

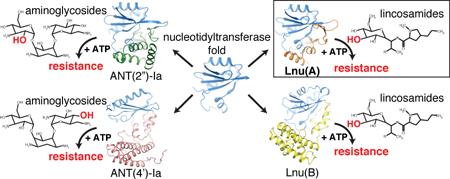
Abstract
One of the main mechanisms of resistance to lincosamide and aminoglycoside antibiotics is their inactivation by O-nucleotidylyltransferases (NTases). Significant sequence variation of lincomycin nucleotidylyltransferase (Lnu) and aminoglycoside nucleotidylyltransferase (ANT) enzymes plus lack of detailed information about the molecular basis for specificity of these enzymes toward chemically distinct antibiotic scaffolds hinders development of a general strategy to curb this resistance mechanism. We conducted an extensive sequence analysis identifying 129 putative antibiotic NTases constituting six distinct subfamilies represented by Lnu(A), (B), (C), (D), (F)/(G) plus ANT(2″) enzymes. Since only the Lnu(B) enzyme has been previously studied in detail, we biochemically characterized the Lnu(A) and Lnu(D) enzymes, with the former representing the most sequence-distinct Lnu ortholog. We also determined the crystal structure of the Lnu(A) enzyme in complex with a lincosamide. These data suggested that while sharing the N-terminal nucleotidylyltransferase domain (NTD), the groups of antibiotic NTases feature structurally distinct C-terminal domains (CTD) adapted to accommodate antibiotics. Comparative structural analysis among antibiotic NTases rationalized their specificity toward lincosamides vs. aminoglycosides through active site plasticity, which allows retention of general catalytic activity, while accepting alterations at multiple, specific positions contributed by both domains. Based on this structural analysis, we suggest that antibiotic NTases evolved from an ancestral nucleotidylyltransferase along independent paths according to the identified groups, characterized by structural changes in the active site and recruitment of structurally-diverse CTDs. These data show the complexity of enzyme-driven antibiotic resistance and provides a basis for broadly-active inhibitors by identifying the key unifying features of antibiotic NTases.
Keywords: antibiotic resistance, lincosamide, aminoglycoside, nucleotidylyltransferase, crystal structure, enzyme evolution
Graphical abstract
INTRODUCTION
Antibiotic resistance is an ongoing and serious clinical problem that threatens the use of existing and complicates the development of novel antibiotics [1]. The lincosamide and aminoglycosides (Fig. 1) represent prime examples of key antibacterials that have been compromised by resistance. Produced by various Streptomyces species, including S. lincolnensis, lincosamide antibiotics such as lincomycin (LCM) and clindamycin (CLI) bind to 23S rRNA in the 50S subunit of the ribosome and thereby interfere with the peptidyltransferase reaction [2]. Lincosamides are used for the treatment of infections caused by Gram-positive bacteria such as Staphylococcus aureus and Streptococcus pneumoniae [3]. The chemically distinct aminoglycosides also target the ribosome and affect translation fidelity in Gram-and Gram-negative pathogens [4].
Fig. 1. Chemical structures of lincosamides and aminoglycosides inactivated by O-nucleotidyltransferase enzymes.

A) Chemical structure of lincosamides. B) Chemical structure of kanamycin B, a representative 4,6-disubstituted 2-deoxystreptamine aminoglycoside. The O-nucleotidylation sites and nucleotidylylation product on both antibiotic classes are indicated with arrows and labelled by catalyzing enzyme family.
Despite their diverse chemical nature, both lincosamides and aminoglycosides are affected by enzyme-catalyzed O-nucleotidylylation that dramatically reduces antibiotic affinity for their target thus rendering bacteria resistant. Lincosamide resistance is conferred by O-nucleotidylyltransferases encoded by lnu (also called lin) genes. Mobile genetic elements carrying lnu genes have been identified in clinical isolates of various bacterial genera including Staphylococcus, Streptococcus, Enterococcus, Bacteriodes, Clostridium and Enterobacter. The identified lincosamide NTases have been designated as Lnu(A) through Lnu(G) and LinAN2 [5–11]. Detailed biochemical and structural data of Lnu enzymes are available only for the Lnu(B) (also known as LinB) protein [12]. This study revealed that the core structural architecture of Lnu enzymes is the nucleotidylyltransferase domain (NTD), a five- or six-stranded β-sheet flanked by three or more α-helices [13]. This fold comprises one face of an active site cleft. Aminoglycoside O-nucleotidylyltransferase encoding genes (known as ant or aad) have been identified in multiple genera of bacteria including pathogens such as Pseudomonas, Acinetobacter and Enterobacter [14]. ANT enzymes are classified into groups based on the position on the antibiotic molecule that comprises the modification site. Accordingly, ANT(2″), ANT(3″), ANT(4″), ANT(4′), ANT(6) and ANT(9) subfamilies of ANT enzymes are currently recognized, with ANT(2″) enzymes being the most widespread.
The Lnu and ANT enzymes were initially linked via a low level of observed sequence similarity [11], but the molecular basis for this sequence and functional similarity was only later revealed through the structural and functional characterization of ANT(2″)-Ia [15]. This work which showed that similarly with Lnu(B), the ANT(2″)-Ia enzyme features a NTD [15]. Crystal structures of other ANT enzymes (ANT(4′)-Ia, ANT(4′)-IIb and ANT(6)-Ia, PDB codes (1KNY [16], 4EBJ/4EBK and 2PBE, respectively) also revealed structurally similar NTDs, suggesting this to be the common domains among all antibiotic NTases. Detailed comparison of the Lnu(B) and ANT enzymes also showed that these enzymes share the key active site residues involved in catalysis, the orientation of ATP as the nucleotidyl donor and chelation of Mg2+ required for their activity [12,15]. NTase is thought to catalyse the nucleophilic attack by the hydroxyl group comprising the substrate modification site on the α-phosphate of ATP [15,17]. Similar structural features were also characterised for palm domain of DNA polymerase β prompting the speculation that Lnu(B) may have evolved from DNA polymerase enzymes [12].
The active center in Lnu(B) and ANT(2″)-Ia enzymes is localized to the cleft between the NTD and an additional C-terminal domain (CTD). In contrast to NTDs, the structures of Lnu(B) and ANT(2″)-Ia CTDs differ dramatically. Nevertheless, CTDs in both of these enzymes contributed to ATP binding and positioning of the antibiotic substrates appropriately for 3′-O-adenylylation (as in Lnu(B) and lincomycin) or 2″-O-adenylylation (as in ANT(2″)-Ia and kanamycin) [12,15]. Given the wide sequence diversity among antibiotic NTases, such functionally important diversity between Lnu(B) and ANT(2″)-Ia enzymes raises intriguing possibilities of additional structural architectures present in other Lnu and ANT enzymes. Furthermore, the molecular and evolutionary basis for how mechanistically related NTases adapted to recognize multiple antibiotic classes remains unexplained.
Here, we address the structural diversity and evolution of antibiotic NTases. Our extensive sequence analysis led to the identification of six distinct groups of potential antibiotic NTases. Biochemical characterization of Lnu(A) and Lnu(D) enzymes confirmed that despite the diversity in sequence they catalyze the formation of the same inactivation product with similar reaction rates. Furthermore, structural analysis of the Lnu(A) enzyme, which represented the most distant branch of Lnu enzymes compared to the previously-studied Lnu(B), revealed a conserved NTD domain combined with a structurally novel CTD, distinct from those characterized for the Lnu(B) and ANT enzymes. These data revealed unifying features responsible for catalysis of the same chemical reaction while highlighting the specific structural modifications responsible for determining lincosamide versus aminoglycoside specificity, and suggested multiple evolutionary emergence events for antibiotic NTases.
