

Abstract
Acquired aplastic anemia (aAA) is a non-malignant disease caused by autoimmune destruction of early hematopoietic cells. Clonal hematopoiesis is a late complication, seen in 20–25% of older patients. We hypothesized that clonal hematopoiesis in aAA is a more general phenomenon, which can arise early in disease even in younger patients. To evaluate clonal hematopoiesis in aAA, we used comparative whole exome sequencing of paired bone marrow and skin in 22 patients. We found somatic mutations in sixteen patients (72.7%) with a median disease duration of 1 year; twelve (66.7%) were patients with pediatriconset aAA. Fifty-eight mutations in 51 unique genes were primarily in pathways of immunity and transcriptional regulation. Most frequently mutated was PIGA, with 7 mutations. Only two mutations were in genes recurrently-mutated in MDS. Two patients had oligoclonal loss of HLA alleles, linking immune escape to clone emergence. Two patients had activating mutations in key signaling pathways (STAT5B(p.N642H), CAMK2G(p.T306M)). Our results suggest that clonal hematopoiesis in aAA is common, with two mechanisms emerging― immune escape and increased proliferation. Our findings expand conceptual understanding of this non-neoplastic blood disorder. Future prospective studies of clonal hematopoiesis in aAA will be critical for understanding outcomes, and for designing personalized treatment strategies.
Keywords: clonal hematopoiesis, aplastic anemia, bone marrow failure, myelodysplastic syndrome, MDS
Introduction
Acquired aplastic anemia (aAA) is a life-threatening blood disorder, affecting children and adults, caused by immune destruction of hematopoietic stem and progenitor cells and the subsequent failure of the bone marrow to sustain normal blood production [1]. Refractory disease and transformation to myelodysplastic syndrome (MDS) are particularly vexing problems in aAA; 10–15% of patients treated with immunosuppressive therapy (IST) evolve to MDS or acute leukemia [2]. Although aAA is considered to be non-malignant, there is a well-described association with clonal blood disorders. Nearly half of aAA patients have clonal populations of cells lacking cell surface proteins linked to a glycosylphosphatidylinositol (GPI) anchor due to somatic loss-of-function mutations in the PIG-A gene; these are called Paroxysmal Nocturnal Hemoglobinuria (PNH) clones due to their susceptibility to complement-mediated lysis [3–4]. More recent reports indicate that ∼10% of aAA patients have acquired copy number-neutral loss of heterozygosity (CN-LOH) in chromosome arm 6p, postulated to emerge by immune selection against specific HLA alleles [5–7]. There are emerging data from targeted sequencing of genes recurrently-mutated in MDS indicating that up to 24% of aAA patients carry somatic mutations in ASXL1, DNMT3A, and BCOR, and may be at a greater risk of malignant transformation [8–11]. Taken together, the available data indicate that somatic mutations in genes other than PIGA are limited to a minority of generally older aAA patients. Importantly, beyond targeted sequencing studies, the full spectrum of clonal hematopoiesis in aAA remains undefined, with little data on clonal hematopoiesis in the pediatric population.
Based on the known association of aAA and clonal blood disorders, we hypothesized that clonal hematopoiesis in aAA may be a more general phenomenon, present in the majority of patients, including children and young adults, and can emerge early in the course of the disease. To comprehensively evaluate the landscape of clonal hematopoiesis in aAA, we used an unbiased approach of comparative whole exome sequencing (WES) of paired bone marrow and skin fibroblast DNA, combined with genome-wide single nucleotide polymorphism array (SNP-A) profiling in twenty two aAA patients. We found clonal hematopoiesis in three quarters of patients, including two thirds of patients with pediatriconset disease. Our results show that even in the younger patients, hematopoiesis in aAA is frequently characterized by somatic mutations, which are distinct from mutations in MDS, and instead bear signatures of immune escape and proliferative signaling, and extend beyond the known association with Paroxysmal Nocturnal Hemoglobinuria.
Materials and Methods
Patients and Study Oversight
The Penn-CHOP Bone Marrow Failure Syndrome (BMFS) cohort is an open prospective/retrospective cohort for the study of molecular mechanisms of BMFS, approved by the Institutional Review Boards of Children’s Hospital of Philadelphia (CHOP) and of the University of Pennsylvania (Penn). Written informed consent from all study participants or their legal guardians was obtained prior to study participation in accordance with the Declaration of Helsinki. All patients with aAA, referred to the Penn-CHOP Comprehensive BMFS Center between 2009 and 2014, who had a stored bone marrow aspirate and skin biopsy material were eligible for this analysis. The diagnosis of aAA was established according to the International Study of Agranulocytosis and Aplastic Anemia[12], and required exclusion of congenital BMFS and other conditions mimicking aAA . Patients with morphological evidence of dysplasia according to the 2008 World Health Organization (WHO) classification[13] were excluded. Complete medical histories, peripheral blood counts, bone marrow histology and cytogenetic analysis were available for all patients. In accordance with the American Academy of Pediatrics Council on Child and Adolescent Health, pediatric-onset aAA was defined as a diagnosis of aAA under the age of 22[14].
Cytogenetics and Hematopathology
Cytogenetic analysis and fluorescence in situ hybridization (FISH) were performed according to standard methods. Bone marrow histology was evaluated by a clinical hematopathologist in a blinded fashion, as patients were entered into the study only after completion of the diagnostic review. In accordance with department policy, all controversial cases were subject to a clinical consensus conference.
SNP-A Analysis
Illumina Infinium SNP-A genotyping of bone marrow aspirate DNA was performed using Illumina Quad610, Illumina Omni1 Quad, or Illumina CytoSNP 850 Beadchips at the CHOP Center for Applied Genomics according to the manufacturer’s protocol. Arrays were analyzed in GenomeStudio (Illumina, Inc., San Diego, CA), which allows direct visualization of B-Allele Frequency and log R ratio. SNP-A data have been deposited in Gene Expression Omnibus (accession GSE48484).
WES
WES was performed on DNA extracted from the patients’ bone marrow aspirate and paired skin fibroblasts using Qiagen DNeasy Blood & Tissue Kit (Qiagen Inc., Valencia, CA) at the BGI@CHOP High Throughput Sequencing Center. Skin fibroblasts were expanded in culture for 3–4 passages prior to DNA extraction. Exome libraries were constructed with Agilent SureSelect All Exon V4 + UTRs kit (Agilent Technologies, Santa Clara, CA). Paired-end WES to 150X average depth was performed using the Illumina HiSeq 2500 platform, according to the manufacturer’s recommendations. Somatic variant calling on bone marrow-skin biopsy pairs was performed with VarScan2, an algorithm optimized for detection of somatic mutations[15], using parameters –min-coverage 4,–min-var-freq 0.08,–p-value 0.05,–strand-filter 1–min-avg-qual 20. Filtering and annotation of somatic mutations was performed using SNP & Variation Suite v8.0 (Golden Helix, Inc., Bozeman, MT). All mutations were manually curated in Integrative Genomics Viewer[16], and were classified into Tiers as described previously[17]. All chromosome coordinates were based on hg19 (NCBI build 37).
Sanger sequencing
All putative Tier 1 and Tier 2 somatic mutations[17] identified with WES were validated with Sanger sequencing. Briefly, amplicons containing putative somatic mutations were subjected to bi-directional sequencing using a 3730 DNA Analyzer (Applied Biosystems, South San Francisco, CA). Mutations were confirmed as somatic if they were present in bone marrow and absent in paired skin DNA.
HLA Typing
Paired-end next-generation sequencing (2×251 bp) of the Human Leukocyte Antigen (HLA) region was performed on the Illumina MiSeq platform at >10,000X average depth as previously described[18]. Sequence alignment and HLA typing were performed using NGSengine™ (GenDx Utrecht, Netherlands) software in the CHOP CLIA-approved Immunogenetics Laboratory.
Telomere Length Measurement
Fifteen patients had lymphocyte telomere length (TL) measurements performed by the CLIA certified TL testing center (Repeat Diagnostics, Inc., North Vancouver, Canada). Five patients, who did not have a clinical TL measurement, but had available material, had TL measured using the FITC-conjugated (C3TA2)3 peptide nucleic acid probe, as previously described[19]. Relative TL were obtained by comparison to a control cell line (GM03671C; Coriell Institute, Camden, NJ), which was assigned a TL of 100%. TL were normalized to the clinical lab TL measurement, and were compared to age-matched normal controls (Repeat Diagnostics).
T Cell Receptor (TCR)-γ Gene Rearrangement
TCR γ gene rearrangement analysis was performed by the Penn CLIA-approved Molecular Diagnostics Laboratory, using fluorescent-based amplification of the consensus V and J regions of TCR γ, as previously described.[20]
Cell Sorting
Lymphoid- and myeloid-enriched cell fractions were obtained from peripheral blood by immunomagnetic selection for CD3 and CD19-positive cells. Briefly, peripheral blood was subjected to red cell lysis, incubated with anti-CD3 and anti-CD19 microbeads (Miltenyi Biotec, San Diego, CA), and separated using “LS” columns into the myeloid-enriched (CD3- and CD19-depleted) and lymphocyte (CD3+ and CD19+ selected) fractions. Purity was verified by flow cytometry.
GO Pathway Analysis
Gene Ontology (GO) enrichment analysis was performed using WebGestalt Gene Analysis Toolkit[21], using a Benjamini-Hochberg adjustment for multiple testing[22], and a significant p-value <0.05.
Statistics
Association analysis of clinical characteristics and presence of detectable clonal hematopoiesis was performed using Fisher’s exact test for categorical variables, and Mann-Whitney U Test for continuous variables, with a two-tailed significance level of 0.05. Multivariate logistic regression was also performed.
Results
Patient Population
We analyzed a cohort of 22 patients with aAA; 18 had pediatric-onset aAA, and 4 had adult-onset aAA (Table 1). Median age at diagnosis was 14.5 years (range 1.5–61). The majority of patients (21 of 22) were diagnosed with severe or very severe aAA; one patient had moderate aAA. Nineteen patients (86.4%) were treated with immunosuppressive therapy (IST) prior to analysis, and two had a history of eltrombopag therapy. Consistent with the known high frequency of PNH in aAA, 45.5% of patients had a PNH clone of over 1% by flow cytometry. 25% of patients (5 of 20 patients with available telomere length measurement) had telomeres under the 10th percentile. The majority of patients (21 of 22) lacked acquired karyotypic abnormalities; one patient had an acquired deletion of chromosome arm 13q. None of the patients had morphologic evidence of dysplasia by bone marrow histopathology.
Table 1.
Patient Characteristics
| Overall (n=22) | Pediatric-Onset (n=18) | Adult-Onset (n=4) | ||
|---|---|---|---|---|
| Patient Characteristics | ||||
| Gender, female (%)/male (%) | 13 (59) / 9 (41) | 9 (50) / 9 (50) | 4 (100) / 0 (0) | |
| Age at diagnosis (years), median (range) | 14.5 (1.5–61) | 12 (1.5–19) | 45.5 (33–61) | |
| Age at sequencing (years), median (range) | 18.5 (2–61) | 17 (2–34) | 48.5 (36–61) | |
| Disease duration (years), median (range) | 1 (0.1–29) | 1 (0.1–29) | 1.7 (0.25–6) | |
| Duration of follow-up (months), median (range) | 14.8 (0–38.2) | 13.3 (0–38.2) | 20.3 (0–25.6) | |
| Disease severity (n) | ||||
| Moderate | 1 | 0 | 1 | |
| Severe | 16 | 15 | 1 | |
| Very Severe | 5 | 3 | 2 | |
| Post-immunosuppression (IST), n (%) | 19 (86.4) | 16 (88.9) | 3 (75.0) | |
| Time from IST (years), median (range) | 1 (0–18) | 1 (0–18) | 0.25 (0.25–3) | |
| Eltrombopag use (n) | 2 | 1 | 1 | |
| Transformation to MDS (n) | 0 | 0 | 0 | |
| Telomere length | ||||
| ≥ 10th Percentile of Age-Matched Controls | 15 | 11 | 4 | |
| < 10th Percentile of Age-Matched Controls | 5 | 5 | 0 | |
| N/A | 2 | 2 | 0 | |
| % of patients with PNH clone (>1%) | 45.5 | 44.4 | 50.0 | |
| Summary of Acquired Genetic Changes | ||||
| Acquired CN-LOH, n (%) | 6 (27.3) | 5 (27.8) | 1 (25.0) | |
| 6pLOH, n (%) | 3 (13.6) | 3 (16.7) | 0 (0.0) | |
| Patients with somatic mutations, n (%) | 16 (72.7) | 12 (66.7) | 4 (100.0) | |
| Tier 1 and 2 gene mutations per patient, median (range) | 2 (0–11) | 1 (0–6) | 8.5 (1–11) | |
| Acquired cytogenetic abnormality, n | 1 | 0 | 1 | |
Pediatric-Onset aAA: diagnosis under 22 years of age; Adult-onset aAA: diagnosis at 22 years and older.
Tier 1: nonsynonymous coding, consensus splice site, or RNA genes; Tier 2: regions with regulatory potential, such as 5’ and 3’ untranslated regions (UTR).
The Majority of Pediatric and Adult aAA Patients Have Clonal Hematopoiesis
We used a combination of metaphase cytogenetics, whole genome SNP-A analysis, and WES to comprehensively evaluate the bone marrow of aAA patients for evidence of clonal hematopoiesis (Table 1–Table 3). To ensure accurate identification of somatic mutations, we identified de novo mutations in the patients’ bone marrow DNA, which were absent in the patients’ constitutional DNA (skin fibroblasts). Identified somatic mutations were classified into Tiers as described previously [17], where non-synonymous coding, consensus splice site, or RNA genes were classified as Tier 1, and regions with regulatory potential, such as 5’ and 3’ untranslated regions (UTR) were classified as Tier 2. All putative Tier 1 and 2 somatic mutations identified by WES were confirmed by bi-directional Sanger sequencing, and, for the HLA alleles, by targeted next-generation deep sequencing.
Table 3.
Clonal Hematopoiesis in Pediatric- and Adult-Onset aAA
| Patient Number |
Acquired Structural Abnormalities | PNH Clone | Acquired Mutations | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | Chr | Clone | Chr | Position | Ref/Alt | Depth | Freq | Classification | Gene | Exon | Protein | Lineage | ||
| Pediatric-Onset aAA | ||||||||||||||
| 281.01 | CN-LOH | 6pterp22.1 | ∼30% | None | 6 | 29911127 | C/G | 56 | 9% | Stopgain | HLA-A | 3 | p.Tyr142* | - |
| CN-LOH | 6pterp12.1 | ∼10% | ||||||||||||
| 118.01 | None | 92–98% | X | 15339787 | TCTGGAGTC ATCCATCTCA AGAAAATGA GGAAGAGGA AGTTGAAAA/− |
364 | 13% | Frameshift Del | PIGA | 6 | p.Val432fs | - | ||
| 5 | 147889435 | C/G | 262 | 28% | Nonsyn SNV | HTR4 | 5 | p.Lys220Asn | - | |||||
| 16 | 84218534 | C/T | 87 | 27% | Nonsyn SNV | TAF1C | 2 | p.Asp21Asn | - | |||||
| 9 | 8518131 | G/T | 250 | 25% | Synonymous | PTPRD | 9 | p.= | - | |||||
| 16 | 14761829 | G/T | 134 | 21% | UTR3 | BFAR | n/a | n/a | - | |||||
| 19 | 547333 | C/T | 76 | 18% | Nonsyn SNV | GZMM | 2 | p.Arg37Cys | - | |||||
| 263.01 | None | 3%1 | 19 | 15882928 | G/A | 49 | 12% | NCExonic | CYP4F24P | n/a | n/a | - | ||
| 56.01 | CN-LOH | 6pterp21.31 | ∼10% | none | 2 | 136575226 | C/T | 216 | 11% | Synonymous | LCT | 6 | p.= | - |
| CN-LOH | 6pterp21.1 | ∼15% | ||||||||||||
| CN-LOH | 6pterp12.1 | ∼45 | ||||||||||||
| CN-LOH | 6pterp11.1 | ∼15 | ||||||||||||
| 54.01 | CN-LOH | 6pterp21.31 | ∼10% | none | 1 | 248903467 | C/T | 20 | 40% | Upstream | LYPD8 | n/a | n/a | M |
| CN-LOH | 6pterp21.1 | ∼10% | 6 | 31323108 | A/− | 341 | 16% | Frameshift Del | HLA-B | 4 | p.Leu294.fs | n/a | ||
| 58.01 | None | 18–25% | X | 15349337 | C/T | 42 | 36% | Splicing | PIGA | 2 | ? | n/a | ||
| 1 | 100908529 | G/A | 130 | 22% | Nonsyn SNV | CDC14A | 7 | p.Val166Met | M | |||||
| 7 | 25162020 | C/T | 354 | 18% | UTR3 | CYCS | n/a | n/a | M | |||||
| X | 71427503 | G/A | 261 | 18% | Stopgain | ERCC6L | 2 | p.Arg372* | M | |||||
| 312.01 | CN-LOH | 15q21.1qter | ∼5–8% | 17–35%1 | X | 134022913 | A/C | 67 | 9% | UTR3 | MOSPD1 | n/a | n/a | - |
| CN-LOH | 15q12qter | ∼5–8% | X | 134022943 | A/G | 55 | 11% | UTR3 | MOSPD1 | n/a | n/a | - | ||
| 252.01 | None | 90% | 18 | 43432246 | AC/− | 59 | 17% | UTR3 | EPG5 | n/a | n/a | - | ||
| X | 15343166 | CACGATCGC CATGCAGAA TG/− |
230 | 13% | Frameshift Del | PIGA | 3 | pAla313fs | - | |||||
| X | 39932819 | C/T | 104 | 13% | Nonsyn SNV | BCOR | 4 | p.Val594Ile | - | |||||
| 8 | 10286083 | A/T | 154 | 12% | UTR3 | MSRA | n/a | n/a | - | |||||
| 145.01 | None | 9% | 19 | 56701720 | C/A | 33 | 55% | Stopgain | ZSCAN5B | 4 | p.Glu322* | M | ||
| 6 | 137465187 | C/T | 530 | 30% | UTR3 | IL22RA2 | n/a | n/a | M | |||||
| X | 15344108 | A/− | 89 | 21% | Frameshift Del | PIGA | 3 | p.Phe259fs | M | |||||
| 20.01 | CN-LOH | 5q15qter | ∼15–20% | 16%1 | 19 | 990233 | G/A | 271 | 16% | Nonsyn SNV | WDR18 | 4 | p.Ala156Thr | - |
| 17 | 65850376 | G/C | 358 | 13% | Nonsyn SNV | BPTF | 2 | p.Asp312His | - | |||||
| 10 | 75601956 | G/A | 162 | 12% | Nonsyn SNV | CAMK2G | 12 | p.Thr306Met | - | |||||
| 6 | 165741064 | T/− | 140 | 12% | UTR3 | PDE10A | n/a | n/a | - | |||||
| 12 | 40153078 | G/T | 213 | 8% | UTR3 | SLC2A13 | n/a | n/a | - | |||||
| 385.01 | None | n/a | X | 15349789 | T/A | 74 | 22% | Nonsyn SNV | PIGA | 2 | p.Lys88Asn | - | ||
| 12 | 55615758 | G/A | 103 | 12% | Synonymous | OR10A7 | 1 | p.= | - | |||||
| 18 | 28911768 | A/G | 74 | 9% | Nonsyn SNV | DSG1 | 6 | p.Met208Val | - | |||||
| 5 | 154194771 | T/G | 497 | 9% | UTR3 | LARP1 | n/a | n/a | - | |||||
| 10 | 119305251 | C/T | 57 | 9% | Nonsyn SNV | EMX2 | 2 | p.Ala172Val | - | |||||
| 434.01 | None | none | 7 | 150929476 | T/G | 19 | 58% | Upstream | CHPF2 | n/a | n/a | Both | ||
| Adult-Onset aAA | ||||||||||||||
| 364.01 | None | none | 17 | 40359729 | T/G | 60 | 23% | Nonsyn SNV | STAT5B | 16 | p.Asn642His | M | ||
| 3 | 125247758 | C/T | 122 | 20% | UTR3 | OSBPL11 | n/a | n/a | M | |||||
| 11 | 31447832 | AGA/− | 134 | 17% | Splicing | DNAJC24 | 4 | ? | M | |||||
| 7 | 116438157 | A/G | 328 | 16% | UTR3 | MET | n/a | n/a | M | |||||
| 20 | 43753010 | C/A | 186 | 16% | Nonsyn SNV | WFDC12 | 1 | p.Gly27Cys | M | |||||
| 20 | 36146442 | G/A | 363 | 16% | UTR3 | BLCAP | n/a | n/a | n/a | |||||
| 6 | 76425706 | A/G | 144 | 16% | UTR3 | SENP6 | n/a | n/a | M | |||||
| X | 49861453 | G/T | 227 | 15% | UTR3 | CLCN5 | n/a | n/a | M | |||||
| 9 | 14085693 | T/A | 378 | 15% | UTR3 | NFIB | n/a | n/a | n/a | |||||
| 16 | 30544011 | G/T | 86 | 14% | UTR3 | ZNF747 | n/a | n/a | M | |||||
| 11 | 62748488 | G/A | 89 | 11% | Nonsyn SNV | SLC22A6 | 6 | p.Arg336Cys | M | |||||
| 76.01 | None | 80–90% | 20 | 35624870 | G/A | 113 | 30% | UTR3 | RBL1 | n/a | n/a | - | ||
| X | 15349874 | -/A | 118 | 28% | Frameshift Ins | PIGA | 2 | p.Ile61fs | - | |||||
| X | 71425778 | G/T | 157 | 27% | Nonsyn SNV | ERCC6L | 2 | p.Pro947Thr | - | |||||
| 11 | 111784673 | G/T | 114 | 26% | UTR3 | HSPB2 | n/a | n/a | - | |||||
| 12 | 49220845 | C/T | 58 | 24% | Nonsyn SNV | CACNB3 | 12 | p.Thr347Met | - | |||||
| 20 | 44685832 | T/G | 81 | 19% | Nonsyn SNV | SLC12A5 | 25 | p.Met1073Arg | - | |||||
| 1 | 20825987 | T/A | 48 | 17% | UTR3 | MUL1 | n/a | n/a | - | |||||
| 11 | 118036457 | C/T | 52 | 14% | UTR3 | SCN2B | n/a | n/a | - | |||||
| 4 | 145629462 | A/G | 146 | 9% | Nonsyn SNV | HHIP | 7 | p.Arg434Gly | - | |||||
| 356.01 | None | none | 6 | 137322940 | C/T | 184 | 49% | Nonsyn SNV | IL20RA | 7 | p.Glu473Lys | Both | ||
| 14 | 75746727 | G/A | 175 | 43% | Nonsyn SNV | FOS | 2 | p.Gly97Arg | Both | |||||
| 20 | 31022573 | GT/− | 121 | 40% | Frameshift Del | ASXL1 | 12 | p.Cys687fs | Both | |||||
| 4 | 119952055 | G/A | 218 | 39% | Nonsyn SNV | SYNPO2 | 4 | p.Glu709Lys | Both | |||||
| 20 | 25459761 | G/A | 177 | 37% | Nonsyn SNV | NINL | 16 | p.Arg667Cys | Both | |||||
| 22 | 42383706 | G/A | 54 | 34% | Nonsyn SNV | SETP3 | 5 | p.Arg165His | Both | |||||
| 7 | 97736216 | C/T | 46 | 27% | UTR5 | LMTK2 | n/a | n/a | Both | |||||
| X | 152848098 | A/G | 121 | 18% | UTR3 | ATP2B3 | n/a | n/a | M | |||||
| 390.01 | CN-LOH | 6WC | ∼5% | 6% | X | 15339766 | GAGGAA/− | 260 | 1% | Del | PIGA | 6 | P.F438del.L439del | - |
| del | 13q13.3q21.1 | 10% | ||||||||||||
Chr, chromosome. PNH Clone, percentage of CD55, CD59-negative granulocytes as measured by flow cytometry of peripheral blood. Ref/Alt, reference sequence/mutated sequence. Depth, read depth of WES at a given location. Freq, mutant allele frequency in bone marrow, calculated as proportion of mutant allele reads at a given position. Lineage, lineage-specificity of somatic mutation (myeloid (M), lymphoid (L), or both).
In agreement with published studies of PIGA gene sequencing in PNH [1, 2], three patients had flow-cytometric evidence of PNH for which we were unable to identify a mutation; these were presumed to be caused by multiple independent mutations in PIGA below the level of detection of WES and Sanger sequencing [3].
Six patients (27.3%) were found to have clones with acquired CN-LOH; CN-LOH for four patients was reported previously [6]. The most common region affected by acquired CN-LOH was chromosome arm 6p, seen in three patients (13.6%). Sixteen patients (72.7%) had evidence of clonal hematopoiesis with confirmed somatic mutations in the bone marrow. Independent evidence of clonal hematopoiesis was present in all sixteen patients even after exclusion of PIGA mutations. The median number of Tier 1 and 2 mutations per patient was 2 (range 0–11). Twelve of 18 patients (66.7%) with pediatric-onset aAA had somatic mutations, with a median of 1 (range 0–6) Tier 1 and 2 mutations per patient; one patient had a single identified somatic mutation in an upstream region of the CHPF2 gene. Six patients (27.3%), all with pediatric-onset aAA, had no detectable clonal hematopoiesis even after expanding analysis to include synonymous, intronic and intergenic mutations.
Six of 16 patients with detectable somatic mutations had peripheral blood material available for cell sorting; in these patients, mutations were evaluated by Sanger sequencing in the immunomagnetically-sorted myeloid and lymphoid cell lineages at a later time point (median interval between two analyses of 1.1 years, range 0.4–2.4 years). In four of the six patients (66.7%), somatic mutations were detected only in the CD3- and CD19-depleted myeloid cell fraction, but not in the lymphoid fraction, indicating that the mutation most likely occurred in a progenitor population with predominant contribution to the myeloid lineage. Two patients had mutations detectable in both myeloid and lymphoid lineages, suggesting that the mutation likely occurred at a stem cell or a multipotent progenitor level, with a potential to contribute to both lineages.
Association analysis of clinical characteristics of the 16 patients with detectable clonal hematopoiesis compared to the 6 patients without detectable clonal hematopoiesis (Figure 1) revealed that patients with clonal hematopoiesis were more likely to be older at diagnosis, with a median age of 16.5 years (range 3–61), as compared to 3.5 years (range 1.5–15) (p=0.012); similarly, they were more likely to be older at the time of WES (median 20 years (range 4–61) compared to 4.5 years (range 2–20)) (p=0.011). Clonal hematopoiesis was detected early in disease, with a median time from diagnosis of 1 year (range 0.08–29). There was no significant association with disease duration, time from immunosuppression, disease severity or telomere lengths. In the multivariate logistic regression including age at diagnosis, disease duration and telomere length simultaneously, age at diagnosis showed a trend toward significant association with clonal hematopoiesis (p=0.0752).
Figure 1. Clinical Characteristics Associated with Presence or Absence of Clonal Hematopoiesis in aAA.

A. The distribution of the total number of identified somatic mutations per patient as it relates to the patients’ age and disease duration. For each patient, listed on the X-axis, the age is plotted as a vertical line, with the beginning of the line corresponding to age at diagnosis and an arrowhead depicting age at WES. The number above the line corresponds to the total number of somatic mutations. B. Association analysis of clinical characteristics with presence or absence of clonal hematopoiesis in aAA.
Acquired Mutations in aAA Carry Signatures of Immune Escape and Proliferative Signaling, and Are Distinct from Mutations in MDS
A total of 58 Tier 1 (n=35) and Tier 2 mutations (n=23) in 51 unique genes were identified (Tables 3–4). Excluding PIGA mutations, previously reported to be associated with aAA[3–4] and recently reported to arise both as early or late subclonal events in classical PNH[23], there were a total of 50 non-PIGA unique gene mutations. Other recurrent mutations were loss-of-function mutations in HLA class I genes (n=2), and mutations in ERCC6L (n=2). Two patients had mutations in genes recurrently affected in MDS, ASXL1 (n=1) and BCOR (n=1); neither patient had evidence of myelodysplasia at the time of analysis.
Table 4.
Gene Ontology (GO) pathway analysis of genes disrupted by Tier 1 mutations in aAA
| GO Category | Category Name | GO ID | R | raw P | adj P |
|---|---|---|---|---|---|
| Biological Process | |||||
| antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-independent |
GO:0002480 | 125 | 1E-04 | 0.0229 | |
| interferon-gamma-mediated signaling pathway | GO:0060333 | 22.51 | 3E-04 | 0.0229 | |
| response to bacterium | GO:0009617 | 8.06 | 3E-04 | 0.0229 | |
| innate immune response | GO:0045087 | 6.26 | 3E-04 | 0.0229 | |
| detection of bacterium | GO:0016045 | 62.52 | 5E-04 | 0.0254 | |
| cellular response to interferon-gamma | GO:0071346 | 18.76 | 5E-04 | 0.0254 | |
| response to interferon-gamma | GO:0034341 | 15.49 | 9E-04 | 0.0381 | |
| detection of biotic stimulus | GO:0009595 | 43.28 | 0.001 | 0.0381 | |
| response to cytokine stimulus | GO:0034097 | 5.56 | 0.002 | 0.0576 | |
| immune response | GO:0006955 | 3.68 | 0.002 | 0.064 | |
| Molecular Function | |||||
| MHC class I receptor activity | GO:0032393 | 61.88 | 5E-04 | 0.036 | |
| transcription regulatory region DNA binding | GO:0044212 | 7.17 | 0.002 | 0.0414 | |
| regulatory region DNA binding | GO:0000975 | 7 | 0.002 | 0.0414 | |
| regulatory region nucleic acid binding | GO:0001067 | 7 | 0.002 | 0.0414 | |
| heat shock protein binding | GO:0031072 | 13.06 | 0.01 | 0.1469 | |
| transcription regulatory region sequence-specific DNA binding | GO:0000976 | 8.22 | 0.024 | 0.194 | |
| secondary active transmembrane transporter activity | GO:0015291 | 6.12 | 0.042 | 0.194 | |
| structure-specific DNA binding | GO:0043566 | 6.03 | 0.043 | 0.194 | |
| sequence-specific DNA binding | GO:0043565 | 3.46 | 0.027 | 0.194 | |
| DNA binding | GO:0003677 | 2.04 | 0.033 | 0.194 | |
R, Ratio of enrichment; raw p, p value from hypergeometric test, adj P, p value adjusted by the multiple test adjustment (Benjamini-Hochberg).
Activating Mutations in Signaling Pathways
Although none of the other mutations were recurrent within our cohort, several mutations affected functional domains and would be predicted to serve as drivers. Two such mutations involved critical residues in cellular signaling proteins, both previously reported to be activating [24–25](Figure 2).
Figure 2. Acquired mutations in the bone marrow of aAA patients.
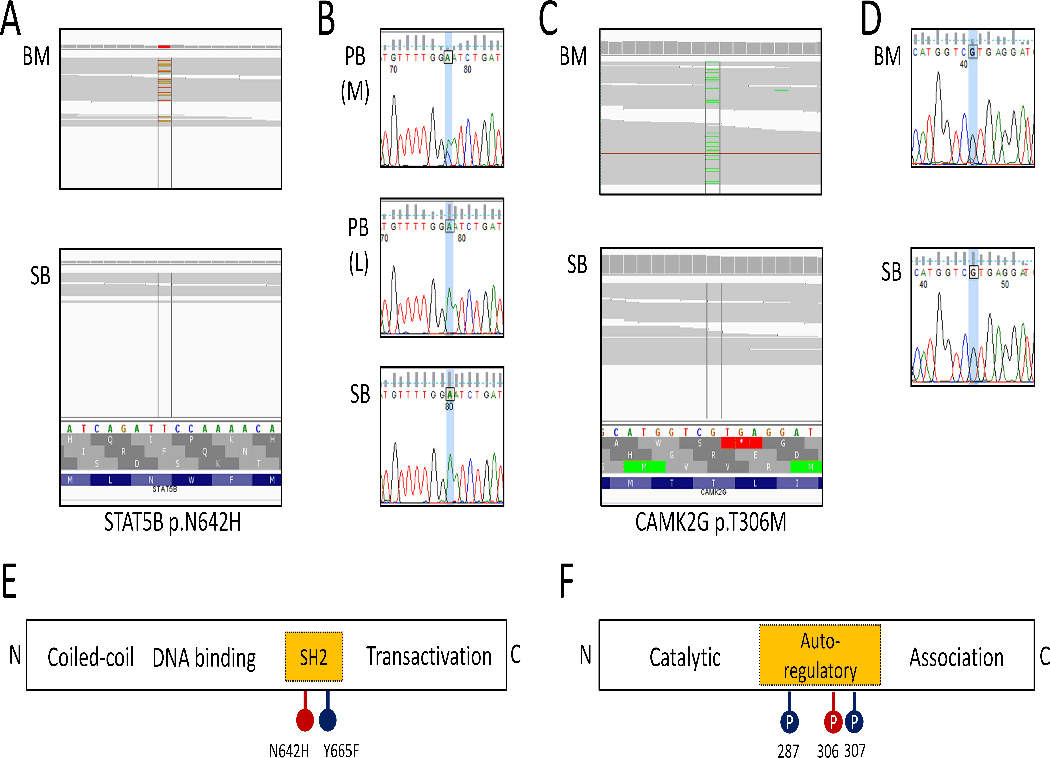
A. IGV screenshots of WES of bone marrow(BM) and constitutional DNA (skin biopsy, SB) showing a g.40359729T>G (p.N642H) mutation in STAT5B gene present in the bone marrow but not in the paired constitutional DNA. B. Sanger sequencing chromatograph showing that the acquired g.40359729T>G (p.N642H) mutation is detected in the immunomagnetically sorted myeloid cell fraction of peripheral blood, PB (M), but is absent from the lymphocyte fraction, PB (L), and skin fibroblast DNA (SB); the chromatograph shows reverse complement sequence. C. IGV screenshots of WES of bone marrow (BM) and constitutional DNA (skin biopsy, SB) showing a g.75601956G>A (p.T306M) mutation in CAMK2G present in the bone marrow but not in the paired constitutional DNA. D. Sanger sequencing chromatograph validating that the acquired g.75601956G>A (p.T306M) mutation in CAMK2G is detected in the bone marrow (BM), but is absent in the skin fibroblast DNA (SB). E. A schematic of the STAT5B protein illustrating the location of previously reported activating mutations in the SH2 domain. F. A schematic of the CAMK2G protein showing the locations of regulatory phosphorylation sites.
Patient 364.01 (diagnosed with aAA in young adulthood, with refractory disease despite IST and a trial of eltrombopag) was found to have 11 Tier 1 and Tier 2 somatic mutations, with a median clone size of 16% (range 11–23%)(Table 3). There was no evidence of myelodysplasia, and no acquired abnormalities detected by metaphase cytogenetics and SNP-A. A somatic p.N642H mutation in the exon 16 SH2 domain of the signal transducer and activator of transcription 5B (STAT5B) gene was identified in the patient’s bone marrow, with the dominant clone size (calculated as a ratio of mutated to reference allele) of 23%, roughly corresponding to half of the patient’s cells carrying a heterozygous mutation (Fig 2A). The STAT5B p.N642H mutation is a recurrent, activating mutation found in some large granular lymphocyte leukemias [24]. In contrast, this patient had no lymphocytosis, and no clonal T cell receptor rearrangement. Sanger sequencing of the mutated STAT5B region in immunomagnetically-sorted peripheral blood revealed that the p.N642H STAT5B mutation was restricted to the myeloid lineage, and was not detected in lymphocytes (Fig 2B).
Patient 20.01 (diagnosed with aAA in adolescence, with partial response to IST, analyzed at relapse while on cyclosporine therapy) was found to have 6 somatic mutations with a median clone size of 12% (range 8–16%), as well as acquired CN-LOH in chromosome arm 5q with an estimated clone size of 15–20%. A somatic p.T306M mutation in the calcium2+/calmodulin-dependent protein kinase II γ (CAMK2G) gene in chromosome band 10q22.2 comprised a 12% clone, corresponding to ∼25% of cells carrying this heterozygous mutation (Fig 2B). T306 of CAMK2G is a known phosphorylation site within the autoregulatory domain, loss of which was previously shown to be activating [26].
Oligoclonal Hematopoiesis with Loss of HLA Class I Alleles
Fourteen percent of patients (3 of 22) had acquired CN-LOH for chromosome arm 6p encompassing the HLA locus (Table 3); CN-LOH for two patients was reported previously [6]. All three patients had evidence of multiple breakpoints for the CN-LOH within the 6p region, indicating emergence of different CN-LOH clones within the same patient. Importantly, two patients with acquired CN-LOH were also found to have somatic loss-of-function mutations within HLA class I genes (Figure 3).
Figure 3. Oligoclonal Loss of HLA class I alleles in aAA.

A. SNP-A genotyping of patient 281.01 depicted as two scatter plots. The top plot shows B-allele Frequency (BAF, a relative frequency of the minor allele); the bottom plot shows Log R Ratio (LRR, a measure of normalized total signal intensity for both alleles). The chromosomal location is depicted on the X-axis. In a region with acquired CN-LOH, the copy number (indicated by the LRR) remains constant, while there is a decreased frequency of the heterozygous alleles (indicated by the change in the left part of the BAF plot). The location of the HLA-A gene is shown by the arrowhead. B) IGV screenshots of WES of bone marrow (BM) and constitutional DNA (skin biopsy, SB) showing the HLA-A g.29911127C>G (p.Tyr142X) mutation in the bone marrow but not in the paired constitutional DNA. C. Next generation sequencing of the HLA locus, showing the location of the nonsense HLA-A g.29911127C>G (p.Tyr142*) mutation (arrow) occurring in cis to the HLA-A*33:03:01 allele. D. SNP-A genotyping of patient 54.01. The top plot shows B-allele Frequency (BAF, a relative frequency of the minor allele); the bottom plot shows Log R Ratio (LRR, a measure of normalized total signal intensity for both alleles). In a region with acquired CN-LOH, the copy number (indicated by the LRR) remains constant, while there is a decreased frequency of the heterozygous alleles (indicated by the change in the left part of the BAF plot). The location of the HLA-B gene is shown by the arrowhead. E. IGV screenshots of WES of bone marrow (BM) and constitutional DNA (skin biopsy, SB) showing the frameshift HLA-A g.31323108delA (p.Leu294fs) mutation in the bone marrow but not in the paired constitutional DNA. F. Next generation sequencing of the HLA locus, showing the location of the HLA-B g.31323108delA (p.Leu294fs) mutation (arrow) occurring in cis to the HLA-B*14:02:01 allele. G. A model schematic depicting recurrent loss of HLA class I alleles in patients with aAA. Cytotoxic T lymphocytes (CTL) cause autoimmune depletion of hematopoietic stem and progenitor cells (HSPC) due auto-antigen presentation in the context of particular HLA class I alleles. Thus, cells with loss of particular HLA class I alleles either via an inactivating mutation, or by loss of heterozygosity favoring the opposite allele, lead to a growth advantage and resultant clonal expansion of the mutant hematopoietic cells. H. Table showing HLA typing results of the peripheral blood for patients 281.01 and 54.01.
SNP-A analysis of patient 281.01 revealed two dominant 6p CN-LOH clones, both encompassing the HLA-A gene (Fig 3). HLA-typing by targeted deep sequencing of patient’s peripheral blood and skin DNA revealed a bias towards the HLA*A 24:02 allele (60%) in the blood, consistent with the loss of the HLA*A 33:03 locus due to LOH (Fig 3A, 3G). Additionally, WES uncovered a somatic nonsense p.Tyr142* mutation in the HLA-A gene in the patient’s bone marrow (Fig 3B). Targeted deep sequencing and HLA typing confirmed that the p.Tyr142* mutation occurred on the HLA*A 33:03 allele, leading to an additional 7% loss of HLA*A 33:03 due to mutational inactivation (Fig 3C).
Similarly, patient 54.01 had two 6p CN-LOH clones detected by SNP-A analysis; the homozygous region encompassed the HLA-B gene (Fig 3D). HLA typing of peripheral blood and skin DNA indicated a bias toward the HLA*B 44:03 allele in the blood (57%), consistent with the loss of the HLA*B 14:02 allele due to LOH (Fig 3F–G). WES revealed a somatic frameshift mutation p.L294fs (Fig 3D), causing an additional 13% loss of the HLA*B 14:02 allele by mutational inactivation (Fig 3D–H).
Significant Enrichment in Pathways Regulating Immunity and Transcription
Gene Ontology (GO) pathway analysis of the 28 genes disrupted by Tier 1 mutations revealed a significant enrichment in pathways regulating immunity and transcription (Table 4). GO categories with significant enrichment included antigen processing and presentation via MHC class I and MHC class I receptor activity, interferon-gamma-mediated signaling and cellular response to interferon-gamma, response to and detection of bacterium, innate immune response, detection of biotic stimulus, as well as transcription and regulatory region, DNA and nucleic acid binding.
Discussion
Using comparative WES and genome-wide SNP-A analysis, we have identified clonal hematopoiesis in the majority of aAA patients, including 67% of patients with pediatric-onset aAA. Our results demonstrate that clonal hematopoiesis in aAA is much more common than previously described, and can emerge early in the disease course―half of the patients with mutations in our cohort were within 1 year of diagnosis. Our data indicate that in the younger patient population, somatic mutations in aAA overlap only to a small degree with mutations typical of MDS. Instead, mutations in aAA are enriched in genes regulating immunity and transcription, with immune escape and proliferative signaling emerging as the main drivers of clonal hematopoiesis.
The finding of early emergence of clonal hematopoiesis in the majority of young aAA patients is likely to redefine how we view aAA, and helps put into context results of candidate gene sequencing in aAA [8–9, 27]. Our data indicate that a large proportion of blood production in aAA patients comes from clonal or oligoclonal hematopoiesis. It is important to note that a haploid mutant allele frequency of 15% as measured by WES corresponds to a 30% diploid clone harboring a heterozygous mutation, and to an even larger proportion when accounting for cellular heterogeneity of bone marrow aspirates. Using an unbiased approach of WES in a young patient cohort, we found mutations in MDS-associated genes only in a small subset of patients (n=2 (9%), one of whom was a 52-year-old adult). The high prevalence and diversity of somatic mutations in aAA patients in our study is similar to findings from WES of thirteen patients with PNH [23], which reflects the overlap in the underlying immune pathogenesis and selective pressure in these disorders, and highlights the role of PIGA as one of a number of drivers of clonal evolution in aAA and PNH. Another important corollary of our findings is that, in the absence of morphologic dysplasia, the presence of somatic mutations cannot be used as a diagnostic tool to distinguish hypoplastic MDS from aAA. Although it is possible that some clones in aAA may be transient, as was previously described for chromosomal abnormalities in aAA [28], our data show that somatic mutations can persist overtime contributing to a significant portion of the patients’ hematopoiesis. Taken together, our results suggest that an individual patient’s clinical course is likely to be influenced by their unique somatic alterations, which, for most patients, differ from mutations of hematologic malignancies.
The diversity of mutations in aAA presents a challenge in distinguishing driver from passenger mutations; driver mutations are expected to involve recurrently-mutated genes or pathways, and to confer a proliferative advantage on the mutant clone. Aside from mutations previously implicated in clonal hematopoiesis, MDS, and leukemia (PIGA, BCOR and ASXL1), only one gene (ERCC6L) was recurrently affected in our cohort. Among the other mutations, those with translational consequences in pathways of hematopoietic growth and immunity were considered potential drivers: the strongest candidates were mutations in HLA alleles, STAT5B, and CAMK2G.
Although selection against specific HLA alleles has been proposed to drive 6p CN-LOH emergence[5–7], it has been difficult to exonerate other genes that are in linkage disequilibrium with the HLA locus and are also relevant to hematopoiesis and immunity (e.g. TNF or complement)[29]. Our finding of two patients with oligoclonal loss of HLA class I alleles, in whom specific HLA alleles were lost by two different mechanisms—loss-of-function mutation and 6p CN-LOH—strongly argues that in these patients loss of HLA alleles drives oligoclonal hematopoiesis (Figure 3). The recently reported nonsense mutation in the HLA*B 40:02 allele in a 7-year-old aAA patient [30] supports our findings, and provides further evidence that HLA class I loss may be a common and previously underappreciated mechanism behind clonal hematopoiesis in aAA. It is intriguing that oligoclonal hematopoiesis has also been observed with PIGA mutations, where multiple inactivating mutations can co-occur [23, 31]; this may be another instance of a more general phenomenon of immune selection leading to oligoclonal hematopoiesis. Future studies using targeted deep sequencing of HLA alleles will help to define the frequency of HLA loss and its prognostic implications [32].
Two other candidate driver mutations in our study—CAMK2G p.T306M and STAT5B p.N642H—affected key proliferative pathways. The first, CAMK2G p.T306M, involves a multifunctional serine-threonine protein kinase critical for Ca2+-based second messenger signaling and for proliferation of myeloid leukemias[33–34]. CAMK2G is inactivated by autophosphorylation of regulatory residues T306 and T307 (numbered T305 and T306 in the α-isoform), which greatly lower its affinity for Ca2+/calmodulin. Mutagenesis of T306 and T307 residues has been shown to abrogate kinase inhibition and increase its activity [26]. It is of interest that this mutation occurred in a patient on long-term cyclosporine therapy; a possible explanation could be that the mutation was selected for by cyclosporine, which activates CAMK2 though inhibition of calcineurin, a Ca2+/Calmodulin-dependent protein phosphatase [35]. The second mutation, STAT5B p.N642H, was previously demonstrated to increase activation of transcriptional activity by the STAT5B gene [24]. Although the lack of historical banked tissue from this patient precludes the evaluation of the timeline of clone emergence, it is intriguing to consider its potential relationship to the patient’s eltrombopag therapy. Enhanced STAT5B signaling conferred by the p.N642H mutation could render STAT5B–mutant cells hypersensitive to thrombopoietin, leading to clonal expansion. Longitudinal studies of aAA patients treated with thrombopoietin agonists and other therapies will be important to define effects of therapy on clonal emergence and its prognostic implications.
Our study has limitations. This is a cross-sectional study of a relatively small patient population with limited follow-up and, thus, cannot assess the prognostic implications of specific mutations. However, aAA is a rare disease; and at a median follow-up of 15 months (range 0–38), none of the patients had neoplastic transformation. Due to material availability, clonal architecture analysis was only performed for two patients with oligoclonal loss of HLA I alleles. Future longitudinal studies of multi-institutional patient cohorts, incorporating clonogenic assays and deep sequencing will be needed to determine the frequency and recurrence of mutations, to correlate mutational spectrum with treatment response and disease outcomes, and to evaluate clonal architecture and clonal dynamics. Our study does not include comparative WES of healthy controls. However, recent studies using WES of control populations have shown that only 2–3% of individuals and up to 10% of people over 65 years have clonal mutations; in contrast, healthy children and young adults have none or only rare and non-clonal somatic mutations[36–43]. Thus, clonal hematopoietic expansion in pediatric and young adult aAA patients is strikingly different from healthy hematopoiesis. It is intriguing to consider potential differences in the mutational spectrum between the pediatric patients and older adults, who are more likely to carry age-related somatic mutations associated with malignancy; future longitudinal studies comparing the mutational profiles and disease outcomes in patients of different age groups will help to better understand the prognostic implications of clonal hematopoiesis and its interaction with age. Finally, the low background mutation rate in younger patients may have caused us to underestimate the frequency of clonal hematopoiesis in children. Our study did not identify subclonal mutations with haploid allele frequencies below ∼10% as measured by WES (which corresponds to a ∼20% diploid clone size). Importantly, all of the identified somatic mutations comprise a large (>20%) fraction of patients’ hematopoietic production, and are thus likely to influence their hematologic presentation, response to therapy, and disease course.
In conclusion, we have shown that clonal hematopoiesis is present in the majority of aAA patients, including two thirds of patients with pediatric-onset aAA, and can occur early in disease. Our data demonstrate that in the younger patient population somatic mutations in aAA have only a partial overlap with mutations of MDS, and instead bear signatures of immune escape and proliferative signaling. Our finding of frequent clonal hematopoiesis in aAA expands the conceptual understanding of the pathophysiology of this non-neoplastic blood disorder, and demonstrates that clonal hematopoiesis in aAA extends beyond the known association with PIGA mutations. Prospective studies of clonal hematopoiesis in aAA and its relationship to aAA therapies and patient outcomes will be critical for understanding the determinants of disease response and transformation, and for designing personalized treatment strategies in aAA.
Table 2.
Clinical and Genetic Findings in Pediatric- and Adult-Onset aAA
| Patient Number |
Age Diagn. (yrs) |
Age WES (yrs) |
Sex | AA Duration (yrs) |
Severity | IST (Y/N) |
Time After IST (yrs) |
TL | TCR | Marrow Cellularity (%) |
WBC (103/µl)/ANC (cells/µl) |
Hgb (g/dL) |
Plt (103/µl) |
Disease Status at WES |
Karyotype | CN- LOH |
PNH Clone |
# Tier 1 & 2 Muts. |
Tier 1 Mutations | Tier 2 Mutations |
Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pediatric-Onset aAA | |||||||||||||||||||||
| 362.01 | 1.5 | 2 | F | 1.2 | SAA | Y | 1.2 | N | n/a | 50–60 | 4.6/2521 | 9.6 | 146 | PR | Normal | None | None | 0 | None | None | |
| 387.01 | 2 | 3 | M | 0.5 | SAA | Y | 0.5 | N | Nl | 60–70 | 5.5/1282 | 7.9 | 52 | PR | Normal | None | None | 0 | None | None | |
| 439.01 | 3 | 4 | F | 0.8 | SAA | Y | 0.3 | N | Nl | 30–50 | 4.2/546 | 9.1 | 60 | PR | 47,XX, +der(15)c[20] |
None | None | 0 | None | None | |
| 281.01 | 3 | 4 | M | 0.7 | SAA | N | n/a | L | n/a | 80–90 | 5.7/1140 | 11.1 | 37 | PR | Normal | 6p | None | 1 | HLA-A (Y142X) | None | |
| 3.01 | 4 | 5 | M | 1.0 | SAA | Y | 1.0 | N | n/a | 70–80 | 5.5/3267 | 10.7 | 39 | PR | Normal | None | None | 0 | None | None | |
| 118.01 | 5 | 34 | F | 29.0 | SAA | Y | 18.0 | n/a | Clonal | n/a | 3.6/2500 | 8 | 39 | PR | Normal | None | Large | 5 |
HTR4 (K220N), TAF1C (D21N), GZMM (R37C), PIGA (V432fs) |
BFAR (3’UTR) |
PTPRD (syn) |
| 263.01 | 5 | 5 | F | 0.5 | SAA | Y | 0.5 | N | n/a | 30–40 | 3.0/990 | 9.6 | 15 | PR | Normal | None | Subclinical | 2 | None, PIGA# |
CYP4F24P (NCExonic) |
|
| 56.01 | 6 | 14 | M | 8 | VSA A | Y | 8 | L | n/a | 80–90 | 5.1/1647 | 10.9 | 35 | Rel. | Normal | 6p | None | 0 | None | None |
LCT (syn) |
| 54.01 | 10 | 21 | M | 11.0 | SAA | Y | 7.0 | L | Nl | 15 | 2.7/713 | 12.2 | 100 | PR | Normal | 6p | None | 1 | HLA-B (Leu294fs) | None |
LYPD8 (upstr) |
| 45.01 | 14 | 20 | M | 6.0 | SAA | Y | 6.0 | L | Nl | 10–70 | 3.7/1628 | 15.5 | 114 | PR | Normal | None | None | 0 | None | None | |
| 58.01 | 14 | 25 | F | 11.0 | SAA | Y | 10.0 | N | n/a | 50–60 | 4.0/1750 | 9.9 | 246 | CR | Normal | None | Moderate | 4 |
CDC14A(V166M), ERCC6L(R372X), PIGA (ex 2 splice) |
CYCS (3’UTR) | |
| 376.01 | 15 | 15 | F | 0.5 | SAA | Y | 0.5 | N | Nl | 60–70 | 3.6/1418 | 11.5 | 115 | PR | Normal | None | None | 0 | None | None | |
| 312.01 | 15 | 16 | M | 1.0 | SAA | Y* | 1.0 | N | Clonal | 50–60 | 5.1/3254 | 15.7 | 151 | Rel. | Normal | 15q | Moderate | 2 | PIGA# |
MOSPD1 (3’UTR) |
|
| 252.01 | 16 | 19 | F | 2.0 | SAA | Y | 2.0 | n/a | n/a | 80–90 | 3.3/1752 | 10.3 | 196 | CR | Normal | None | Large | 4 |
BCOR (V594I), PIGA (A313fs) |
MSRA (3’UTR), EPG5 (3’UTR) |
|
| 145.01 | 17 | 18 | F | 0.6 | VSA A | Y | 0.3 | N | Indetr. | 30–40 | 4.5/1080 | 9.7 | 14 | NR | Normal | None | Subclinical | 3 |
ZSCAN5B (Q322X), PIGA (F259fs) |
IL22RA2 (3’UTR) | |
| 020.01 | 17 | 26 | F | 9.0 | SAA | Y | 6.0 | L | Nl | 40 | 4.8/1890 | 11.2 | 41 | Rel. | Normal | 5q | Moderate | 6 |
CAMK2G (T306M), BPTF (D312H), WDR18 (A156T), PIGA# |
PDE10A (3’UTR), SLC2A13 (3’UTR) |
|
| 385.01 | 18 | 18 | M | 0.1 | VSA A | N | 0.0 | N | Nl | 0–50 | 3.4/942 | 10.9 | 24 | Diagn. | Normal | None | n/a | 4 |
EMX2 (A172V), DSG1 (M208V), PIGA (K88N) |
LARP (3’UTR) |
OR10A
7 (syn) |
| 434.01 | 19 | 19 | M | 0.3 | SAA | Y | 0.3 | N | Nl | 80 | 3.8/2481 | 12.8 | 162 | CR | Normal | None | None | 0 | None | None |
CHPF2 (upstr) |
| Adult-Onset aAA | |||||||||||||||||||||
| 364.01 | 33 | 36 | F | 3.0 | SAA | Y | 3.0 | N | Nl | 60 | 7/3730 | 8.7 | 62 | Refr. | Normal | None | None | 11 |
SLC22A6 (R336C), STAT5B (N642H), WFDC12 (G27C), DNAJC24 (ex4 splice) |
OSBPL11 (3’UTR), SENP6(3’UTR), MET(3’UTR), NFIB(3’UTR), ZNF747(3’UTR ), BLCAP(3’UTR), CLCN5 (3’UTR) |
|
| 76.01 | 38 | 44 | F | 6.0 | NSA A | N | n/a | N | Nl | 75 | 3.2/2260 | 8.1 | 124 | NSAA | Normal | None | Large | 9 |
CACNB3 (T347M), SLC12A5(M1073R), HHIP (R434G), ERCC6L (P947T), PIGA (I61fs) |
MUL1 (3’UTR), HSPB2 (3’UTR), SCN2B (3’UTR), RBL1 (3’UTR) |
|
| 356.01 | 53 | 53 | F | 0.3 | VSA A | Y | 0.3 | N | Nl | 35 | 4.1/2390 | 10.2 | 190 | PR | Normal | None | None | 8 |
SYNPO2(Q709K), IL20RA(Q473K), FOS(G97R), NINL(R667C), SEPT3(R165H), ASXL1(C687fs) |
LMTK2 (5’UTR), ATP2B3 (3’UTR) |
|
| 390.01 | 61 | 61 | F | 0.4 | VSA A | Y | 0.3 | N | Nl | 25 | 3.2/1920 | 8.3 | 32 | PR | 46,XX,del(13) (q12q14)[2]/ 46,XX[18] |
6 WC | Subclinical | 1 | PIGA(F438del.L439del) | None | |
SAA, severe aAA; VSAA, very severe aAA; NSAA, non-severe aAA. TL, telomere lengths (N: ≥10th percentile of normal, L: <10th percentile of normal). Marrow cellularity, bone marrow cellularity at the time of WES, performed at the corresponding disease duration (AA Duration), time from immunosuppression (Time After IST), and Disease Status (CR, complete response; PR, partial response; Rel., relapse, Refr., refractory aAA, Diagn., diagnosis). TCR, T-cell receptor rearrangement (Nl: polyclonal; Clonal; Inder.: indeterminate). PNH Clone (None (<1%), Subclinical (1–10%), Moderate (10–50%), Large (>50%)).
: In agreement with published studies of PIGA gene sequencing in PNH [1, 2], three patients had flow-cytometric evidence of PNH for which we were unable to identify a mutation by WES or bi-directional Sanger sequencing of the PIGA exons and their immediately flanking regions. These were likely caused by multiple independent mutations in PIGA below the level of detection of WES and Sanger sequencing [3].
Acknowledgments
We thank all patients and their referring physicians for participation in our studies. We are grateful to Jian-Meng Fan, Ho-Sun Lam, Peter Nicholas, Donna Wilmoth and Laura Tooke for technical assistance and to Mahdi Sarmady and members of the Mason and Bessler laboratories for helpful discussions. This work was supported by NHLBI/NIH 5-T32-HL-07439-34, NHLBI/NIH K12 HL097064, AACR-Amgen, Inc. Fellowship in Clinical/Translational Cancer Research, and AA & MDS International Foundation Research Grant to D.B., NIH/NIGMS T32-GM008638 grant to J.R., NIH/NCATS UL1TR000003 to UPENN/CHOP ITMAT/CTRC and T.O., and NCI/NIH R01 CA105312, Buck Family Endowed Chair in Hematology, and NIH/NIDDK R24DK103001 to MB.
References
- 1.Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. N Engl J Med. 1997;336:1365–1372. doi: 10.1056/NEJM199705083361906. [DOI] [PubMed] [Google Scholar]
- 2.Socie G, Henry-Amar M, Bacigalupo A, Hows J, Tichelli A, Ljungman P, McCann SR, Frickhofen N, