. The first destination harboring APP’s activity is the plasma membrane, where APP ectodomain is detectable throughout the surface of the soma and processes, particularly in non-permeabilized neurons. There, APP could function as receptor for extracellular ligands to regulate cell adhesion, survival, growth, path finding [2]. The plasma membrane is also a major site of α-secretase cleavage [19], and shedding of sAPPα. It is also a transit station before reentry of APP into the cell along the endocytic pathway [6], carrying APP – via the sorting/recycling endosome – back to the plasma membrane, to the TGN/Golgi, late endosome/lysosome, or to the ER/nuclear envelope. The function of APP along the retrograde/recycling trafficking route could be related to signaling – via signaling endosomes, for example. In the nucleus, APP participates in the regulation of gene expression via its AICD fragment [9]. The presence of CTFs in the nuclear membrane is also likely. Thus, the neuronal nucleus is a destination for APP. At the ER, the site of APP synthesis, APP could act as sensor of impeded axonal transport [20]. In this respect, it was proposed that phosphorylation and amyloidogenic processing of APP, triggered by cargo accumulation in the soma, initiates a cascade of events aimed at restoring normal transport. A destination of APP, frequently ignored, is the ER-mitochondria contact site, where APP and/or APP fragments accumulate [21]. As already mentioned, mitochondria themselves may contain APP and APP fragments [8].
Initially considered to be primarily transported into axons, APP is now known to be targeted into dendrites as well. Full-length APP localizes at the pre- and post-synaptic sites, where one proposed function is to form membrane tethers across the synapse [22]. APP fragments, including the N-terminal sAPP, concentrate at process endings [14], and may play a role in synaptic function [23]. While the APP transported into neurites was proposed to be proteolytically processed at the synapse, there is evidence that the NTFs and CTFs are transported into the processes, and reach the terminals independently, where they localize to distinct regions of the growth cone [14, 18]. In cultured neuronal cells, APP – mostly fragments – enters the fine, filopodia-like processes, another destination of APP [14]. Phosphorylated APP species preferentially accumulate in filopodia [14], and it is likely that posttranslational modifications in addition to phosphorylation, such as modification by the peptidyl-prolyl cis-trans isomerase, Pin1, or cytoplasmic glycosylation by O-GlcNAcase control the trafficking and the targeting/anchoring to specific destinations of APP. Thus, it is likely that APP reaches its destinations in neurites both as full-length APP, and as fragments cleaved elsewhere in the neuron (Fig. 1C).
What is transported: full-length APP, or APP fragments?
There is evidence that APP is cleaved both in the neuronal soma, and in the neurites. The prevailing view is that APP is initially transported to the plasma membrane as full-length protein [6]; cleavage of APP then occurs locally, along endocytic/recycling pathways, upon reuptake of cell surface localized APP. In this scenario, the APP fragments generated in the soma are retained and degraded in the soma; consequently, all APP transported into the neurites would be full-length APP, with cleavage into fragments occurring mainly at synaptic regions. However, as described above, APP-derived fragments are detected separately in the processes (both with immunocytochemistry and dual tagged APP constructs), and accumulate at distinct sites at the terminals (Fig. 1B). In some cases, bona fide sAPP and Aβ were detected along the neurites with C-terminal-end-specific antibodies to sAPP and Aβ40/42, unequivocally identifying these fragments [14, 18]. Moreover, Aβ and N-truncated pyroglutamate Aβ was detected in dense-core vesicles [24]; only the Aβ peptide, not full-length APP, is modified by glutaminyl cyclase. Supporting independent transport of APP NTFs and CTFs, a study showed that APP deletion mutants, lacking either the N-or C-terminus, are correctly transported to the axonal and dendritic compartments [25].
With regard to the plethora of fragments that are transported into neurites, their exact nature is not easily determined. It appears that the complexity of the proteolytic processing of APP is much higher than anticipated. In view that both full-length APP and APP fragments are transported in neurites, the precise site of processing, the site(s) of sorting of the generated fragments, and the directionality of their transport relative to soma remain to be established.
Where is APP cleaved in physiological and pathological conditions?
It is largely considered that β-secretase cleavage of APP occurs in endosomes and late Golgi compartments, including the TGN [6]. Many studies indicate that γ-secretase cleavage occurs at the plasma membrane, and the endosomal/lysosomal system, including phagosomes and autophagosomes, both in the soma and the neurites [26–32]. Regions in the vicinity of synapses, both pre- and post-synaptic, are viewed as hot spots of Aβ production [7], mostly in response to synaptic activity. Synaptic activity and conditions of AD appear to facilitate convergence of APP with the secretases in acidic compartments [33]. In the soma, much of the APP is processed along the endocytic route, and the cleaved fragments – primarily the NTFs – are then transported into neurites [34, 35] (Fig. 2A). However, data accumulated over the years indicate that both β- and γ-secretase cleavage could also occur in early secretory compartments [20, 26, 36]: the ER, the ERGIC, and Golgi. The main argument against the possibility of APP processing in early secretory compartments is that neither BACE1 (the β-secretase in neurons), nor the γ-secretase complex, could be active here [6]. Yet, at steady-state BACE1 primarily localizes in the soma, including the ER. Moreover, among the major BACE1 interactors are the reticulons [37], which are largely restricted to the tubular ER [38]. Also, all subunits of the γ-secretase complex colocalize at the ER. With regard to the invoked necessity of posttranslational modifications of secretase subunits required for activity, known to occur in post ER compartments, we stress that there is robust bidirectional (i.e., anterograde and retrograde) trafficking both between the plasma membrane and the TGN, and between Golgi and ER. Active secretases could thus relocate to the ER, ERGIC, and cis-Golgi. While colocalization of APP with the secretases during transport into neurites under normal conditions is still debated, APP certainly colocalizes with the secretases in the soma, in early secretory compartments. Although their colocalization is not proof of cleavage, in conditions of dysregulated transport of APP and secretases, as likely occur in AD [5], significant active β- and γ-secretases could accumulate in the ER/ERGIC compartments. The argument that BACE1 would not be active at a pH higher than that of endosomes is also not realistic; maximal activity of the enzyme is not required for APP cleavage. In addition, phosphorylation of APP – which occurs to a large extent at the ER [20], and is increased in AD [39] and stress [40] – could favor APP cleavage at the slightly higher pH in the ER lumen. Recent studies reported that, in the aging brain, APP fragments accumulate in perinuclear compartments in the soma [41, 42]. In cultured neuronal cells, bona fide APP fragments are occasionally detected in a perinuclear compartment of the ER that associates with neurofilaments [43] (Fig. 2B). We conclude that APP cleavage is a dynamic, continuously changing process, which occurs at multiple sites within the neuron, not only in endocytic compartments.
Figure 2.
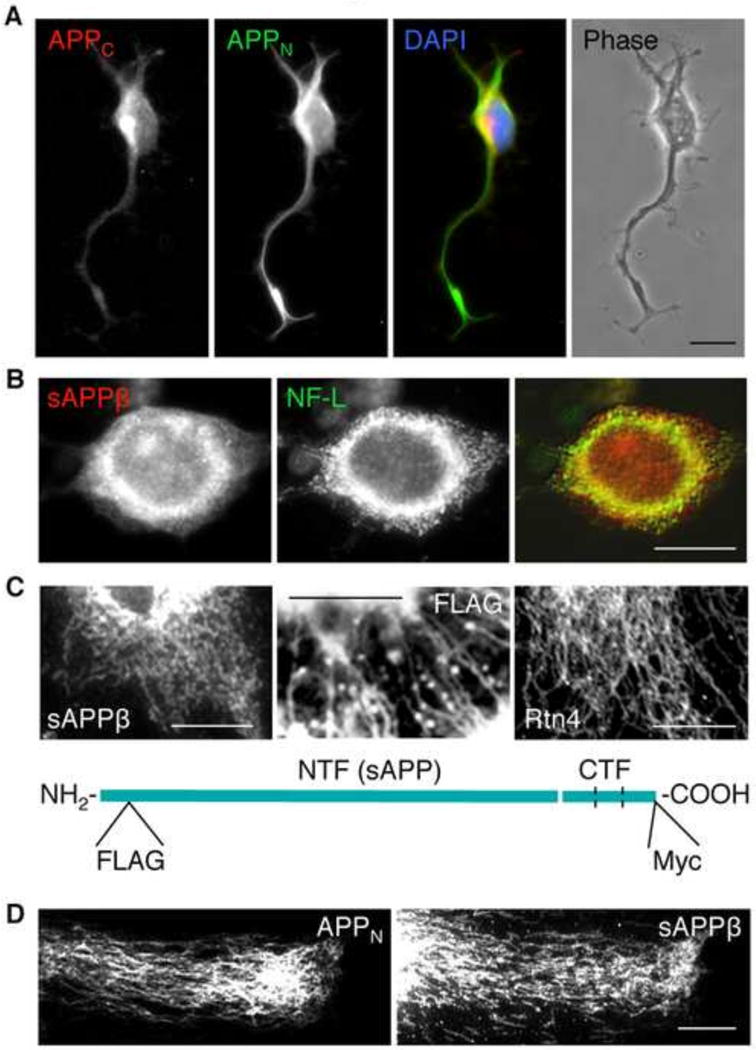
The APP NTFs are present in tubular structures extending from the soma to neurite terminals. (A) Accumulation of APP N-terminal (labeling with antibody 22C11), but not C-terminal epitopes (labeling with antibody Y188, Epitomics) is detected at neurite terminals of CAD cells. Bar = 20 μm.
(B) In the soma, sAPPβ, detected with a C-terminal-end-specific antibody (IBL), colocalizes with neurofilaments (antibody to NF-L; Sigma). Bar = 20 μm.
(C) Filamentous distribution of sAPPβ (detected with a C-terminal-end-specific antibody), and of FLAG-tagged APP NTFs, in the soma of CAD cells transfected with FLAG-APP-Myc. These distributions are similar to that of Rtn4, a bona fide constituent of the tubular ER. The diagram shows the positions of the tags in FLAG-APP-Myc. Bar = 20 μm.
(D) Filamentous distribution of APP NTFs, detected in flat processes of CAD cells with antibodies to a N-terminal epitope (22C11; APPN) and to sAPPβ. A nexus of filaments, colocalizing with the ER marker, Rtn4, is often detected at terminals. Bar = 20 μm.
The motors and the roads for the transport of APP and APP fragments
It is assumed – although not always clearly demonstrated – that, within the soma, short distance transport of APP to and from the plasma membrane, and between various organelles, relies on the microtubule motors, kinesin-1 and cytoplasmic dynein. As for the long distance transport, studies indicate that anterograde transport of APP in axons is powered by kinesin-1 [13, 44, 45], moving along a mixed population of acetylated and nonacetylated microtubules [14, 18]. Retrograde transport uses cytoplasmic dynein [46, 47]. These motors appear to power transport of both full-length APP and APP fragments; the CTFs actually enter neurites with low efficiency [14, 18, 48] (see also Fig. 2A). There is less information on the transport of APP within the dendrites; however, it is likely that the same microtubule motors, kinesin-1 and cytoplasmic dynein, are used. Other kinesin motors could also deliver APP to the processes. If one assumes that APP itself recruits the motor (see below), there is at least one other kinesin that could bind to APP: the kinesin-2 member, KIF17, a primarily dendritic and ciliary motor, which could be indirectly recruited to APP via the APP binding protein, Mint1/X11α [49–51].
The CTFs, mostly phosphorylated, have been detected at the growth cone, in filopodia, and along neuronal processes, in filiform extensions along the neurites of cultured neuronal cells [14]. These cellular structures are devoid of microtubules, but contain actin filaments. While APP could be recruited to transport entities penetrating in actin rich extensions in many ways, we mention that Myosin-X, a filopodial motor [52], contains an ERM domain capable of directly binding the consensus YENPTY- motif in the APP cytoplasmic domain. Thus, transport of APP to various sites in the neuron could use a several microtubule and actin motors.
Is APP a motor-cargo linker, or only a cargo protein?
Two papers at the turn of the millennium proposed that APP serves as a cargo linker for the major anterograde motor, kinesin-1, by directly binding to the light chains, and thus recruiting the motor to a variety of cargo vesicles [29, 44]. Other studies support a slightly amended model where the binding of kinesin-1 to APP is not direct, but mediated by bridging proteins, such as JNK-interacting protein-1 (JIP-1) [53, 54]. We note that only a small fraction of APP (which is phosphorylated at Thr668) may do so under physiological conditions [45]. Although transport of APP can occur in the absence of JIP-1 [55], this scaffolding protein is involved in regulating bidirectional transport of APP [46]. While there is strong support for a role of APP in axonal transport [47, 56], likely as a motor-cargo linker [57], as described above, other mechanisms for motor recruitment to APP carrying vesicles have been reported. For example, it was shown that calsyntenin, a well-characterized kinesin-1 binding protein, recruits this motor to some APP-containing vesicles [58].
In our view, there is not a unique way by which APP – and its fragments – are transported into the processes. Redundant mechanisms likely coexist, and many certainly have not yet been discovered. We would like to point to a caveat in the interpretation of data derived from the use of C-terminally-tagged APP, when the tag is a GFP variant. GFP tags could alter the biology of APP, including transport, by interfering with protein binding to its cytoplasmiuc domain; as such, they bias against transport mechanisms where APP serves as anchor for the motor, and favor transport mechanisms where APP is not involved in motor recruitment. Also, the level of expression of APP – if too high – alters normal recruitment of motors to APP, potentially altering the APP:motor ratio. In conclusion, APP containing transport entities are likely targeted to the neurites by mechanisms involving both APP-dependent and independent motor recruitment.
Conventional and unconventional mechanisms of APP transport
Information about the pathways and mechanisms of APP transport have been obtained with three approaches: (1) Assessment of the progression of the radioactive wave of pulse-labeled APP along axonal tracts, such as the optic nerve and perphorant path [59, 60]; (2) Direct visualization of transport of fluorescently tagged APP in cultured, transfected neurons [13]; and (3) Extraction of information on transport from the localization of endogenous or exogenously expressed, tagged APP along its delivery routes [14, 18]. Transport of APP tagged with YFP at the extreme C-terminus, showed striking, unidirectional motility of elongated structures, moving at speeds up to 10 μm/s, which are not characteristic for kinesin-1 motility [13]. This strange behavior of APP-YFP is not consistent with the results of immunodetection of endogenous APP with antibodies to its cytoplasmic domain, which show a discrete distribution of punctiform structures. On the other hand, the distribution of the small C- and N-terminal tags of a dual-tagged FLAG-APP-Myc is remarkably similar to the distribution of C- and N-terminal epitopes of endogenous APP in flat processes of neuronal cells in culture [14, 18]. Interestingly, these distributions are strikingly different: The labeling of the C-terminal region of APP shows discrete, vesicle-like distributions – typical for kinesin-1 transported vesicles – throughout the neurites, including the growth cone. By contrast, the N-terminal labeling – likely representing NTFs – shows a peculiar, filamentary distribution that extends from the soma to neurite terminals [18] (Figs. 1B and 2C, D). This distribution of the NTFs is not typical for kinesin-1 cargos, and suggests that the NTFs and CTFs use different mechanisms of transport in neurites. The distribution of the NTFs is reminiscent of extensive tubular networks, such as that of the ER (Fig. 2C), an organelle that extends protrusions deep into the neurites. Because mitochondria also contain APP (see above), these two organelles – the tubular ER and the mitochondria – likely play active roles in the transport of APP and fragments. The possibility that the tubular ER performs long distance transport is an area of intensive research.
Factors that regulate the transport of APP also regulate its processing, and vice-versa
Transport of APP from its site of synthesis in the soma to its destinations throughout the neuron is regulated at each step: (1) sorting and recruitment of APP into the cargo; (2) recruitment of the molecular motor to the cargo, and its activation; (3) motile properties of the molecular motor; (4) availability of tracks (microtubules or actin filaments); (5) presence of “road blocks” that prevent free movement of cargo along the tracks; (6) availability of ATP; (7) level of expression of APP; (8) nature and degree of proteolytic processing of APP at the site of synthesis and at transit stations along the transport routes. The AD linked mutations in APP, PS1 and PS2 (subunits of γ-secretase complex), or in other genes that increase the susceptibility to AD may act at many of these levels, and alter APP transport. Because APP fragments appear to be transported separately, the modifiers of APP processing are likely to alter APP’s downstream transport. Of particular importance is the phosphorylation of APP at Thr668 (numbering in APP695) [61, 62], since this modification facilitates the amyloidogenic processing of APP by both β- and γ-secretase [39, 63]. This phosphorylation also specifies the recruitment of kinesin-1 via JIP-1 [45], and possibly the recruitment of myosin motors, which is determinant for targeting pAPP to distal ends in the growth cone [14]. Phosphorylation of AICD could be required for targeting to the nucleus [64]. Overall, this phosphorylation alters the interactome of APP’s cytoplasmic domain [15]. Most importantly, in mouse models of AD the phosphorylation of APP at Thr668 is required for the development of neuronal pathology [63, 65–68]. The effects on APP transport of phosphorylation at other residues in the cytoplasmic domain of APP, of glycosylation by O-GlcNAcase, or isomeration of proline by Pin1, are little investigated.
A recent study found that a variant of KLC1, KLC1vE, confers increased susceptibility to amyloidogenic cleavage of APP, by an unknown mechanism [69]. The recruitment of kinesin-1 to APP via KLC1vE could alter trafficking of APP through the compartments responsible for amyloidogenic cleavage. Alternatively, KLC1vE could more efficiently recruit JNK to APP, and facilitate the phosphorylation conducive of secretase cleavage [70]. These scenarios highlight the numerous ways by which APP transport and processing are entangled.
What is transport telling us about the function of APP?
A fraction of APP is transported to the synapse as full-length protein, where it could mediate cell-to-cell interactions across the synapse, and thus regulate the function of the synapse. Full-length APP is required for this function. Other cellular processes may also rely only on full-length APP [71]. However, both the full-length APP and the cleavage fragments appear to be required for other functions of APP. For example, full-length APP serves as a sensor for impeded axonal transport, and elicits a stress response aimed at restoring transport. Yet, this response relies on the amyloidogenic cleavage of APP and the generation of fragments with defined functions. This homeostatic mechanism has relevance for LOAD [20]. Finally, the fact that APP fragments, in addition to full-length APP, are transported to remote locations in the neuron indicates that the fragments themselves could have functions that are independent of the parent protein [14]. The finding that the NTFs and CTFs are transported in neurites separately, using distinct mechanisms of transport [18], suggests that their functions are also independent of each other. Further, since the transport routes of pAPP differ from those of nonphosphorylated APP [14, 45], it is likely that pAPP species have functions distinct from those of their nonphosphorylated counterparts.
Elucidating Transport of APP: The Challenges Ahead
In spite of extensive research, the pathways and the mechanisms of transport of APP in neurons are only beginning to be understood. APP trafficking is highly dynamic, and subjected to complex regulation by factors that vary according to the physiological challenges of the neuron. As a consequence, APP transport differs between neighboring neurons, both in situ and in cell culture. Under normal conditions, the variability could arise – among others – from the cell-to-cell differences in the levels of expression of APP, degree of regulated proteolytic processing, and degradation. This situation is exacerbated when studying the transport of exogenously expressed APP, whose expression levels could vary by an order of magnitude among cells. The stress of all sorts, which becomes chronic at old age, affects transport and thus alters APP processing, which in turn modifies the transport of APP fragments. The real problem with the elucidation of APP transport routes and transport mechanisms is that intracellular APP is an intractable protein with the current methodology. What do antibodies detect? Full-length APP, or APP fragments? What fragments? What do the tags report: the presence of full-length APP, or presence of APP fragments? Does tagged APP faithfully reproduce the biology of APP, at least with regard to processing and transport? These questions are difficult to answer. Certainly, the generation of improved constructs, with the tags placed in ways that do not interfere with the complex biology of APP, is essential. Finally, is the primary neuron in culture the best system to study APP transport? How relevant are the neuronal cell lines to the normal physiology of the neuron in situ? Although the technology is rapidly improving, imaging APP transport in neurons in live animals still lacks sufficient resolution. Even in cell culture, the small soma and the thin processes of primary neurons limit the unambiguous identification of the transport pathways. Not much more than a moving particle – a dot or a line – can be detected in the neurons transfected with fluorescently tagged APP. Choosing a neuronal cell line that extends high caliber, flat neurites, where the microtubule tracks are clearly distinguished (e.g., the CAD cells) [72], could help the study of APP transport. The analysis of the movement of a fluorescent dot along the neurite, currently done with kymographs, provides general characteristics of motility: direction of movement, velocity, run length, frequency and duration of pauses. This tool becomes powerful in studying the mechanisms of motility only when combined with molecular biology approaches that target proteins suspected to regulate transport, hoping that they will alter the characteristics of transport. Such approaches work best when the research allows for more exploration rather than being limited to a few hypotheses.
Highlights.
APP is cleaved throughout the neuron, including in early secretory compartments
The APP fragments that accumulate in neurites are largely generated in the soma
The APP N- and C-terminal fragments are independently transported to distinct destinations
Post-Golgi and endosome-derived vesicles, and tubular endoplasmic reticulum transport APP in neuritis
The mechanisms of transport of APP N- and C-terminal fragments are fundamentally different
Acknowledgments
Support for the work in the authors’ laboratory comes from the National Science Foundation (award IOS-1347090 to Z.L.M. and V.M.), National Institutes of Health (award AG039668 to Z.L.M.), and New Jersey Health Foundation (awards to Z.L.M. and V.M.). We apologize for not being able to include many important references, due to space limitations.
Abbreviations
- APP
Amyloid-β Precursor Protein
- AD
Alzheimer’s disease
- Aβ
Amyloid-β
- ER
endoplasmic reticulum
- ERGIC
ER-Golgi Intermediate Compartment
- TGN
trans-Golgi network
- AICD
APP Intracellular Domain
- PTB
phosphotyrosine binding
- JIP-1
JNK-interacting protein-1
- APPC and APPN
APP C- and N-terminal epitopes
- NTF and CTF
N- and C-terminal fragment
- RTN4
Reticulon 4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science. 1987;235:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- 2.Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6:27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 4.Walter J, Haass C. Posttranslational modifications of amyloid precursor protein: ectodomain phosphorylation and sulfation. Methods in molecular medicine. 2000;32:149–168. doi: 10.1385/1-59259-195-7:149. [DOI] [PubMed] [Google Scholar]
- 5.Muresan V, Muresan Z. Is abnormal axonal transport a cause, a contributing factor or a consequence of the neuronal pathology in Alzheimer’s disease? Future Neurology. 2009;4:761–773. doi: 10.2217/fnl.09.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBoer SR, Dolios G, Wang R, Sisodia SS. Differential release of beta-amyloid from dendrite- versus axon-targeted APP. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:12313–12327. doi: 10.1523/JNEUROSCI.2255-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 10.Yang J, Li T. The ciliary rootlet interacts with kinesin light chains and may provide a scaffold for kinesin-1 vesicular cargos. Exp Cell Res. 2005;309:379–389. doi: 10.1016/j.yexcr.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. The EMBO journal. 2012;31:4106–4123. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaether C, Skehel P, Dotti CG. Axonal membrane proteins are transported in distinct carriers: a two- color video microscopy study in cultured hippocampal neurons. Mol Biol Cell. 2000;11:1213–1224. doi: 10.1091/mbc.11.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muresan V, Varvel NH, Lamb BT, Muresan Z. The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J Neurosci. 2009;29:3565–3578. doi: 10.1523/JNEUROSCI.2558-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamayev R, Zhou D, D’Adamio L. The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol Neurodegener. 2009;4:28. doi: 10.1186/1750-1326-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldsbury C, Mocanu MM, Thies E, Kaether C, Haass C, Keller P, Biernat J, Mandelkow E, Mandelkow EM. Inhibition of APP Trafficking by Tau Protein Does Not Increase the Generation of Amyloid-beta Peptides. Traffic. 2006;7:873–888. doi: 10.1111/j.1600-0854.2006.00434.x. [DOI] [PubMed] [Google Scholar]
- 17.Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L, Veerle B, Coen K, Munck S, De Strooper B, Schiavo G, Annaert W. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A. 2011;108:E559–568. doi: 10.1073/pnas.1100745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villegas C, Muresan V, Ladescu Muresan Z. Dual-tagged amyloid-beta precursor protein reveals distinct transport pathways of its N- and C-terminal fragments. Hum Mol Genet. 2014;23:1631–1643. doi: 10.1093/hmg/ddt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sisodia SS. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci U S A. 1992;89:6075–6079. doi: 10.1073/pnas.89.13.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muresan V, Muresan Z. A persistent stress response to impeded axonal transport leads to accumulation of amyloid-beta in the endoplasmic reticulum, and is a probable cause of sporadic Alzheimer’s disease. Neurodegenerative Dis. 2012;10:60–63. doi: 10.1159/000332815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schon EA, Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci. 2013;55:26–36. doi: 10.1016/j.mcn.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Wang B, Yang L, Guo Q, Aithmitti N, Songyang Z, Zheng H. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci. 2009;29:10788–10801. doi: 10.1523/JNEUROSCI.2132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Muller UC. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cynis H, Funkelstein L, Toneff T, Mosier C, Ziegler M, Koch B, Demuth HU, Hook V. Pyroglutamate-amyloid-beta and glutaminyl cyclase are colocalized with amyloid-beta in secretory vesicles and undergo activity-dependent, regulated secretion. Neurodegener Dis. 2014;14:85–97. doi: 10.1159/000358430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Back S, Haas P, Tschape JA, Gruebl T, Kirsch J, Muller U, Beyreuther K, Kins S. beta-amyloid precursor protein can be transported independent of any sorting signal to the axonal and dendritic compartment. J Neurosci Res. 2007;85:2580–2590. doi: 10.1002/jnr.21239. [DOI] [PubMed] [Google Scholar]
- 26.Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer’s A beta(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- 27.De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 29.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LSB. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 30.Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- 31.Wilson CA, Doms RW, Zheng H, Lee VM. Presenilins are not required for Abeta42 production in the early secretory pathway. Nat Neurosci. 2002;5:849–855. doi: 10.1038/nn898. [DOI] [PubMed] [Google Scholar]
- 32.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das U, Scott DA, Ganguly A, Koo EH, Tang Y, Roy S. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron. 2013;79:447–460. doi: 10.1016/j.neuron.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muresan Z, Muresan V. Neuritic Deposits of Amyloid-β Peptide in a Subpopulation of Central Nervous System-Derived Neuronal Cells. Mol Cell Biol. 2006;26:4982–4997. doi: 10.1128/MCB.00371-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niederst ED, Reyna SM, Goldstein LS. Axonal Amyloid Precursor Protein and its Fragments Undergo Somatodendritic Endocytosis and Processing. Mol Biol Cell. 2014 doi: 10.1091/mbc.E14-06-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skovronsky DM, Doms RW, Lee VM. Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol. 1998;141:1031–1039. doi: 10.1083/jcb.141.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat Med. 2004;10:959–965. doi: 10.1038/nm1088. [DOI] [PubMed] [Google Scholar]
- 38.Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 39.Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muresan Z, Muresan V. The amyloid-beta precursor protein is phosphorylated via distinct pathways during differentiation, mitosis, stress, and degeneration. Mol Biol Cell. 2007;18:3835–3844. doi: 10.1091/mbc.E06-07-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hatami A, Albay R, 3rd, Monjazeb S, Milton S, Glabe C. Monoclonal Antibodies against Abeta42 Fibrils Distinguish Multiple Aggregation State Polymorphisms in Vitro and in Alzheimer Disease Brain. The Journal of biological chemistry. 2014;289:32131–32143. doi: 10.1074/jbc.M114.594846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pensalfini A, Albay R, 3rd, Rasool S, Wu JW, Hatami A, Arai H, Margol L, Milton S, Poon WW, Corrada MM, Kawas CH, Glabe CG. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol Dis. 2014;71:53–61. doi: 10.1016/j.nbd.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muresan V, Villegas C, Ladescu Muresan Z. Functional Interaction between Amyloid-beta Precursor Protein and Peripherin Neurofilaments: A Shared Pathway Leading to Alzheimer’s Disease and Amyotrophic Lateral Sclerosis? Neurodegener Dis. 2014;13:122–125. doi: 10.1159/000354238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamal A, Stokin GB, Yang Z, Xia C, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I [In Process Citation] Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 45.Muresan Z, Muresan V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-Jun NH2-terminal kinase-interacting protein-1. J Cell Biol. 2005;171:615–625. doi: 10.1083/jcb.200502043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu MM, Holzbaur EL. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. The Journal of cell biology. 2013;202:495–508. doi: 10.1083/jcb.201302078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szpankowski L, Encalada SE, Goldstein LS. Subpixel colocalization reveals amyloid precursor protein-dependent kinesin-1 and dynein association with axonal vesicles. Proc Natl Acad Sci U S A. 2012;109:8582–8587. doi: 10.1073/pnas.1120510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodrigues EM, Weissmiller AM, Goldstein LS. Enhanced beta-secretase processing alters APP axonal transport and leads to axonal defects. Hum Mol Genet. 2012;21:4587–4601. doi: 10.1093/hmg/dds297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guillaud L, Wong R, Hirokawa N. Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat Cell Biol. 2008;10:19–29. doi: 10.1038/ncb1665. [DOI] [PubMed] [Google Scholar]
- 50.Jenkins PM, Hurd TW, Zhang L, McEwen DP, Brown RL, Margolis B, Verhey KJ, Martens JR. Ciliary targeting of olfactory CNG channels requires the CNGB1b subunit and the kinesin-2 motor protein, KIF17. Curr Biol. 2006;16:1211–1216. doi: 10.1016/j.cub.2006.04.034. [DOI] [PubMed] [Google Scholar]
- 51.Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor- containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 52.Berg JS, Cheney RE. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat Cell Biol. 2002;4:246–250. doi: 10.1038/ncb762. [DOI] [PubMed] [Google Scholar]
- 53.Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem. 2003;278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- 54.Matsuda S, Matsuda Y, D’Adamio L. Amyloid beta protein precursor (AbetaPP), but not AbetaPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J Biol Chem. 2003;278:38601–38606. doi: 10.1074/jbc.M304379200. [DOI] [PubMed] [Google Scholar]
- 55.Vagnoni A, Glennon EB, Perkinton MS, Gray EH, Noble W, Miller CC. Loss of c-Jun N-terminal kinase-interacting protein-1 does not affect axonal transport of the amyloid precursor protein or Abeta production. Hum Mol Genet. 2013;22:4646–4652. doi: 10.1093/hmg/ddt313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gallagher JJ, Zhang X, Ziomek GJ, Jacobs RE, Bearer EL. Deficits in axonal transport in hippocampal-based circuitry and the visual pathway in APP knock-out animals witnessed by manganese enhanced MRI. Neuroimage. 2012;60:1856–1866. doi: 10.1016/j.neuroimage.2012.01.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satpute-Krishnan P, DeGiorgis JA, Conley MP, Jang M, Bearer EL. A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc Natl Acad Sci U S A. 2006;103:16532–16537. doi: 10.1073/pnas.0607527103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vagnoni A, Perkinton MS, Gray EH, Francis PT, Noble W, Miller CC. Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Abeta production. Hum Mol Genet. 2012;21:2845–2854. doi: 10.1093/hmg/dds109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amaratunga A, Fine RE. Generation of amyloidogenic C-terminal fragments during rapid axonal transport in vivo of beta-amyloid precursor protein in the optic nerve. J Biol Chem. 1995;270:17268–17272. doi: 10.1074/jbc.270.29.17268. [DOI] [PubMed] [Google Scholar]
- 60.Buxbaum JD, Thinakaran G, Koliatsos V, O’Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ando K, Oishi M, Takeda S, Iijima K, Isohara T, Nairn AC, Kirino Y, Greengard P, Suzuki T. Role of phosphorylation of Alzheimer’s amyloid precursor protein during neuronal differentiation. J Neurosci. 1999;19:4421–4427. doi: 10.1523/JNEUROSCI.19-11-04421.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muresan Z, Muresan V. c-Jun NH2-terminal kinase-interacting protein-3 facilitates phosphorylation and controls localization of amyloid-beta precursor protein. J Neurosci. 2005;25:3741–3751. doi: 10.1523/JNEUROSCI.0152-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mazzitelli S, Xu P, Ferrer I, Davis RJ, Tournier C. The loss of c-Jun N-terminal protein kinase activity prevents the amyloidogenic cleavage of amyloid precursor protein and the formation of amyloid plaques in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:16969–16976. doi: 10.1523/JNEUROSCI.4491-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muresan Z, Muresan V. A phosphorylated, carboxy-terminal fragment of {beta}-amyloid precursor protein localizes to the splicing factor compartment. Hum Mol Genet. 2004;13:475–488. doi: 10.1093/hmg/ddh054. [DOI] [PubMed] [Google Scholar]
- 65.Lombino F, Biundo F, Tamayev R, Arancio O, D’Adamio L. An intracellular threonine of amyloid-beta precursor protein mediates synaptic plasticity deficits and memory loss. PLoS One. 2013;8:e57120. doi: 10.1371/journal.pone.0057120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oules B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Brechot P, Trebak M, Checler F, Benfenati F, Chami M. Ryanodine Receptor Blockade Reduces Amyloid-beta Load and Memory Impairments in Tg2576 Mouse Model of Alzheimer Disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sclip A, Antoniou X, Colombo A, Camici GG, Pozzi L, Cardinetti D, Feligioni M, Veglianese P, Bahlmann FH, Cervo L, Balducci C, Costa C, Tozzi A, Calabresi P, Forloni G, Borsello T. c-Jun N-terminal kinase regulates soluble Abeta oligomers and cognitive impairment in AD mouse model. The Journal of biological chemistry. 2011;286:43871–43880. doi: 10.1074/jbc.M111.297515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, Lim TH, Pastorino L, Kunwar AJ, Walton JC, Nagahara AH, Lu KP, Nelson RJ, Tuszynski MH, Huang K. JNK3 Perpetuates Metabolic Stress Induced by Abeta Peptides. Neuron. 2012;75:824–837. doi: 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morihara T, Hayashi N, Yokokoji M, Akatsu H, Silverman MA, Kimura N, Sato M, Saito Y, Suzuki T, Yanagida K, Kodama TS, Tanaka T, Okochi M, Tagami S, Kazui H, Kudo T, Hashimoto R, Itoh N, Nishitomi K, Yamaguchi-Kabata Y, Tsunoda T, Takamura H, Katayama T, Kimura R, Kamino K, Hashizume Y, Takeda M. Transcriptome analysis of distinct mouse strains reveals kinesin light chain-1 splicing as an amyloid-beta accumulation modifier. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1307345111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gan KJ, Morihara T, Silverman MA. Atlas stumbled: Kinesin light chain-1 variant E triggers a vicious cycle of axonal transport disruption and amyloid-beta generation in Alzheimer’s disease. BioEssays: news and reviews in molecular, cellular and developmental biology. 2014 doi: 10.1002/bies.201400131. [DOI] [PubMed] [Google Scholar]
- 71.Muller UC, Zheng H. Physiological functions of APP family proteins. Cold Spring Harb Perspect Med. 2012;2:a006288. doi: 10.1101/cshperspect.a006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Muresan Z, Muresan V. SWAN Alzheimer Knowledge Base. Alzheimer Research Forum; 2009. CAD cells are a useful model for studies of APP cell biology and Alzheimer’s disease pathology, including accumulation of Aβ within neurites. Available at: http://mind-swanweb1.mgh.harvard.edu/swan/browser/showEntity.action?objectId=urn%3Alsid%3Aswan.org%3Aresearchstatement%3Aca7169f1-ff7d-456c-b3f3-c52bec1e8074. [Google Scholar]