RESULTS
Analysis of Lnu sequences reveals six distinct subfamilies
To assess the sequence variation among Lnu enzymes we conducted an exhaustive search of GenBank for potential antibiotic NTases using characterized Lnu and ANT enzyme sequences as queries. This analysis led to the identification of 129 potential antibiotic NTase sequences present in diverse genera of pathogenic, non-pathogenic and environmental bacteria. We then used this subset of potential NTases enzyme sequences for comparative analysis and phylogenetic reconstruction (Fig. 2).
Fig. 2. Diversity in sequences of Lnu enzymes.

A) Multiple sequence alignment of Lnu and selected sequence homologs from the extensive BLAST search. Sequences are labelled by gi number and genus and are grouped/colored according to the phylogenetic reconstruction in Figure 2B. Secondary elements from crystal structures of Lnu(A), Lnu(B) and ANT(2″)-Ia are shown above each respective sequence and coloured by nucleotidyltransferase domain (light blue) and C-terminal domains (various colours). Boxes around the sequences indicate the three characteristic conserved motifs of the NTD. Stars and arrows above the sequences indicate magnesium-coordinating and antibiotic-interacting residues, respectively. The N- and C-termini of some sequences that are beyond the limits of Lnu(A) (i.e. C-terminal α-helix 7 of ANT(2″)-Ia) are hidden for clarity.
B) Phylogenetic reconstruction of all Lnu and sequence homologs identified through BLAST search. Selected genus and/or species origin of sequences are indicated. Key nodes defining the sequence clusters are labelled with their Bayesian probabilities. The percentages beside the Lnu/ANT(2″) enzyme names are sequence identity values relative to Lnu(A). Scale bar indicates number of amino acid changes per site.
As expected, the N-terminal region corresponding to the NTD in Lnu(B) structure represented the most conserved region across the entire set of retrieved sequences (Fig. 2a). More specifically, we observed high conservation of particular signature motifs in the NTD, including a glycine-rich motif localized to the α2 helix in the Lnu(B) structure [13], a motif with two conserved acidic residues in the β2 strand of the central β sheet, and a conserved acidic residue in β5 near the C-terminus of this domain. We also observed subfamily-specific sequence signatures in the NTD. For example, the glycine-rich motif was invariably represented by the [HY]WLDGGWGVD sequence in Lnu(A) and its closest homologs, while the corresponding region in Lnu(B) and its homologs featured a different, also highly conserved, CMMYGSFTKG motif. Another example was the distinction between the Lnu(A) and the ANT(2″)-Ia groups of sequences due to the invariant SRMEL versus YGFLA motifs localized to the β4 strand.
The region corresponding to the Lnu(B) CTD demonstrated significantly higher sequence divergence across the Lnu family and, consequently, we identified several sequence signatures in this region that were conserved in a subfamily-specific manner (Fig. 2a). Particularly, the Lnu(A) sequence and its homologs contained the Fxx[DE]WF[ST] motif, while the aligned region in Lnu(B) and its closest homologs featured the conserved DTKAM[FL]IY motif, but the ANT(2″)-Ia group of sequences contained a PxGxCPxx sequence. The ANT(2″)-Ia enzymes also possessed extra elements at the C-terminus not found in the Lnu(A) group.
Taking into account the subfamily-specific sequence signatures in the NTD and CTDs and especially the vast sequence diversity of the CTDs, a phylogenetic reconstruction supported the presence of distinct subfamilies of antibiotic NTases (Fig. 2b). The Lnu(A) sequence and its close homologs from Staphylococcus, Streptococcus, Bavariicoccus, Clostridium and Enterococcus formed a defined cluster. This clade also included Lnu(A)′, LinAn2 and Lnu(E) sequences, as was proposed previously [10].
According to our phylogenetic analysis, the Lnu(C)/Lnu(D) sequences belonged to a distinct clade closer aligned with ANT(2″)-Ia and the Lnu(A) groups (Fig. 2b). The ANT(2″)-Ia clade featured sequences from multiple genera including Citrobacter, Pseudomonas and Klebsiella plus sequences from the orders Deinococcales, Burkholderiales, Nostocales and Cytophalages. In line with such phylogenetic distribution, the Lnu(A) enzyme shared 27% and 19% primary sequence identity with Lnu(C)/Lnu(D) and ANT(2″)-Ia enzymes, respectively.
The Lnu(B) clade and the highly similar Lnu(F) and Lnu(G) enzyme groups were closely positioned [8] and occupied the opposite pole of the phylogenic reconstruction as compared to the cluster featuring Lnu(A)/Lnu(E) (Fig. 2b). Accordingly, the Lnu(A) and Lnu(B) enzymes shared only 9% identity which represented the most distantly related sequences within the retrieved sequence set. The phylogeny also placed other NT-fold containing enzymes including ANT(4′) and ANT(6) close to the Lnu(B) enzymes. Notably, these ANT enzyme classes shared on average only 3% sequence identity with Lnu(A).
Interestingly, each Lnu/ANT enzyme subfamily included a group of sequences from Actinobacteria. The Actinobacteria-derived sequences closest to the Lnu(B) and Lnu(F)/Lnu(G) subfamilies are annotated as DNA polymerases β in Genbank; this observation is agreement with the previously noted structural similarity between Lnu(B) and DNA polymerase β [12].
In total, this sequence analysis established that antibiotic NTases form discrete subfamilies and that the inter-subfamily pairwise sequence identity is very low. We speculated that the observed conserved motifs translate into distinct structural architectures and/or functional characteristics along the apparent subfamilies.
Lnu(A), Lnu(B) and Lnu(D) enzymes had common catalytic properties
To test if sequence-distant Lnu subfamilies retain common catalytic properties, we tested selective representatives for O-nucleotidylylation activity against lincosamides. Using the coupled nucleotidylylation-phosphatase activity assay established for Lnu(B) [12], we observed that the Lnu(A) and Lnu(D) enzymes have comparable activities against CLI using ATP as a nucleotidyl group donor (Table 1). Lnu(A), Lnu(B) and Lnu(D) shared comparable catalytic efficiency, low-μM affinity for LCM and CLI substrates, and mid-μM affinity for ATP. The Lnu(B) specificity in nucleotidylylation of the 3′-hydroxyl group of CLI was previously shown [12]. Using 1H-NMR we demonstrated that Lnu(A) and Lnu(D) also catalyzed the 3′-O-nucleotidylylation of LCM and CLI (Fig. S2).
Table 1.
Kinetic parameters of Lnu enzymes at pH 7.5
| Enzyme | kcat (s−1) | KM (μM) | kcat/KM (s−1M−1) |
|---|---|---|---|
| Lnu(A) | |||
| CLI | 0.35 ± 0.01 | 4.2 ± 0.4 | 8×104 |
| LCM | 0.42 ± 0.01 | 7.8 ± 0.5 | 5×104 |
| ATP | 0.54 ± 0.01 | 160.8 ± 9.7 | 3×103 |
| Lnu(D) | |||
| CLI | 0.13 ± 0.003 | 2.7 ± 0.2 | 5×104 |
| LCM | 0.13 ± 0.002 | 2.6 ± 0.3 | 5×104 |
| ATP | 0.063 ± 0.002 | 94.3 ± 15.2 | 7×102 |
| Lnu(B)a | |||
| CLI | 0.31 ± 0.0003 | 3.4 ± 0.44 | 9×104 |
| ATP | 0.6 ± 0.0005 | 217 ± 23 | 2.8×103 |
data for Lnu(B) are from reference 12 and shown for comparison to Lnu(A) and Lnu(D).
Thus, this clearly demonstrated that despite their significant sequence diversity and positions on the deduced phylogeny (Fig. 2), the Lnu(A), Lnu(B) and Lnu(D) enzymes share activity toward lincosamide substrates that would lead to inactivation of these antibiotics.
The crystal structure of Lnu(A) revealed a two-domain architecture featuring a novel CTD
To gain insight into the molecular basis of activity of Lnu enzymes and the effects of the considerable sequence diversity in this family, we undertook structural characterization of the Lnu(A) enzyme. The structure of Lnu(A) was determined using selenomethionine-enriched protein crystal by single anomalous dispersion (SAD) phasing and refined to 2.0 Å (Table 2). The Lnu(A) crystal’s asymmetric unit contained two peptide chains each spanning Lnu(A) residues 4 to 161 which adopted a nearly identical conformation (RMSD of 0.12 Å over 147 Cα atoms). In accordance with the sequence analysis, the N-terminal 96 residues of Lnu(A) formed a canonical NTD, featuring a central 4-stranded β-sheet and an overall secondary structure topology of α1-β1-α2-β2-α3-β3-β4-β5 [13] (Fig. 3a). In agreement with previously-described NTDs, the Lnu(A) β-sheet was curved to form a large active site cavity comprised of residues (D46, D48 and D90) localized to the β2 and β5 strands of the central β-sheet and belonging to the highly conserved sequence signatures (Fig. 2, 3a).
Table 2.
X-ray diffraction data collection statistics
| PDB Code | Lnu(A) apoenzyme 4FO1 | Lnu(A)-LCM complex 4WH5 |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c, Å | 57.4, 61.9, 61.3 | 56.4, 63.4, 60.1 |
| β, ° | 103.2 | 101.6 |
| Resolution, Å | 29.9 – 2.00 | 24.0 – 1.82 |
| Rsyma | 0.129 (0.569)b | 0.043 (0.174) |
| Rpimc | 0.056 (0.262) | 0.026 (0.110) |
| I/σ(I) | 15.8 (3.75) | 16.3 (2.04) |
| Completeness, % | 100 (100) | 99.8 (99.5) |
| Redundancy | 7.7 (7.7) | 3.5 (3.3) |
| Refinement | ||
| Resolution, Å | 29.9 – 2.15 | 23.4 – 1.82 |
| No. of reflections: working, test | 22329, 1109 | 37671, 1996 |
| R-factor/free R-factord | 21.8/26.2 (25.0/35.5) | 19.0/23.0 (25.4/31.2) |
| No. of refined atoms, molecules | ||
| Protein | 2638, 2 | 2671, 2 |
| Substrate | N/A | 54, 2 |
| Solvent | 60 | 5 |
| Water | 225 | 475 |
| B-factors | ||
| Protein | 52.8 | 27.1 |
| Substrate | N/A | 31.1 |
| Solvent | 72.8 | 42.2 |
| Water | 57.9 | 37.7 |
| R.m.s.d. | ||
| Bond lengths, Å | 0.003 | 0.013 |
| Bond angles, ° | 0.763 | 1.424 |
.
Figures in parentheses indicate the values for the outer shells of the data
.
.
N/A = not applicable.
Fig. 3. Structure of Lnu(A).

A) Structure of apo Lnu(A) with secondary structure elements labelled and HEPES molecule (sticks) bound in the active site. A single chain of the dimer is shown. The nucleotidyltransferase (residues 4-96) and C-terminal (residues 133-161) domains are coloured in light blue and orange, respectively. B) Structure of Lnu(A) – LCM complex. The dimer is shown, with one protomer coloured as in Fig. 1a, the second coloured in white. Tyrosine-144, which forms an interaction to the substrate binding cavity, is shown in sticks. Elements from chain B are labelled with an apostrophe. LCM molecules are shown in sticks. Magnesium ions are shown as red spheres.
C-terminal to the NTD (residues 97 to 161), the Lnu(A) structure featured four additional β-strands forming two β-hairpins (β6-β7 and β8-β9) connected by a small α-helix (α4) and a two α-helix bundle (α5, α6) (Fig. 3a). In keeping with the classification of NT-fold containing enzymes [13], we considered this portion of the Lnu(A) structure to be the CTD.
The interface between the two Lnu(A) protomers in the asymmetric unit covered 1032 Å2 suggesting that this association represents a stable dimer (Fig. 3b). In agreement with this observation, size exclusion chromatography showed that Lnu(A) formed dimers and potentially higher oligomers in solution (Table S1). Notably, the homodimeric interface in the Lnu(A) crystal was formed exclusively through interactions between CTDs of the protomers with a majority of interdomain interactions hydrophobic in nature.
The Lnu(A)-LCM complex structure highlighted the specific roles of conserved residues in the NTD and CTDs
Overall, the Lnu(A) protomer structure adopted a “Pacman” shape, with a large (1681 Å3) and prominent acidic cavity between the NTD and CTDs (Supplemental Material Fig. S1). In line with this cavity harboring the Lnu(A) active center, the Lnu(A) apoenzyme structure contained additional electron density corresponding to a HEPES molecule trapped within this cavity (Fig. 3a).
To characterize Lnu(A) interactions with its substrates (Fig. 1a) we determined the structure of Lnu(A) in complex with LCM. This crystal structure was solved by Molecular Replacement using the Lnu(A) apoenzyme structure as a model (Table 2). As expected, well-defined electron density corresponding to the LCM molecule was found in both cavities of the Lnu(A) dimer at the position overlapping with that of HEPES molecule found in the Lnu(A) apoenzyme structure (Fig. 3a, 3b). We also observed electron density that was modeled as two magnesium ions in the Lnu(A)-LCM complex’s active site (Fig. 3b).
The positions of the LCM molecules varied between the two chains of the Lnu(A) dimer (Fig. S3a). While the propylproline (hydrophobic) regions of LCM bound to the two Lnu(A) active sites superimposed well, the methylthiolincosamide (polar) moieties adopted alternative conformations; the two LCM molecules could be related by a rotation about the plane of the propylproline ring and about the C6-N8 bond linking the propylproline and methylthiolincosamide regions. This alternative conformation of LCM, as bound to the Lnu(A)chain A active site, resulted in six protein-LCM hydrogen bonds (Fig. S3b) while the Lnu(A)chain B active site participated in seven hydrogen bonds with LCM (Fig. 4). Such variation in substrate conformation was likely due to the absence of the nucleotide co-substrate that is necessary for catalysis.
Fig. 4. Lincosamide recognition by Lnu(A).

Details of interactions between Lnu(A) and LCM (chain B complex). Residues shown form hydrogen bonds or hydrophobic interactions with LCM and are grouped according to their presence in the NTD (blue arc) or CTD (orange arc). Electron density for LCM shown is a FO–FC omit map contoured at 2.0 σ. Dashes indicate hydrogen or coordination bonds.
Given that the position of lincomycin in the Lnu(A)chain B active site better resembled the binding conformation of clindamycin in the ternary Lnu(B)-clindamycin-AMPCPP crystal structure [12] (see the following section) and that D90 is the putative catalytic acid/base in Lnu(A) by analogy with studies of ANT(2″)-Ia [15], we focused on the detailed analysis of this enzyme-substrate complex as the most probable functional Lnu(A)-lincosamide complex (Fig. 4). This analysis showed that both the NTD and CTD contributed to interactions with the LCM molecule. On the substrate side, the interactions mainly involved the hydroxyl groups on the methylthiolincosamide sugar moiety: the 3′-OH formed hydrogen bonds with the sidechains of D48, R78 and D90 residues and coordinated a magnesium cation. This magnesium cation in the Lnu(A) chain B-LCM complex structure was highly-coordinated (with sidechains of the D46, D48 and D90 residues and three ordered water molecules). Thus the coordination sphere of this magnesium cation was complete; in contrast, the second magnesium cation in the active site was coordinated by only five ligands, including the D46 and D48 side chains and three water molecules.
The Lnu(A) residues F114, W118, F119, I132, A136 and F140 belonged to the CTD of the protein and formed a hydrophobic pocket which accommodated the propylproline moiety of LCM molecule (Fig. 4). In addition, the sidechain of Y144′ (from the other protomer in the Lnu(A) dimer) reached into the active site to form hydrogen bonds with the substrate, consistent with the presence of dimeric Lnu(A) in solution.
Observed interactions between Lnu(A) and the LCM substrate rationalized the sequence conservation patterns we identified in this enzyme and its close homologs (Fig. 2a). Specifically, residues forming the propylproline-binding hydrophobic pocket (i.e. W26, F114, W118, F119, I132 and F140) and those providing hydrogen bonds to the methylthiolincosamide moiety (i.e. D48, D90 and H92) are all highly conserved in the Lnu(A) subfamily.
While we were unsuccessful in obtaining crystals of Lnu(A) bound to ATP or its analogs, the Lnu(A) chain B-LCM complex contained sufficient unoccupied volume (1400 Å3) in the active site to accommodate a nucleotide moiety. This space is appropriately located for the nucleotidylylation reaction near α2, α2-β2 loop and the β2 structural elements of Lnu(A), the Mg2+ ions and the methylthiolincosamide group of LCM. Nucleotide binding likely displaces at least some of the waters contributing to coordination of the Mg2+ ions.
Overall, our structural data showed that the Lnu(A) NTD mediated interactions important for orientation of active site magnesium ions and the polar region of the LCM substrate, while the CTD provided the hydrophobic environment for the corresponding region of the antibiotic.
Comparative analysis of Lnu(A) and Lnu(B) structures defined the role of the distinct C-terminal domains
Having characterized the bi-domain molecular structure of the Lnu(A) enzyme and the specific roles played by its NTD and CTD in antibiotic recognition, we next conducted comparative structural analysis between Lnu(A) and Lnu(B). In agreement with primary sequence conservation, Lnu(A) and Lnu(B) protomer structures superposed well via their NTDs with RMSD of 3.5 Å over matching Cα atoms (Lnu(A) residues 4-96 and Lnu(B) residues 2-94) (Fig. 5). As expected, the superimposable regions in the Lnu(A) and Lnu(B) NTD domains included the central β-sheet and accompanying α-helices with near-perfect superposition of residues involved in coordination of Mg2+ cations (Fig. 5). In contrast, the CTDs of Lnu(A) and Lnu(B) showed no structural similarity. The two β-hairpins and two α-helices in the CTD of Lnu(A) had no equivalents in Lnu(B), which is predominantly α-helical.
Fig. 5. Comparison of antibiotic recognition modes by Lnu(A), Lnu(B) and ANT(2″)-Ia.

A) Overall structures of Lnu(A)-LCM, Lnu(B)-CLI (PDB ID: 3JZ0) and ANT(2″)-Ia-KAN (PDB ID: 4WQK) complexes. The nucleotidyltransferase domains (NTD) are coloured light blue and the C-terminal domains (CTD) in orange, yellow and green, respectively, according to the enzyme. The antibiotic substrates are shown in sticks, the magnesium ions as red spheres and the nucleotidylation sites on the antibiotic molecules are labelled with arrows. The N- and C-termini are indicated with N and C, respectively.
B) Detailed comparison of enzyme-antibiotic interactions. Views are in the same orientation as Fig. 5A but zoomed in and amino acid sidechains that form hydrogen bonds or hydrophobic interactions with antibiotics are shown in sticks. For Lnu(B), the bound AMPCPP molecule is shown in ball-and-stick in light grey.
Despite significant structural differences, both Lnu(A) and Lnu(B) accommodated lincosamide substrates in the generally similar area of their central active site cavities formed between the NTD and CTDs (Fig. 5). Most importantly, in the catalytically competent position, the 3′-OH group of the lincosamide molecule representing the modification site occupied virtually the same position in both Lnu(A) and Lnu(B) structures even though much of the remainder of the lincosamide molecules adopted substantially different relative conformation in the Lnu(A) and Lnu(B) active sites. In particular, the propylproline moieties of the LCM and CLI substrates occupied distinct hydrophobic binding pockets in Lnu(A) and Lnu(B) due to conformational differences in the lincosamide molecules (Fig. 5b). In addition, Lnu(B) residues Y27 and Y44 cradled the central region of the CLI molecule, allowing for its twisted conformation while the equivalent residues in Lnu(A) - D28 and D50 are not suited for such a role (Fig. 5b). Finally, CTD residues participating in the hydrophobic interactions with the substrate did not match to the same specific locations in Lnu(A) and Lnu(B) structures (i.e. Lnu(B) F104, I110, also reflected in the primary sequence alignment, Fig. 2a).
The comparison of the Lnu(B)-CLI-AMPCPP with Lnu(A)-LCM complexes suggested a position for nucleotide binding in Lnu(A) that is consistent with the location previously noted as being empty and accessible for binding ATP (i.e. near the methylthiolincosamide moiety of LCM and the Mg2+ ions in Fig. 5b). Interestingly, there are distinctions in the mode of nucleotide recognition by these two Lnu enzymes: the residues in Lnu(B) that interact with the phosphates of AMPCPP are not observed in Lnu(A). The second chain in the dimer of Lnu(B) contributed R165 and R170 from its CTD for this role [12] but these residues are absent in Lnu(A), since the dimerization interface of this enzyme is different. Instead, this role could be fulfilled by Lnu(A) R42 or R45 which are found in the NTD (not shown). The precise binding location of ATP requires experimental evidence.
Thus this new structural data pointed to the presence of at least two distinct lincosamide recognition mechanisms supported by structurally different Lnu enzymes, and likely details of nucleotidyl (i.e. phosphates) recognition modes. Nonetheless, these structural distinctions in ligand recognition modes are compatible with the same catalytic reaction.
NTase specificity towards distinct antibiotic classes is conferred by key alterations of the substrate binding cleft chemical environment
Having established that Lnu enzymes show structural diversity especially in their CTDs, we were interested in a structural comparison with the ANT(2″) class of antibiotic NTases. Recently we described the crystal structure of ANT(2″)-Ia [15] (PDB IDs: 4WQK, 4WQL).
We showed that the overall structural similarity between Lnu(A) and ANT(2″)-Ia enzymes extended into the catalytic apparatus (including the superimposable positions of residues involved in coordinating Mg2+ cations, catalysis and positioning of the substrate hydroxyl group for nucleotidylylation, i.e. Lnu(A) D48, D48, D90 vs. ANT(2″)-Ia D44, D46 and D86 and the near-equivalent positioning of the substrate nucleotidylylation sites (Fig. 5a and [15]). Such close structural similarity between Lnu(A) and ANT(2″)-Ia enzymes invited a detailed comparative analysis of their active centers in order to identify the molecular features responsible for differential antibiotic substrate specificity.
This analysis revealed that despite largely similar general structural features, the ANT(2″)-Ia and Lnu(A) enzymes displayed significant variation across multiple residues contributing to their active sites (Fig. 5b). These changes resulted in dramatically different local chemical environment closely tailored to the chemical natures of the antibiotic substrate. Specifically, active site residues from the NTD do not show conservation between the two enzymes: Lnu(A) S77 and H92 residues from the β4 and β5 strands, forming the “top” of the antibiotic binding site, corresponded to ANT(2″)-Ia Y74 and E88. The H92 residue in Lnu(A) formed a hydrogen bond to the 4′-OH of lincomycin (Fig. 4, Fig. 5b) but the corresponding E88 residue in ANT(2″)-Ia adopts a conformation to position the central 2-deoxystreptamine ring of kanamycin (Fig. 5b). The Y74 residue of ANT(2″)-Ia provides a stacking interaction with the 2-deoxystreptamine ring of kanamycin (Fig. 5b) but the corresponding residue in Lnu(A), S77, is incapable of forming such an interaction to an antibiotic substrate. Additionally, there are specific sequence substitutions in the CTD of these two enzymes. Lnu(A) F140 and S142, residues belonging to the α5 helix and contributing to the “bottom” of the active site cavity, corresponded to D131 and Y134 in the ANT(2″)-Ia structure (Fig. 5b). F140 from Lnu(A) participates in the hydrophobic pocket for binding of the propylproline moiety of lincomycin but the corresponding residue of ANT(2″)-Ia, D131, is positioned to interact with the 2-deoxystreptamine ring of kanamycin. The ANT(2″)-Ia Y134 residue is stacked against the prime ring of kanamycin while the equivalent S142 residue in Lnu(A) is not capable of such an interaction. Furthermore, the hydrophobic pocket in Lnu(A) for binding of the propylproline region of lincomycin does not exist in ANT(2″)-Ia due to numerous other sequence substitutions. Along with these key sequence substitutions in the nucleotidylyltransferase active centers of Lnu(A) and ANT(2″)-Ia, the two enzymes differ in conformation of the loop between α5 and α6, and ANT(2″)-Ia contains an additional C-terminal α-helix (α7) that packs against these helices (Fig. 5a). The loop in Lnu(A) contains the residue Y144′ which contributes to lincomycin binding across the dimer interface (Fig. 4); ANT(2″)-Ia does not contain any equivalent residue. As well, this region participates in the dimerization of Lnu(A) (Fig. 3b) and the presence of this α-helix would obstruct dimerization of ANT(2″)-Ia; this is in agreement with the observation of exclusively monomeric ANT(2″)-Ia in solution [15]. These represent additional significant structural adaptations that differentiate substrate specificity.
Generally, this comparative analysis pointed to the specific sequence adaptations in the nucleotidylyltransferase active site in addition to structural modularity of the CTDs as the primary determinants allowing the structurally similar ANT(2″)-Ia and Lnu(A) enzymes to expedite specificity toward different antibiotic scaffolds. Extending this analysis to the other representatives of ANT and Lnu enzyme families we suggest that the common N-terminal domain in these enzymes provides the necessary molecular features for the nucleotidylylation reaction. However, adaptation to specific substrates could be realized through subtle modulations of the chemical environment in the central cavity as well as recruitment of structurally diverse C-terminal motifs.
DISCUSSION
Antibiotic-modifying enzymes are key sources of resistance to antibacterial drugs in the clinic [18]. Although development of specific inhibitors targeting such enzymes has been shown to restore the susceptibility of the pathogens to antibiotics, a strategy best exemplified by β-lactamase inhibitors (reviewed in [19]), our advancement on this path is complicated by the lack of comprehensive molecular information about resistance mechanisms. Resistance-conferring enzymes often comprise large, diverse and widespread protein families with understudied molecular complexity. Gaining molecular insight on this diversity, as well as on the evolution and origins of resistance enzymes, is necessary to facilitate the rational design of new, less resistance-prone antibacterials and development of tailored inhibitors of modifying enzymes.
In this study, we investigated the sequence, structure-function, and evolutionary variation among antibiotic NTases that confer resistance to lincosamides and aminoglycosides. Previous reports on identification of lnu and ant genes for these enzymes pointed to the presence of a widespread and diverse family, which, however, has remained mostly uncharacterized. Through extensive sequence similarity search of GenBank, we were able to expand the lincosamide NTase family to over 120 potential enzymes from a set of 8 experimentally-validated Lnu enzymes. Phylogenetic reconstruction among identified members of this family revealed the presence of distinct subfamilies, represented by the Lnu(A)/Lnu(E), Lnu(B), Lnu(F)/Lnu(G) and Lnu(C)/Lnu(D) enzymes, respectively. Our phylogenetic analysis also confirmed the relationship between lincosamide and aminoglycoside NTases, with the most similar group of latter enzymes represented by the clinically relevant variant ANT(2″)-Ia. Other aminoglycoside NTase groups represented by ANT(4′)-Ia, ANT(4″)-Ib and ANT(6)-Ia also could be placed on our phylogenetic reconstruction near the Lnu(B) and Lnu(F) enzyme clades.
Functional characterization of Lnu(A) and Lnu(D) established that a high variation in sequence does not translate into different chemistry, and that both orthologs catalyze the same reaction as the previously-characterized Lnu(B) enzyme. Structural characterization of Lnu(A) allowed for rationalization of this functional similarity despite the structural diversity of Lnu enzymes. The crystal structure showed a highly conserved N-terminal NTD and this domain featured conserved residues that are directly implicated in catalysis in similar positions to those in Lnu(B) and ANT(2″)-Ia [12,15]. These residues were involved in the proper positioning of the modified 3′-OH group of the lincomycin substrate, the chelation of magnesium cations necessary for coordinating nucleotide and providing a potential catalytic base [15] for the reaction.
Accordingly, the lincosamide and aminoglycoside substrates adopted a similar general position in the active site clefts of Lnu(A), Lnu(B) and ANT(2″)-Ia, formed between the NTD and CTDs. The shared molecular features of the NTDs of the antibiotic NTases likely account for their comparable catalytic properties despite the drastic structural diversification of the CTDs of Lnu(A) and Lnu(B) and the multiple active site sequence variations compared to ANT(2″)-Ia. Our structural analysis also showed that significant sequence deviation in the CTD of the Lnu(A) enzyme translated into a dramatic structural diversification of this domain compared to Lnu(B). Thus, antibiotic NTases demonstrated significant diversity within the general theme of a two-lobe structural architecture; a highly conserved NTD is combined with structurally diverse CTDs able to support substrate O-nucleotidylylation. Since Lnu(C)/Lnu(D) and Lnu(F)/Lnu(G) groups of antibiotic NTases also show significant sequence variation in their C-terminal portion, we anticipate that these enzymes feature yet another structurally distinct CTD fold.
The high level of sequence dissimilarity suggested ancient divergence of the different Lnu and ANT enzyme groups identified by our phylogenetic analysis, particularly for the subfamilies at opposite poles of the tree. The distinct Lnu and ANT subfamilies likely originated from ancient nucleotidylyltransferases which facilitated transfer of an AMP group to metabolite substrates that did not necessarily possess anti-bacterial activity and subsequently diverged along independent paths (i.e. the Lnu(B) group evolved from DNA polymerase β, while the Lnu(A) or ANT(2″)-Ia groups evolved from an as-yet unknown ancestor). This concept was put forward in a larger analysis of the nucleotidylyltransferase superfamily [13]. The Actinobacteria NTase homologs identified by phylogenetic reconstruction for each of the clusters of bona fide antibiotic-active Lnu and ANT(2″) sequences could represent the environmental “reservoirs” from which antibiotic NTase enzymes independently evolved. In the face of antibiotic exposure, selective pressure would favour genetic events leading to alterations providing these ancestral enzymes to adapt their activity towards the distinct lincosamide and aminoglycoside antibiotic scaffolds, such as the features uncovered by structural analysis of the modern Lnu and ANT enzymes.
This hypothesis reflects with the “protoresistance” concept that has been put forward to explain the evolution of many different antibiotic-modifying enzymes, including aminoglycoside phosphotransferases (APH), streptogramin B acetyltransferases, β-lactamases, macrolide glycosyltransferases [20] and for the Lnu(B) enzyme [12]. Each of these resistance enzyme families show structural similarity with non-resistance-conferring enzymes; accordingly, the targeted antibiotic substrates possess chemical similarities with the non-antibiotic substrates of these ancestors. Thus, ancestral Lnu and ANT enzymes may have operated on “lincosamide-like” and “aminoglycoside-like” compounds, respectively. The substrates of these ancestral enzymes likely shared chemical similarity in their hexose rings that contacted the nucleotidylylation apparatus, but were chemically distinct elsewhere.
Horizontal transfer of antibiotic resistance genes from environmental to pathogenic bacteria is a well-established phenomenon that could explain this evolution of NTases [21]. The transfer of lincosamide resistance genes from Actinobacteria has been suggested as the most probable pathway for rise of lincosamide resistance [22]. Yet, to date there has been no detection of a Streptomyces lincolnensis-derived Lnu enzyme, or O-nucleotidylylation inactivation of lincosamides in this antibiotic producer. However, the presence of LCM O-NTase in the other Streptomyces species such as S. coelicolor has been reported [23]. Our genomic analysis also identified such enzymes, including the S. coelicolor gene with GenBank accession number NP_624450 (gi 21218671 in Fig. 1a) that may encode a similar enzyme. Detailed characterization of activity of this and other Actinobacteria-derived NTases may provide the missing links in the reconstruction of the emergence and evolution of antibiotic NTases.
Structural characterization of the Lnu(A) enzyme pointed to the presence of additional homologous enzymes that may have similar NTase activity. Notably, a similarity search identified the structures of the hypothetical protein TM1012 from Thermatoga maritima and a putative NTase JHP933 from Helicobacter pylori as close homologs of Lnu(A) (Table 3, Fig. S4b) [24,25]. TM1012 matched particularly closely, with both proteins sharing nearly identical NTD and CTDs (Fig. S4a). Although comparative analysis indicated that JHP0933 and TM1012 proteins lack some of the conserved lincosamide-interacting residues, these proteins could be small molecule NTases with as-yet unknown substrate specificity. The relevance of the structural homology between these enzymes and Lnu(A) would be advanced by characterisation of the substrate specificity of these putative NTases.
Table 3.
Representative top structural homologs of Lnu(A)
| PDB IDa | Protein name | Z-score | RMSD (# Cα atoms) | % identity |
|---|---|---|---|---|
| 4WQK | ANT(2″)-Ia | 16.2 | 2.3 (145) | 18 |
| 2FCL | T. maritima hypothetical protein TM1012 | 12.3 | 3.3 (157) | 15 |
| 4OK0 | H. pylori putative NT JHP933 | 9.3 | 2.8 (125) | 16 |
| 3UQ0 | (euk.)b DNA pol λ | 7.7 | 3.2 (101) | 12 |
| 4F5Q | (euk.) DNA pol β | 7.6 | 3.9 (105) | 10 |
| 4NKT | (euk.) poly (U) polymerase | 7.3 | 3.9 (100) | 12 |
| 1UEV | (arch.) CCA-adding enzyme | 7.0 | 4.4 (118) | 7 |
| 4M04 | (euk.) DNA poly μ | 6.8 | 3.9 (103) | 13 |
| 4K99 | (euk.) cGMP-AMP synthase | 6.5 | 4.4 (116) | 8 |
| 3NYB | (euk.) pola (A) RNA polymerase | 6.4 | 3.4 (86) | 8 |
| 3JZ0 | E. faecium Lnu(B) | 6.4 | 3.7 (107) | 11 |
| 1JMS | (euk.) terminal DNA NT | 6.3 | 4.0 (106) | 10 |
| 4ATB | (euk.) NF90 | 5.9 | 4.7 (114) | 7 |
| 1MIV | B. stearothermophilus CCA-adding enzyme | 5.9 | 3.8 (113) | 12 |
| 2NRK | E. faecalis GrpB | 5.7 | 4.0 (94) | 10 |
| 1ZEL | M. tuberculosis RV2827C | 5.4 | 3.7 (105) | 13 |
| 1NO5 | H. pylori HI0073 NT | 3.5 | 4.8 (102) | 11 |
| 2PBE | B. subtilis predicted ANT(6)-Ia | 4.4 | 3.2 (88) | 9 |
| 3L9D | S. mutans Smu.1046C | 4.1 | 3.6 (94) | 6 |
| 4EBK | ANT(4′)-IIb | 3.6 | 4.2 (78) | 9 |
| 1KNY | ANT(4′)-Ia | 3.1 | 3.6 (73) | 12 |
multiple PDB IDs of the same protein were often retrieved; indicated stats are from those PDB IDs with the highest Z-score).
euk. = eukaryotic; arch. = archeal.
Finally, the observed conservation of the NTD catalytic apparatus between otherwise diverse Lnu(A), Lnu(B) and ANT(2″)-Ia enzymes points to a possible strategy for development of broad-spectrum small molecule entities targeting this class of antibiotic resistance enzyme. Indeed, the recent characterization of α-hydroxytropolone compounds that inhibit the activity of ANT(2″)-Ia implied that these compounds compete for binding to the Mg2+-ATP binding site of this enzyme [26]. In this regard, the presented structural information will be important in evaluating the suitability of these compounds for inhibition of this lincosamide resistance enzyme. In particular, a comparison of the Mg2+-AMPCPP-coordinating residues of Lnu(B) [12] with the equivalent region of Lnu(A) showed very close similarity in the position and coordination of the Mg2+ ions on one face of the ATP binding site but we could not observe any apparent structural similarity in the ATP phosphate tail binding site or on the region contributed by the CTDs. Thus, future antibiotic NTase inhibitor discovery efforts should focus on manipulation of the binding of Mg2+ ions.
In summary, the data presented here underline the modularity and versatility of the NTase fold in mediating resistance to chemically diverse antibiotic compounds. According to our analysis, antibiotic active NTases evolve through molecular modulation of the catalytic site and engagement of structurally diverse C-terminal elements. Our findings illustrate the immense challenge facing antibacterial drug discovery and optimization of antibiotics, as selective pressure exerted by antibiotics can lead to multiple adaptive responses and modification of pre-existing catalytic capabilities towards resistance functionalities.
MATERIALS AND METHODS
Cloning, expression and purification of Lnu(A) and Lnu(D)
For biochemical analysis, the lnu(A) sequence from Staphylococcus haemolyticus plasmid pIP855 [5] and the lnu(D) sequnce from Streptococcus uberis [11] were synthesized with codon sequence optimized for Escherichia coli expression and subcloned into the pET28a expression vector (Novagen). For structural analysis, the lnu(A) sequence was cloned by ligase-independent cloning into the p15Tv-LIC vector [27], providing a N-terminal His6-tag fusion followed by a TEV protease cleavage site.
For biochemical analysis, lnu(A) and lnu(D) were expressed in E. coli BL21(DE3), grown to an OD600 of 0.6 at 37°C, chilled to 16°C and induced overnight with 500 μM isopropyl β-D-thiogalactopyranoside. Cells were harvested via centrifugation at 5,000g and pellets stored at −20°C. For structural analysis, selenomethionine (Se-Met)-substituted lnu(A) was expressed using the standard M9 high yield growth procedure according to the manufacturer’s instructions(Shanghai Medicilon), with E. coli BL21(DE3) codon plus cells. All enzymes were purified by Ni-affinity chromatography. Cells were resuspended in binding buffer (50 mM HEPES pH 7.5, 100/300 mM NaCl, 10 mM imidazole, and 2% glycerol (v/v)), lysed using a cell disrupter or with a sonicator and cell debris were removed via centrifugation at 30000g. Cleared lysate was loaded onto a 5 mL Ni-NTA column (QIAGEN) pre-equilibrated with binding buffer, extensively washed with binding buffer containing 30 mM imidazole and proteins were eluted using the above buffer with 250 mM imidazole. For structural analysis, the His6 tag was removed by cleavage with TEV protease overnight at 4°C in dialysis with buffer 0.3 M NaCl, 50 mM HEPES, pH 7.5, 5% glycerol and 0.5 mM tris[2-carboxyethyl]phosphine, followed by binding to Ni-NTA resin and capture of flow-through. Fractions containing the protein of interest were identified by SDS-polyacrylamide gel electrophoresis and further purified via gel filtration on a HiLoad 16/60 Superdex75 prep grade column (10 mM HEPES pH 7.5, 50 mM KCl).
Lnu(A) and Lnu(D) kinetic analysis
Enzyme activity was measured using a continuous EnzChek® Pyrophosphate assay following the Invitrogen protocol. The reaction was set up in 96-well flat bottom plates (Nalge NUNC Int.) and reaction progress was monitored for 10 min at 360 nm. The total volume of the reaction mixture was 250 μL, and the reaction buffer was 50 mM HEPES (pH 7.5), 100 mM KCl and 2 mM MgCl2. The reaction was initiated using 10 μl of either clindamycin (CLI), lincomycin (LCM), or ATP. When monitoring CLI or LCM dependence, 500 μM ATP was used. Final concentrations of CLI ranged between 0.78 and 50 μM and those of LCM between 0.78 and 100 μM. For ATP characterization, 30 μM CLI was used and the ATP concentration ranged from 15 μM to 1 mM. Four μg of protein were used per reaction. LCM and CLI were purchased from Sigma Aldrich. The kinetic data were analyzed using Grafit 4.021 (Erithacus Software, Staines, UK) and GraphPad Prism software (GraphPad Software Inc., La Jolla, USA).
Lnu(A) and Lnu(D) reaction product synthesis and purification
The reaction mix contained 50 mM HEPES pH 7.5, 100 mM KCl, 2 mM MgCl2, 10 mM ATP, 10 mg CLI and 1 mg/mL enzyme. The final reaction of volume 4 mL was incubated overnight at room temperature, lyophilized, manually purified using SepPak-C18 column equilibrated with 100% methanol and then water. Sample was resuspended in water and eluted in 5 mL steps: water 50% acetonitrile in water, and 100% acetonitrile. The products were eluted in 50% acetonitrile.
NMR analysis of Lnu(A) and Lnu(D) reaction products
1D and 2D NMR experiments were performed using a Bruker AVIII 700 MHz instrument equipped with a cryoprobe in deuterated water. Chemical shifts are reported in parts per million and calibrated using the residual tetramethylsilane solvent signal at 4.65 ppm.
Characterization of Lnu(A) and Lnu(D) oligomeric state via analytical gel filtration
The oligomeric state of the Lnu(A) and Lnu(D) enzymes in solution was determined via analytical gel filtration using a Superdex 200 10/300 GL column as per the manufacturer’s instructions (GE Healthcare). The column was calibrated using a gel filtration LMW calibration kit. The elution buffer for the standards and the enzymes was 50 mM sodium phosphate (pH 7.2) 150 mM NaCl. Additionally, Lnu(A) experiments were repeated in buffer containing 20 mM HEPES (pH 7.5) and 100 mM KCl.
Lnu(A) crystallization, X-ray diffraction data collection and structure determination
Se-Met Lnu(A) (apoenzyme) was crystallized at room temperature using the hanging drop method, with 2 μL of 13 mg/mL protein solution mixed with 2 μL of reservoir solution (1.4 M trisodium citrate dihydrate, 0.1 M HEPES pH 7.3). Se-Met Lnu(A)-LCM was obtained by co-crystallization using the same reservoir solution as the apoenzyme crystal supplemented with 5 mM LCM. Both crystals were cryoprotected with paratone oil.
For the Se-Met Lnu(A) (apo) crystal, diffraction data at 100 K were collected at the Advanced Photon Source (APS), Argonne National Laboratory, Life Sciences Collaborative Access Team beamline 21-ID-F, fitted with a MarMosaic 225 CCD, at the selenium absorption peak (0.97872 Å). For the Lnu(A)-LCM complex crystal, diffraction data at 100 K were collected at the Structural Genomics Consortium on a Rigaku Micromax-007 HF rotating copper anode source with a Rigaku Saturn A200 CCD. All X-ray data were reduced with HKL-3000 [28]. The Lnu(A) (apo) structure was solved by single anomalous dispersion (SAD) phasing using Phenix.autosol [29] which identified six of the eight selenium sites in the asymmetric unit (two protein chains, each with four selenomethionine residues). An initial model of the protein was built using Phenix.autobuild, followed by rounds of manual model building and refinement with Coot [30] and Phenix.refine. The structure of the Lnu(A)-LCM complex was solved by molecular replacement using the Lnu(A) (apo) as a search model in Phenix.phaser. The presence of LCM molecules was validated using omit maps: all atoms of the ligand plus other atoms within 5 Å of the ligand were deleted, followed by simulated annealing (Cartesian) using Phenix.refine with default parameters, followed by model building into residual positive FO – FC density. For both structures, B-factors were refined as anisotropic for Lnu(A) atoms and isotropic for non-protein atoms. TLS parameterization (groups were chain A residues 4-94, 95-161, and chain B residues 4-94, 95-161) was included in final rounds of refinement for both structures. The final atomic models of Lnu(A) (apo) and Lnu(A)-LCM complex included residues 4-161 of each of the two chains in the asymmetric unit for each structure. Average B-factor and bond angle/length rmsd values were calculated using Phenix. All geometry was verified using the Phenix and Coot validation tools plus the PDB Adit server. The structures have good backbone geometry with the following percentage of residues in the most favored, additional allowed, generously allowed and disallowed regions, respectively, of the Ramachandran plots: Lnu(A) (apo): 92.5, 7.5, 0 and 0%; Lnu(A)-LCM: 94.0, 5.2, 0.7 and 0%.
Lnu(A) structure analysis
Structure similarity searches of the Protein Data Bank were performed using the PDBeFold and Dali servers [31,32]. Structure superpositions were performed with the Mustang algorithm [33]. Interactions between Lnu(A) and LCM were identified in Coot or PyMOL [34]. Protein-protein interfaces were determined using the PDBePISA server [35]. Electrostatic potential surfaces were calculated using PyMOL and APBS [36]. The Lnu(A) active site cavity was identified and analyzed using the CASTp server [37].
Lnu protein sequence analysis
BLAST searches of the NCBI non-redundant database were conducted using Lnu(A), Lnu(B), Lnu(C), Lnu(D), Lnu(E), Lnu(F), Lnu(G), Lnu(A)′, LinAn2 and ANT(2″)-Ia sequences as queries and hits matching over 75% of the length of the hit and query sequences were retained. Partial, incomplete, redundant sequences or those from uncultured bacteria were discarded. Sequences were aligned with the Muscle algorithm within the Jalview package [38], followed by manual verification, using a structural superposition between Lnu(A) and Lnu(B) generated by Mustang [33] as a guide. The multiple sequence alignment was visualized and edited with Jalview [38]. Phylogenetic reconstruction was performed using MrBayes [39] and the tree was visualized with FigTree (http://tree.bio.ed.ac.uk/software/figtree).
Supplementary Material
Highlights.
Lnu O-nucleotidylyltransferases (NTases) confer resistance to lincosamides
Antibiotic NTases form six sequence-diverse groups
Lnu(A) structure reveals key molecular features conferring antibiotic specificity
Structural analysis shows antibiotic NTases contain distinct C-terminal domains
These findings illustrate the active site plasticity of antibiotic NTases
Acknowledgments
We thank V. Yim and O. Egorova for technical assistance. We thank A. Dong, Structural Genomics Consortium and Z. Wawrzak, Life Sciences Collaborative Access Team/Advanced Photon Source, for x-ray data collection and/or structure solution. The structures presented were solved by the Center for Structural Genomics of Infectious Diseases (CSGID, http://csgid.org); this project has been funded in whole or in part with U.S. Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272200700058C (2007 to September 27, 2012) and HHSN272201200026C (starting Sept. 1, 2012).
Abbreviations used
- ANT
aminoglycoside nucleotidyltransferase
- CLI
clindamycin
- CTD
C-terminal domain
- KAN
kanamycin
- LCM
lincomycin
- Lnu
lincosamide O-nucleotidyltransferase
- NT
nucleotidyltransferase
- NTase
NTase enzyme
- NTD
nucleotidyltransferase domain
Footnotes
Accession numbers
The structure factors and atomic coordinates for Lnu(A) (apoenzyme) and Lnu(A)-LCM complex structures were deposited into the Protein Data Bank with the PDB ID codes 4FO1 and 4WH5, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization (WHO) Antimicrobial resistance: global report on surveillance. 2014 [Google Scholar]
- 2.Schlünzen F, Zarivach R, Harms J, Bashan A, Tocilj A. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–21. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 3.Spizek J, Novotná J, Řezanka T. Lincosamides: chemical structure, biosynthesis, mechanism of action, resistance, and applications. Adv Appl Microbiol. 2004;56:121–54. doi: 10.1016/S0065-2164(04)56004-5. [DOI] [PubMed] [Google Scholar]
- 4.Houghton JL, Green KD, Chen W, Garneau-Tsodikova S. The future of aminoglycosides: the end or renaissance? Chembiochem. 2010;11:880–902. doi: 10.1002/cbic.200900779. [DOI] [PubMed] [Google Scholar]
- 5.Leclercq R, Carlier C, Duval J, Courvalin P. Plasmid-mediated resistance to lincomycin by inactivation in Staphylococcus haemolyticus. Antimicrob Agents Chemother. 1985;28:421–4. doi: 10.1128/aac.28.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brisson-Noël A, Delrieu P, Samain D, Courvalin P. Inactivation of lincosaminide antibiotics in Staphylococcus. Identification of lincosaminide O-nucleotidyltransferases and comparison of the corresponding resistance genes. J Biol Chem. 1988;263:15880–7. [PubMed] [Google Scholar]
- 7.Zhao Q, Wendlandt S, Li H, Li J, Wu C, Shen J, et al. Identification of the novel lincosamide resistance gene lnu(E) truncated by ISEnfa5-cfr-ISEnfa5 insertion in Streptococcus suis: de novo synthesis and confirmation of functional activity in Staphylococcus aureus. Antimicrob Agents Chemother. 2014;58:1785–8. doi: 10.1128/AAC.02007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levings RS, Hall RM, Lightfoot D, Djordjevic SP. linG, a new integron-associated gene cassette encoding a lincosamide nucleotidyltransferase. Antimicrob Agents Chemother. 2006;50:3514–5. doi: 10.1128/AAC.00817-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts MC. Update on macrolide–lincosamide–streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol Lett. 2008;282:147–59. doi: 10.1111/j.1574-6968.2008.01145.x. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Shoemaker NB, Wang GR, Salyers AA. Characterization of a Bacteroides mobilizable transposon, NBU2, which carries a functional lincomycin resistance gene. J Bacteriol. 2000;182:3559–71. doi: 10.1128/jb.182.12.3559-3571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petinaki E, Guerin-Faublee V, Pichereau V, Villers C, Achard A, Malbruny B, et al. Lincomycin Resistance Gene lnu(D) in Streptococcus uberis. Antimicrob Agents Chemother. 2008;52:626–30. doi: 10.1128/AAC.01126-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morar M, Bhullar K, Hughes DW, Junop M, Wright GD. Structure and Mechanism of the Lincosamide Antibiotic Adenylyltransferase LinB. Structure. 2009;17:1649–59. doi: 10.1016/j.str.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 13.Kuchta K, Knizewski L, Wyrwicz LS, Rychlewski L, Ginalski K. Comprehensive classification of nucleotidyltransferase fold proteins: identification of novel families and their representatives in human. Nucleic Acids Res. 2009;37:7701–14. doi: 10.1093/nar/gkp854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramirez M. Aminoglycoside modifying enzymes. Drug Resistance Updates. 2010 doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox G, Stogios PJ, Savchenko A, Wright GD. Structural and Molecular Basis for Resistance to Aminoglycoside Antibiotics by the Adenylyltransferase ANT(2″)-Ia. MBio. 2015;6:e02180–14. doi: 10.1128/mBio.02180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen LC, Benning MM, Holden HM. Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyltransferase. Biochemistry. 1995;34:13305–11. doi: 10.1021/bi00041a005. [DOI] [PubMed] [Google Scholar]
- 17.Batra VK, Shock DD, Prasad R, Beard WA, Hou EW, Pedersen LC, et al. Structure of DNA polymerase beta with a benzo[c]phenanthrene diol epoxide-adducted template exhibits mutagenic features. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17231–6. doi: 10.1073/pnas.0605069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13:42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 19.Gill EE, Franco OL, Hancock REW. Antibiotic adjuvants: diverse strategies for controlling drug-resistant pathogens. Chem Biol Drug Des. 2015;85:56–78. doi: 10.1111/cbdd.12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morar M, Wright GD. The genomic enzymology of antibiotic resistance. Annu Rev Genet. 2010;44:25–51. doi: 10.1146/annurev-genet-102209-163517. [DOI] [PubMed] [Google Scholar]
- 21.Wright GD. Antibiotic resistance in the environment: a link to the clinic? Curr Opin Microbiol. 2010;13:589–94. doi: 10.1016/j.mib.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Walker MS, Walker JB. Streptomycin biosynthesis and metabolism. Enzymatic phosphorylation of dihydrostreptobiosamine moieties of dihydro-streptomycin-(streptidino) phosphate and dihydrostreptomycin by Streptomyces extracts. J Biol Chem. 1970;245:6683–9. [PubMed] [Google Scholar]
- 23.Marshall VP, McGee JE, Cialdella JI, Baczynskyj L, Chirby DG, Yurek DA, et al. Purification of lincosaminide O-nucleotidyltransferase from Streptomyces coelicolor Müller. J Antibiot. 1991;44:895–900. doi: 10.7164/antibiotics.44.895. [DOI] [PubMed] [Google Scholar]
- 24.Yoon JY, Lee SJ, Kim DJ, Lee B-J, Yang JK, Suh SW. Crystal structure of JHP933 from Helicobacter pylori J99 shows two-domain architecture with a DUF1814 family nucleotidyltransferase domain and a helical bundle domain. Proteins. 2014;82:2275–81. doi: 10.1002/prot.24572. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Ye X, Su Y, Sun L, She F, Wu Y. Crystal Structure Confirmation of JHP933 as a Nucleotidyltransferase Superfamily Protein from Helicobacter pylori Strain J99. PLoS ONE. 2014;9:e104609. doi: 10.1371/journal.pone.0104609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirsch DR, Cox G, D’Erasmo MP, Shakya T, Meck C, Mohd N, et al. Inhibition of the ANT(2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic α-hydroxytropolones. Bioorg Med Chem Lett. 2014;24:4943–7. doi: 10.1016/j.bmcl.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, Mark ID. A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol Biol. 2009;498:105–15. doi: 10.1007/978-1-59745-196-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–66. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 29.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–68. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 32.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. MUSTANG: a multiple structural alignment algorithm. Proteins. 2006;64:559–74. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- 34.DeLano W. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. n.d. [Google Scholar]
- 35.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–8. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.