

Summary
Autophagy is an important component of the immune response. However, the functions of autophagy in human diseases are much less understood. We studied biological consequences of autophagy deficiency in mice lacking the essential autophagy genes Atg7 or Atg5 in myeloid cells. Surprisingly, these mice presented with spontaneous sterile lung inflammation, characterized by marked recruitment of inflammatory cells, submucosal thickening, goblet cell metaplasia and increased collagen content. Lung inflammation was associated with increase in several pro-inflammatory cytokines in the bronchoalveolar lavage and in serum. This inflammation was largely driven by interleukin 18 as a result of constitutive inflammasome activation. Following intraperitoneal LPS injection, autophagy deficient mice had higher levels of pro-inflammatory cytokines in lungs and in serum, as well as increased mortality than control mice. Intranasal bleomycin challenge exacerbated lung inflammation in autophagy deficient mice and produced more severe fibrotic changes than in control mice. These results uncover a new and important role for autophagy as negative regulator of lung inflammation.
Introduction
Autophagy has been recognized as an important regulator in many biological processes (1). As such, there is increasing interest in understanding the roles of autophagy in human diseases. Recently, mutations in autophagy genes were linked to several disorders (2). However, the role of autophagy in organ physiology and pathology needs to be elucidated as a prelude to better understanding of its role in human diseases. In the lung, autophagy is likely critical for several important biological pathways implicated in lung diseases. Collectively, inflammatory lung injury syndromes account for the majority of morbidity and mortality associated with lung diseases and critical illness. One significant gap in our knowledge is the role that each cell type plays in orchestrating the injury. This issue is complicated by the extensive interactions between different cells in the lungs and the alveolar space. The use of cell- specific genetically engineered mouse models could greatly enhance our understanding of the roles of various cell types in lung injury pathogenesis. Macrophages are important cells in host defense. Resident macrophages in different tissues, including lung alveolar macrophages, ensure continuous immune surveillance and play a critical role in activating the cascade of immune responses. Autophagy deficiency in macrophages has been shown to result in increased inflammasome activity (2). Further, activation of autophagy limits IL-1β production by targeting ubiquitinated inflammasomes for destruction (3). However, the consequences of autophagy deficiency in vivo are much less understood. Here, we describe the development of spontaneous sterile lung inflammation in mice lacking Atg7 or Atg5 in myeloid cells. This inflammation was largely driven by interleukin (IL)-18 subsequent to constitutive inflammasome activation. This study reveals essential role of autophagy as a negative regulator of lung inflammation and identify IL-18 as a critical mediator in lung injury due to autophagy deficiency.
Materials and Methods
Reagents and antibodies
LPS (E. coli O111:B4), DNase, collagenase, bleomycin sulfate and recombinant mouse macrophage colony-stimulating factor (M-CSF) were from Sigma. Percoll™ was from GE Health Care. IL-1β receptor antagonist (Anakinra) was from Biovitrium. IL-17 neutralizing antibody (MAB421), rat IgG2a IL-17 isotype control (MAB006) and rat IgG1 IL-18 isotype control (MAB005) were from R&D systems. IL-18 neutralizing antibody (D048-3) was from MBL. LC3B antibody was previously described (4). ATG7 antibody was from Rockland Immunochemicals, Inc. (600-401-487). SQSTM1 (P62) antibody was from American Research Products (03-GP62-C-1).
Mice
We purchased LysM-cre mice from Jackson laboratory. Atg7flox was obtained from M. Komatsu. To specifically delete Atg7 and Atg5 from myeloid cells, we crossed Atg7flox and Atg5flox mice to LysM-cre, both on a C57BL/6 background (5). Age and sex matched Atgf/f/LysM-cre or wild type (Atgf/f) mice were examined in each experiment. Mice were housed within a specific pathogen free vivarium. Atg7f/f/LysM-cre mice were re-derived into a higher barrier 4 vivarium. Institutional Animal Care and Use Committee approved the research protocol.
LPS and bleomycin challenges
Intraperitoneal administration of LPS was done using a dose of 10 mg/kg for male and 20 mg/kg for female (6). Intranasal challenge of bleomycin was done as previously described (7). Briefly, bleomycin was dissolved in PBS at a concentration of 2.5 U/kg. Mice were anesthetized with isoflurane and bleomycin in 50 μl PBS or PBS alone was instilled to the airway by inhalation through the nasal openings.
Histology and immunohistochemistry
Mice were sacrificed and organs were harvested, fixed and embedded in paraffin blocks. Paraffin embedded organs were deparaffinized, rehydrated, sectioned and stained for hematoxylin and eosin. Sections from fixed inflated lung were stained for periodic acid Schiff for detection of mucin glycoprotein, or Masson’s Trichrome for collagen. For immunohistochemistry of lung sections, rat monoclonal Cd45/B220 (B cells marker) from BD Pharmingen, rat monoclonal anti F4/80 (macrophages marker) from Abcam, or MUC5AC clone 45M1 from Thermo scientific, were used as primary antibodies.
Quantification of immunohistological findings
The number of B220, PAS and muc5ac positive cells were counted on non-overlapping high power fields (magnification 400X) of lung parenchyma (for B220) or airway epithelium (for muc5ac and PAS) beginning at the periphery of the section. Two separate stained sections for each antibody were counted per mouse and the mean number of positive cells was reported. For peribronchial trichrome staining, area was outlined and quantified using a light microscope and ImageJ software from National Institutes of Health. Results are expressed as the area of trichrome staining per micrometer length of basement membrane of bronchioles. To estimate the level of lung fibrosis following bleomycin challenge, the Ashcroft score was determined as previously reported (8). The amount of soluble collagen in BAL and lung tissue homogenates was quantified using the Sircol assay from Biocolor, Carrick, UK. Lung inflammation score was quantified as previously described (9).
Evaluation of cellular composition of BAL
Mice were sacrificed by CO2 asphyxiation and BAL was done by instilling 0.8 ml PBS in trachea and then withdrawing it. Differential cell count of BAL was done on cytospins by evaluating 200 cells based on characteristic morphology.
Bone marrow derived macrophages
Bone marrow cells from 6–12 week old mice were cultured in DMEM containing 32 ng/ml mouse M-CSF for 7 days to differentiate into macrophages.
Flow cytometry
Lung leukocytes were isolated as previously described (10). Lung leukocytes as well as single cell suspensions from spleen, thymus and bone marrow were stained by antibodies against CD16/CD32, CD45,-APC, Lys6G-PE Lys6G/Ly6c-PE, CD45/B220-FITC, CD3e-PE-Cy7, CD4-PE, or CD8a-FITC, all from BD Pharmingen, or F4/80-Pe-Cy7 from eBioscience. Dead cells were excluded by Sytox blue from Invitrogen. Data were collected with FACS LSR Fortessa from BD Bioscience and analyzed using FlowJo software from Tree Star. Total number of leukocytes was obtained by multiplying total number of cells by the frequency of CD45+ cells obtained from flow cytometry analysis. Total cell numbers from single cell suspensions of lungs, spleens, thymus bone marrow and BAL were counted using hemocytometer. Bone marrow proliferation was assessed using Click-iT EDU Flow Cytometry Assay Kit from Molecular Probes. Mice were injected intraperitoneally for 3 hours with 200 μg of 5-ethynyl-2′-deoxyuridine (EDU), sacrificed and bone marrow cells were analyzed for proliferative activity.
ELISA
IL-1β, IL-17, TGF-β, IL-6, and IL-33 were quantified using Quantikine ELISA kits from R&D Systems. Mouse IL-18 ELISA kit from MBL international was used to measure serum and BAL IL-18. Serum IgE was measured using BD optEIA ELISA kit from BD bioscience. Levels of 32 cytokines and chemokines (Eotaxin, G-CSF, GM-CSF, IFN-γ, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, IL-1α, IL-1β, IL-2, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, IP-10, KC-like, LIF, LIX, M-CSF, MCP-1, MIG, MIP-1α, MIP-1β, MIP-2, RANTES, TNF-α, VEGF) were simultaneously measured in mouse serum using a Milliplex Mouse Cytokine/Chemokine 32 plex assay from Millipore on a luminex-based multi analyte plate from BioPlex; Bio-Rad.
ELISpot
Single cell suspensions from the lungs and spleens were obtained after flushing the systemic and pulmonary circulation with PBS, tissues were cut into small portions and then dispersed through 40 μm nylon filters. Red blood cells were lysed in ammonium-chloride-potassium (ACK) lysing buffer and remaining cells were washed twice and resuspended to 107cells/ml (11) The numbers of IL-1β producing cells were quantified by aliquoting 106cells/100μl to a 96 well microtiter plates precoated with monoclonal anti-mouse IL-1β from Millipore. The numbers of IL-17, IL-4, and IFN-γ producing cells were quantified by ELISpot Mouse Development Module. IL-1β antibodies, ELISpot kits, and color development reagents were from R&D Systems.
IL-1 receptor blockade and antibody neutralization of IL-17 or IL-18
In survival studies, following LPS administration, 25 mg/kg Anakinra was injected subcutaneously every 4 hours for 12 hours (12). Antibodies against IL-18 (15 μg) or isotype control were injected intaperitoneal every 4 hours for 12 hours. To determine the effect of IL-1β on spleen weight, 100 mg/kg Anakinra was subcutaneously injected 30 minutes before and 3 hours after LPS. For neutralizing the lung inflammation, Anakinra was injected subcutaneously daily for 8 days at a dose of 200 mg/kg. Control mice, not receiving Ankinra, were injected subcutaneously with PBS. Neutralizing IL-17 antibody (100 μg) or isotype control was injected intraperitoneal every other day for three times. IL-18 antibody (25–50 μg) or isotype control were injected intaperitoneal every other day for two or three times. Mice were sacrificed 24 hours following the last injection.
Statistical analysis
Paired results were compared using student’s t-test. A two-way analysis of variance followed by Bonferroni correction was used for multiple comparisons. Survival was evaluated with the log rank test. All analyses were carried out with GraphPad Prism Version 5.00 Software.
Results
Autophagy deficiency in myeloid cells results in spontaneous lung inflammation
Mice with deficiency of key autophagy genes Atg5 or Atg7 die in the first day after birth (5). To generate myeloid-specific Atg7 knockout mice, we crossed mice bearing an Atg7flox allele with LysM-Cre transgenic mice, which express Cre recombinase specifically in cells of myeloid origin, including macrophages and neutrophils (4). Mice were raised in a specific pathogen free barrier facility. The resulting mice, Atg7f/fCre+ (henceforth termed Atg7Δ) were viable with normal growth pattern similar to age-matched wild type controls (Atg7f/f). Consistent with conditional Atg7 knockout in macrophages, bone marrow-derived macrophages (BMDM) from Atg7Δ exhibited marked reduction of ATG7 (Fig. 1A). Further, accumulation of SQSTM1 (also known as p62), an autophagy substrate, and the diminished lipidation of the microtubule associated protein 1 light chain 3B (LC3B) from type I to II confirmed autophagy deficiency in these cells (13). Examination of spleen, liver, gastrointestinal tract, and brain from Atg7Δ and Atg7f/f mice revealed no gross abnormalities. Surprisingly, Atg7Δ mice developed spontaneous lung inflammation characterized by increased infiltration of inflammatory cells in lung tissues, submucosal thickening and increased collagen deposition. Lung inflammation was observed at two month-old mice and persisted in mice evaluated up to ten months of age (Figs 1B–D). Flow cytometry analysis of single cell suspension from lung leukocytes showed that Atg7Δ mice had a higher number of total leukocytes, as indicated by the leukocyte common antigen (LCA)/CD45 antibody staining (10), including higher number of neutrophils, as indicated by the Ly6G antibody staining for neutrophils (Figs 1E–F). Immune staining of lung tissue revealed that Atg7Δ mice accumulated macrophages and B220+ cells and concomitant serum analysis indicated elevated IgE levels (Fig. S1A–C).
Fig. 1. Atg7Δ mice presents with spontaneous lung inflammation.

Bone marrow-derived macrophages (BMDM) from wild type (Atg7f/f) or Atg7Δ mice were analyzed by immunoblotting (A). Lung tissues from two (B), six (C) or ten (D) month old mice were analyzed by hematoxylin and eosin (H&E) staining. Flow cytometry analysis of single cell suspensions of the lungs was done using the pan leukocytic marker anti-CD45 (E) or anti-Ly6G (F). Quantification graphs are shown. Bronchoalveolar lavage (BAL) from 2–4 months old Atg7Δ or control Atg7f/f mice was analyzed for total cell count (G) or differential cell count (H). Mϕ, macrophages; Lymph, lymphocytes; PMN, polymorphonuclear neutrophils. Data are mean ± SEM, n=3 to 8 mice/genotype, *p<0.05, ** p<0.01 Scale bar, 50 μm.
Analysis of bronchoalveolar lavage (BAL) revealed that Atg7Δ mice exhibited accumulation of leukocytes, compared to wild-type Atg7f/f (Figs 1G). Differential cell count of BAL showed that Atg7Δ mice had higher percentages of neutrophils and lymphocytes (Figs 1H). Further, Atg7Δ mice had a higher number of leukocytes in bone marrow, including macrophages and B lymphocytes (Fig. S1D–F). To identify a possible cause for increased number of cells in bone marrow from Atg7Δ mice, we investigated the proliferation of total leukocyte population in bone marrow after intraperitoneal injection of EDU. Flow cytometry analysis of these cells showed that leukocytes of Atg7Δ mice incorporated more EDU, indicating increased proliferative activity (Fig. S1G). The above findings indicated that autophagy in myeloid cells was essential for the maintenance of normal lung homeostasis.
Mice were housed in pathogen free environment and repeated screenings failed to show any evidence of infection. To further rule out the possibility that an occult infectious agent contributed to the observed phenotype, we re-derived Atg7Δ and control Atg7f/f mice into a higher barrier 4 (B-4) facility. Histological analysis of lung tissues of re-derived mice confirmed sustained lung inflammation in Atg7Δ mice, compared to control mice (Fig. S2A–C). Further, BAL of re-derived Atg7Δ mice had increased total leukocyte count and differential cell counts of neutrophils and lymphocytes. These results confirm the sterile phenotype of lung inflammation observed in Atg7Δ mice.
Mucus metaplasia in the lungs of Atg7Δ mice
We next investigated evidence of tissue injury and remodeling as a result of lung inflammation in Atg7Δ mice. Goblet cell metaplasia is a common pathological feature that occurs in several inflammatory lung diseases. Atg7Δ mice exhibited increased mucus production and accumulation of mucin in the airway epithelia (Fig. 2). These findings were revealed by periodic acid Schiff (PAS) staining and confirmed by immunohistochemical analysis using mucin specific Muc5ac antibody. Quantification of PAS and Muc5ac positive cells in the airway epithelium revealed higher numbers of positive cells in Atg7Δ mice compared to wild type Atg7f/f mice. These results indicated that lung inflammation in Atg7Δ mice was associated with goblet cell metaplasia.
Fig. 2. Mucus hypersecretion and goblet cell metaplasia in Atg7Δ mice.

Periodic acid Schiff staining was performed on lungs from two (A), six (B) or ten (C) months old control Atg7f/f or Atg7Δ mice. Quantitative analyses of results are shown (D). Lung tissues from six-month-old mice were immunostained for muc5ac, and the quantitation of positive cells is shown (E). Data are shown as mean ± SEM, n=4 to 8 mice/genotype, *p<0.05 **p<0.01. Scale bar, 50 μm.
Lung inflammation and mucus metaplasia in Atg5Δ mice
To rule out the possibility that the phenotype observed in Atg7Δ mice was caused by loss of non-autophagic functions of Atg7, we generated myeloid-specific Atg5Δ mice by breeding mice bearing an Atg5flox allele with LysM-Cre transgenic mice. Similar to data obtained from Atg7Δ, lung tissues of Atg5Δ mice revealed increase in the infiltration of leukocytes (Fig. S2D–I). Atg5Δ mice also exhibited mucus cell metaplasia in the airway epithelia, and had a higher number of total leukocytes, particularly neutrophils, in lung tissues and BAL. These results confirmed the data obtained from Atg7Δ mice and further suggested that autophagy deficiency, and not autophagy-independent gene-specific function, was responsible for the lung phenotype observed.
Constitutive inflammasome activation in Atg7Δ mice
Tissue damage and disruption of cellular homeostasis are the hallmarks of inflammation. Inflammasomes are protein complexes that play a critical role in the recognition of danger-associated molecular patterns from damaged tissue or dying cells (14). Secretion of the active form of the potent pro-inflammatory cytokines IL-1β and IL-18 occurs as a result of inflammasome complex assembly and activation. Deficiency of autophagy has been linked to hyperactivity of the inflammasome (2). We reasoned that lung inflammation in Atg7Δ mice might be caused by a constitutive inflammasome activity. To test this hypothesis, we examined single cell suspensions from the lungs of Atg7Δ mice by ELISpot. The number of cells producing IL-1β was markedly elevated in lungs from Atg7Δ mice (Fig. 3A). Inflammasome activation has been shown to potentiate Th17 cell-dominant immune responses (15). Further, pulmonary inflammation induced by direct intranasal IL-1β challenge in mice is mediated by IL-17 (16). To identify the nature of the T helper (TH) immune response mediating the spontaneous inflammation in the lung, we used ELISpot to determine the number of cells that produce interferon-γ (IFN-γ) (for TH1), IL-4 (for TH2) or IL-17 (for TH17). Atg7Δ mice yielded higher number of IL-17 cells in lungs (Fig. 3B). The numbers of IFN-γ or IL-4 producing cells, in lungs and spleens, were similar between Atg7f/f and Atg7Δ mice (data not shown). IL-18, another inflammasome product, has been shown to be important in lung inflammation associated with Influenza A virus infection (17). In our study, BAL fluid showed that Atg7Δ mice had higher levels of IL-1β and IL-18, compared to barely detectable levels in Atg7f/f mice (Fig. 3C). Importantly, in Atg7Δ mice, IL-18 concentration was about 7 fold higher than IL-1β, suggesting an important role for IL-8 in lung inflammation observed in these mice. IL-1β and IL-18 were also higher in serum of Atg7Δ mice, compared to control mice (Fig. 3D).
Fig. 3. Cytokine upregulation in Atg7Δ mice.

Lungs from Atg7f/f mice or Atg7Δ mice were analyzed by ELISpot. Representative images and quantitative analyses for IL-1β (A) or IL-17 (B) are shown. BAL (C) and serum (D) from 2–4 month old Atg7Δ or control Atg7f/f mice were analyzed for IL-1β, IL-18, TGF-β and IL-6. Data are shown as mean ± SEM, n = 4–12 mice/genotype, *p<0.05 **p<0.01.
Increased IL-18, IL-1β and IL-17 in the lungs of Atg7Δ mice suggested that pulmonary neutrophilia was driven by the increase in these cytokines (18, 19). A recent study has shown that IL-17 plays a role in bleomycin induced lung inflammation and fibrosis in mice, and that induction of collagen by IL-17 was TGF-β dependent (20). To investigate possible biological consequences of inflammasome activation and subsequent increase in IL-17 in the lungs of Atg7Δ mice, we examined latent TGF-β levels. BAL from Atg7Δ mice contained higher amounts of TGF-β than in control Atg7f/f mice (Fig. 3C). It has been previously shown that TGF-β and IL-6 can promote the differentiation of naïve CD4 cells to IL-17 producing T cells (21). Analysis of BAL from Atg7Δ mice indicated substantial increase in IL-6 compared to BAL from Atg7f/f mice (Fig. 3C).
Lung inflammation in Atg7Δ mice is primarily mediated by IL-18
Our data above revealed upregulation of inflammasome associated cytokine IL-1β, IL-17 and IL-18 in Atg7Δ mice. Unlike IL-1β, pro-IL-18 is presorted inside macrophages and it’s readily secreted upon inflammasome activation, without the need for a priming step (14, 22, 23). Thus, we speculated that IL-18 might have an important role in lung inflammation in Atg7Δ mice. We used neutralization strategies to determine the relative contribution of each cytokine to the causation of lung inflammation in these mice. Treatment of Atg7Δ mice with the interleukin-1 receptor antagonist (Anakinra), or the use of neutralizing antibodies against IL-17 had little effect on lung inflammation in Atg7Δ mice. In contrast, the use of antibodies against IL-18 markedly prevented lung inflammation in Atg7Δ mice compared to mice injected with isotype control antibodies (Figs 4A–B). Furthermore, only IL-18 antibody treatment led to substantial reduction in the total number of leukocytes in BAL of Atg7Δ mice (Figs 4C–D). Differential cell count showed that IL-18 antibody treatment almost completely inhibited the recruitment of lymphocytes and neutrophils in the BAL of Atg7Δ mice (Fig. 4E). Despite having no effect on the tissue inflammation score or total number of leukocytes in BAL, IL-17 antibody treatment was effective in reducing the number of neutrophils in BAL. These results suggest that lung inflammation, in Atg7Δ mice, was primarily driven by IL-18. The Lung pathology observed in Atg7Δ mice suggested that autophagy in myeloid cells has an important protective role against lung inflammation. To elucidate such role, we investigated the role of autophagy in two clinically relevant models of lung inflammation, namely acute lung injury secondary to sepsis, and interstitial lung fibrosis secondary to bleomycin exposure.
Fig. 4. IL-18 is required for lung inflammation in Atg7Δ mice.

(A–B) Representative images from Atg7Δ mice treated with saline or Anakinra (upper panels), isotype control or IL-17 antibody treatment (middle panels), or isotype control or IL-18 antibody (lower panels). (A) Lung tissues were analyzed by hematoxylin and eosin staining. (B) BAL was analyzed by HEMA-3 staining. Quantification of the inflammatory score is shown in (C) Total cell count (D) and deferential count (E) were determined. Data are shown as mean ± SEM n=3–13 mice/treatment, *p<0.05 **p<0.01. Scale bar, 50 μm
Enhanced susceptibility of Atg7Δ mice to endotoxemia
Sepsis syndrome can lead to septic shock that is associated with high rate of mortality (12). Sepsis is associated with increased IL-1β and IL-18 production and other systemic inflammatory responses including lung injury. Endotoxins such as LPS play an important role in the above responses. We conducted studies to test the susceptibility of Atg7Δ mice to endotoxins. We injected mice with intraperitoneal LPS and evaluated cytokine production, survival, and degree of lung inflammation. Levels of IL-1β, IL-18 and IL-17 were substantially elevated in Atg7Δ mice, compared to control mice, as early as 4–6 hours following LPS injection (Fig. 5A–C). We next evaluated survival of Atg7f/f and Atg7Δ mice in response to LPS, injected intraperitoneally at a dose of 10 mg/kg for males and 20 mg/kg for females (6). Survival rate was significantly reduced for both male and female Atg7Δ mice, compared to control mice (Fig. 5D–E). In addition, Atg7Δ mice died much earlier than Atg7f/f mice.
Fig. 5. Increased cytokine production and reduced survival in Atg7Δ mice. in response to LPS.
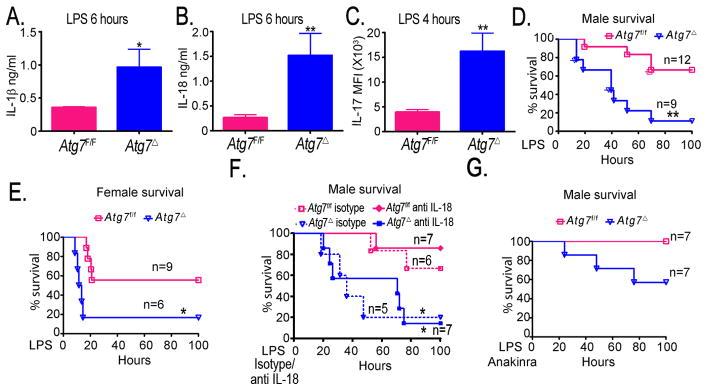
(A–C) Following intraperitoneal LPS injection in mice, serum levels of IL-1β (A) IL-18 (B) and IL-17 (C) were evaluated. (D–F) Kaplan-Meier survival curves of mice were recorded following injection of LPS (D–E) or LPS plus anti IL-18 antibody or isotype control (F) and LPS plus interleukin 1 receptor antagonist Anakinra (G). Data are mean ± SEM, n=3 to 6 mice/genotype for each time point *p<0.05, **p<0.01.
We then wanted to determine the effect of IL-18 neutralization on mice survival in LPS induced sepsis. We co-administered mice with LPS and IL-18 antibodies or with LPS and isotype control (19). IL-18 neutalization, following LPS injection, improved survival of Atg7f/f mice, but had no significant effect on survival of Atg7Δ mice (Fig. 5F). These data suggest that IL-18 is important in endotoxin-induced lethality. However, in autophagy deficient mice, the increase of other inflammatory mediators, including IL-1β, play an important role in endotoxin-induced lethality.
To determine the relative contribution of the increased IL-1β on the enhanced endotoxin-induced lethality in Atg7Δ mice, we co-treated mice with LPS and Anakinra (12). IL-1 receptor blockade attenuated LPS lethality by reducing the frequency of death and delaying its onset in Atg7Δ mice (Fig. 5G). These findings are consistent with an important role for IL-1β in endotoxin-induced death in Atg7Δ mice. The incomplete prevention of LPS lethality probably reflects the upregulation of other pro-inflammatory cytokines downstream of inflammasome activation in Atg7Δ mice. These data suggested that, whereas IL-1β was essentially dispensable for lung inflammation, it played a more important role in endotoxin-induced lethality.
LPS induces higher levels of serum cytokines in Atg7Δ mice
Four hours following LPS injection, the levels of 32 cytokines and chemokines were measured in the serum from Atg7f/f and Atg7Δ mice. Compared to control mice, Atg7Δ mice had increased serum level of IFN-γ, IL-3, IL-13, leukemia inhibitory factor (LIF), regulated on activation normal T cell expressed and secreted (RANTES) and tumor necrosis factor-alpha (TNF-α) in response to LPS (Fig. S3A, and data not shown).
Atg7Δ mice exhibit enhanced lung inflammation after LPS challenge
Acute lung injury is a major cause of morbidity and mortality in patients with sepsis. LPS challenge in mice is often used as a model to study acute lung injury of sepsis. Intraperitoneal injection of LPS leads to increase in the recruitment of macrophage to the BAL and neutrophil sequestration to the lung (24). We, therefore, examined lung inflammation in Atg7f/f and Atg7Δ mice after intraperitoneal LPS injection. Histological analysis of lung tissues showed that Atg7Δ mice displayed a marked increase in the infiltration of inflammatory leukocytes to lungs, compared to Atg7f/f mice (Figs 6A). Further, there was increased goblet cell metaplasia after LPS injection in Atg7Δ mice. PAS staining for the glycoprotein content of mucin revealed that Atg7Δ mice contained much higher PAS positive epithelial cells in the airways (Fig. 6B). Collagen deposition was increased in lungs of Atg7Δ mice, as early as 6 hours following LPS injection and at death, suggesting the development of pro-fibrotic lesions (Fig. 6C–D). BAL of Atg7Δ mice had higher total cell count following LPS injection, compared to Atg7f/f mice (Fig. S3B). Differential cell count of BAL showed that Atg7Δ mice exhibited a significantly higher percentage of lymphocytes and neutrophils following LPS injection (Fig. S3C). In contrast, Atg7f/f mice did not show any relevant changes in the percentages of lymphocytes and neutrophils, as most of the cells recovered in BAL from these mice (Atg7f/f) before and after LPS injection were macrophages. The findings in Atg7f/f control mice are consistent with a recent study that found no changes in the number of neutrophils or in the concentration of IL-1β in the BAL, following intraperitoneal injection of LPS (25). Similarly, in our study, levels of IL-1β, IL-18 and IL-17 in Atg7f/f control mice were largely unchanged six hr post LPS (Fig. 6D–F). In contrast, there was marked increase in IL-1β, IL-18 and IL-17 in BAL of Atg7Δ mice. These data suggested that lack of Atg7 in myeloid cells led to failure of lung homeostatic protective mechanisms in Atg7Δ mice.
Fig. 6. Atg7Δ mice exhibit exaggerated lung inflammation after LPS challenge.

Atg7f/f or Atg7Δ mice were injected intraperitoneally with LPS. At the time of death, lung tissues were collected and stained with H&E (A), periodic acid Schiff (B), or Masson’s trichrome (C). The latter was quantified as mean area (MA) per μm of basement membrane (BM). Soluble collagen in lung homogenates was quantified using Sircol assay at zero and six hr following LPS injection (D). IL-1β (E), IL-18 (F), and IL-17 (G) levels were measured in BAL using ELISA at the indicated time points. Data are mean ± SEM from 3 to 6 mice per genotype at each time point.*p<0.05 **p<0.01. Scale bar, 50 μm.
In addition to increased cytokines and lung inflammation, our studies revealed that Atg7Δ mice rapidly developed marked increase in the size of the spleen within four hr of LPS injection. In contrast, there was no significant change in spleen size in similarly treated Atg7f/f mice (Fig. S3D). A previous study found that mice with conditional disruption of the IKK-β in myeloid cells, which led to increased inflammasome activity, developed splenomegaly and neutrophilia, and that these inflammatory changes were dependent on the presence of IL-1 receptor (18). In our study, concomitant Anakinra treatment partially neutralized the increase in the size of the spleen seen after LPS injection in Atg7Δ mice, whereas it had no significant effect on spleens of Atg7f/f mice (Fig. S3D). These data indicated that autophagy deficiency in myeloid cells predisposed mice to a severe form of lung inflammation and splenomegaly in response to LPS. We then investigated if Atg7Δ mice would also exhibit greater predisposition to pulmonary fibrosis.
Increased collagen production in Atg7Δ mice
One of the most deleterious outcomes of lung inflammation is pulmonary fibrosis, which leads to loss of the normal architecture of the lung and deterioration of respiratory function. Pulmonary fibrosis is characterized by fibroblast proliferation and deposition of extracellular matrix proteins such as collagen (26). Idiopathic pulmonary fibrosis is a fatal disease of unknown etiology characterized by deterioration of the respiratory function, and progressive lung scarring and fibrosis (26). Although murine models are used frequently to study human lung fibrosis, mice are more resistant to the development of permanent fibrotic lesions (27). We hypothesized that lung inflammation will predispose Atg7Δ mice to lung remodeling in the form of increased deposition of extracellular matrix proteins. At the age of 2 months, Atg7Δ mice exhibited a two-fold increase in the content of collagen in lung tissues (Fig. 7D). Soluble collagen in the BAL was also increased by 10 month of age in Atg7Δ mice (Fig. S3E). These results indicated that lung inflammation in Atg7Δ mice was accompanied by lung remodeling, and deposition of extracellular matrix.
Fig. 7. Atg7Δ mice exhibit increased lung responses to bleomycin.

Atg7f/f control mice or Atg7Δ mice were intranasally instilled with PBS or bleomycin for 7 or 14 days. Percent loss of body weight was recorded (A). Lung tissues were analyzed by H&E (B). Ashcroft scoring was used to evaluate the extent of fibrosis (C). Soluble collagen in lung homogenates (D) and BAL (E) was quantified using Sircol assay. Levels of TGF-β (F) and IL-6 (G) were measured using ELISA. Total count of leukocytes was determined (H). Data are shown as mean ± SEM, n= 4–16 mice/genotype, *p<0.05 **p<0.01. Scale bar, 200 μm.
Atg7Δ mice exhibit enhanced susceptibility to bleomycin-induced lung fibrosis
Mice challenged intranasally with bleomycin exhibit inflammation, accumulation of collagen, and fibrosis in the lungs and this model has been used to study the pathogenesis of pulmonary fibrosis (7, 16, 20). Atg7f/f and Atg7Δ mice were subjected to intranasal bleomycin or PBS and lungs were analyzed at 7 and 14 days post challenge. Both Atg7f/f and Atg7Δ mice showed a moderate loss of body weight, but the percentage of weight loss was more in Atg7Δ mice (Fig. 7A). Mice instilled with PBS did not show any weight loss. Histological analysis of lung tissues, at day 14 after bleomycin challenge, showed that Atg7Δ mice presented with severe histopathological features of pulmonary fibrosis, compared to control Atg7f/f mice (Fig. 7B). Consistent with these findings, Atg7Δ mice showed pronounced trichrome staining compared to Atg7f/f mice following bleomycin treatment (Fig. S4A). The severity of lung fibrosis was scored using Ashcroft scaling system (8). Further, quantification of soluble collagen levels in lung tissue homogenate and BAL was performed. Atg7Δ mice developed increased lung fibrosis and higher collagen levels (Fig. 7C–E). Moreover, levels of latent TGF-β and IL-6, considered mediators of fibrosis, were elevated in BAL of Atg7Δ mice compared to Atg7f/f (Fig. 7F–G). Both Atg7f/f and Atg7Δ mice showed a marked increase in the number of total leukocytes in BAL in response to bleomycin. However, total leukocyte count of BAL in Atg7Δ mice was higher (Fig. 7H). The percentages of lymphocytes and neutrophils were higher in Atg7Δ mice after bleomycin treatment (Fig. S4B–D). Further, Atg7Δ, but not wild-type, mice displayed a progressive splenomegaly following bleomycin challenge (Fig. S4E). Collectively, these data suggested an important protective role for autophagy in lung inflammation and fibrosis in response to bleomycin challenge.
Discussion
There is a growing interest in translating autophagy studies into better understanding of human diseases (28). However, the in vivo roles of autophagy in relation to organ pathology are not well understood. Studies addressing such roles are likely to provide breakthroughs into consequences of autophagy aberration on human diseases. In this regard, our studies reveal several novel findings. They showed that autophagy in myeloid cells was required for normal lung homeostasis. In the absence of autophagy, there was constitutive inflammasome activation leading to a spontaneous sterile lung inflammation. This effect was primarily mediated by IL-18 production. An important finding of this study is elucidation of the differential roles of IL-1β and IL-18 in lung injury and sepsis. IL-18 was critical for lung inflammation, whereas IL-1β was more responsible for the system effect of sepsis.
Lung inflammation predisposed autophagy-deficient mice to increased mortality from sepsis and led to enhanced lung fibrosis in response to fibrotic agent. Importantly, our findings were shown in mice deficient in either Atg7 or Atg5. Thus, they strongly suggest that the findings are due to autophagy deficiency and not due to deficiency of autophagy-independent functions of autophagy proteins. In addition, the reproduction of similar phenotype in Atg7Δ mice re-derived in high barrier facility confirmed that lung inflammation in these mice was not due to occult infection predisposed to by autophagy deficiency.
Inflammatory lung injury contributes to the underlying etiology of many lung diseases. In its acute form, lung injury is often associated with sepsis and carries a high mortality rate (29). Other less acute forms of lung injury are thought to contribute to variety of lung diseases including interstitial lung diseases, pneumonia, asthma, chronic obstructive pulmonary disease, cystic fibrosis and lung cancer. In our study, mice with conditional deletion of Atg7 or Atg5 in cells of myeloid origin presented with marked recruitment of inflammatory cells to the lungs. Macrophages, lymphocytes and neutrophils accumulated into the alveolar spaces and lung parenchyma. Further, airway epithelia underwent mucus cell metaplasia, an important feature of lung remodeling. Moreover, lung tissues accumulated much higher amounts of the extracellular matrix protein collagen, another feature that manifests during lung fibrosis and remodeling. In older autophagy-deficient mice, there was increase in collagen in BAL, a feature suggestive of a progressive phenotype. Intraperitoneal injection of LPS induced lung inflammation that was much more enhanced and was associated with collagen deposition in autophagy deficient mice. Lung inflammation in Atg7Δ mice was enhanced after bleomycin challenge. In response to bleomycin, lungs from Atg7Δ mice showed an amplified inflammation, as well as increase in fibrotic mediators, collagen deposition and fibrosis.
Our data indicate that lung inflammation was primarily mediated by IL-18, as indicated by our neutralization results. Despite failure of the interleukin 1 receptor antagonist to neutralize the lung inflammation at basal level of autophagy deficiency, anakinra treatment was effective in antagonizing morbidity and mortality induced by systemic administration of LPS. The neutralization studies suggest that the two cytokines have distinct roles in inflammation. Our data indicates that IL-18 mediates the sterile lung inflammation observed at baseline in Atg7Δ mice, whereas IL-1β effect was more pronounced in sepsis. The underlying mechanisms of such roles might be explained by the fact that IL-18 is constitutively expressed in macrophages, whereas IL-1β requires transcriptional activation by NF-κB (14). Thus, IL-18 might represent a therapeutic target in lung inflammation associated with sepsis.
Lung inflammation in Atg7Δ mice was also evident in airway epithelial cells. Prior studies found that myeloid cell-specific deletion of IκBα resulted in NF-κB activation in both myeloid cells and in epithelial cells with intact IκBα (24). IL-1β or IL-18 secreted by macrophages can bind to their cognate receptors expressed on epithelial cells and lead to transcriptional activation of downstream signals such as NF-κB or mitogen activated protein kinases, and the subsequent expression of more IL-1β/IL-18 as well as other proinflammatory cytokines and chemokines. IL-1β/IL-18 can also lead to recruitment of neutrophils and lymphocytes, which can interact with airway epithelial cells and amplify the inflammation. The finding of mucus cell metaplasia in lungs of Atg7Δ mice is likely the consequence of the interaction between myeloid cells and epithelial cells. It has been shown previously that human IL-1β expression in Clara cells lead to mucus cell metaplasia in mice (30). Recently, IL-18 was found to induce similar pathology via IL-17, IFNγ and IL-13 (31). Neutrophils release several inflammatory and oxidative mediators that can lead to lung tissue damage and features of remodeling including goblet cell metaplasia. Hence, mucus cell metaplasia in Atg7Δ mice could be caused by one or a combination of several inflammatory mediators.
Another important finding in our study is the increase of basal levels of TGF-β and IL-6 in the BAL of Atg7Δ mice. TGF-β has an important role in epithelial to mesenchymal transition and in the pathogenesis of pulmonary fibrosis. TGF-β, together with IL-1β and IL-6, induce Th17 lineage development. IL-17 is a strong neutrophil attractant implicated in the induction of inflammatory responses and lung damage in response to bleomycin, partially by increasing TGF-β (32). In the context of pulmonary fibrosis, a recent report suggested that autophagy might be deficient in subjects with the disease (33). In summary, our study indicated that autophagy in myeloid cells was required to maintain normal lung homeostasis and to prevent excessive inflammatory responses. This study provides the groundwork for further understanding of the role of autophagy in human diseases.
Supplementary Material
Acknowledgments
We thank members of the Eissa laboratory for useful discussions and technical assistance. Atg7f/f and Atg5f/f mice were kind gifts from Dr. M. Kmoatsu and RIKEN BioResource, Japan, respectively.
Footnotes
This study was supported by funding from the National Heart, Lung and Blood Institute and by the Dan L Duncan Cancer Center, Cytometry and Cell Sorting, and Proteomics Cores at Baylor College of Medicine with funding from the NIH (AI036211, CA125123, and RR024574) and NCI (P30CA125123) and the expert assistance of Joel M. Sederstrom, and Dr. Shixia Huang.
References
- 1.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 3.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nature immunology. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu XD, Ko S, Xu Y, Fattah EA, Xiang Q, Jagannath C, Ishii T, Komatsu M, Eissa NT. Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J Biol Chem. 2012;287:19687–19698. doi: 10.1074/jbc.M112.350934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 6.Mabley JG, Horvath EM, Murthy KG, Zsengeller Z, Vaslin A, Benko R, Kollai M, Szabo C. Gender differences in the endotoxin-induced inflammatory and vascular responses: potential role of poly(ADP-ribose) polymerase activation. The Journal of pharmacology and experimental therapeutics. 2005;315:812–820. doi: 10.1124/jpet.105.090480. [DOI] [PubMed] [Google Scholar]
- 7.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, Ryffel B, Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117:3786–3799. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. Journal of clinical pathology. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Jr, Crawford JM, Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. [PubMed] [Google Scholar]
- 10.Jain AV, Zhang Y, Fields WB, McNamara DA, Choe MY, Chen GH, Erb-Downward J, Osterholzer JJ, Toews GB, Huffnagle GB, Olszewski MA. Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary Cryptococcus neoformans infection. Infect Immun. 2009;77:5389–5399. doi: 10.1128/IAI.00809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nature medicine. 2009;15:267–276. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 12.Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO journal. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–874. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, di Padova F, Fick L, Charron S, Lagente V, Eberl G, Le Bert M, Quesniaux VF, Huaux F, Leite-de-Moraes M, Ryffel B, Couillin I. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PloS one. 2011;6:e23185. doi: 10.1371/journal.pone.0023185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, Xavier RJ, Green DR, Lamkanfi M, Dinarello CA, Doherty PC, Kanneganti TD. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nature immunology. 2013;14:480–488. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu LC, Enzler T, Seita J, Timmer AM, Lee CY, Lai TY, Yu GY, Lai LC, Temkin V, Sinzig U, Aung T, Nizet V, Weissman IL, Karin M. IL-1beta-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKbeta. Nature immunology. 2011;12:144–150. doi: 10.1038/ni.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Netea MG, Fantuzzi G, Kullberg BJ, Stuyt RJ, Pulido EJ, McIntyre RC, Jr, Joosten LA, Van der Meer JW, Dinarello CA. Neutralization of IL-18 reduces neutrophil tissue accumulation and protects mice against lethal Escherichia coli and Salmonella typhimurium endotoxemia. J Immunol. 2000;164:2644–2649. doi: 10.4049/jimmunol.164.5.2644. [DOI] [PubMed] [Google Scholar]
- 20.Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, Wang XX, Liu HZ, Sun W, Hu ZW. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. 2011;187:3003–3014. doi: 10.4049/jimmunol.1004081. [DOI] [PubMed] [Google Scholar]
- 21.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell. 2012;150:1235–1248. doi: 10.1016/j.cell.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA. Differences in signaling pathways by IL-1beta and IL-18. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8815–8820. doi: 10.1073/pnas.0402800101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han W, Joo M, Everhart MB, Christman JW, Yull FE, Blackwell TS. Myeloid cells control termination of lung inflammation through the NF-kappaB pathway. American journal of physiology. Lung cellular and molecular physiology. 2009;296:L320–327. doi: 10.1152/ajplung.90485.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King BA, Kingma PS. Surfactant protein D deficiency increases lung injury during endotoxemia. American journal of respiratory cell and molecular biology. 2011;44:709–715. doi: 10.1165/rcmb.2009-0436OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- 27.Chung MP, Monick MM, Hamzeh NY, Butler NS, Powers LS, Hunninghake GW. Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. American journal of respiratory cell and molecular biology. 2003;29:375–380. doi: 10.1165/rcmb.2003-0029OC. [DOI] [PubMed] [Google Scholar]
- 28.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature reviews. Drug discovery. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Jr, Hoffman E, Hubmayr RD, Leppert M, Matalon S, Munford R, Parsons P, Slutsky AS, Tracey KJ, Ward P, Gail DB, Harabin AL. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. American journal of respiratory and critical care medicine. 2003;167:1027–1035. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 30.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. American journal of respiratory cell and molecular biology. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 31.Kang MJ, Choi JM, Kim BH, Lee CM, Cho WK, Choe G, Kim DH, Lee CG, Elias JA. IL-18 induces emphysema and airway and vascular remodeling via IFN-gamma, IL-17A, and IL-13. American journal of respiratory and critical care medicine. 2012;185:1205–1217. doi: 10.1164/rccm.201108-1545OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. The Journal of experimental medicine. 2010;207:535–552. doi: 10.1084/jem.20092121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K. Insufficient autophagy in idiopathic pulmonary fibrosis. American journal of physiology Lung cellular and molecular physiology. 2013;304:L56–69. doi: 10.1152/ajplung.00213.2012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.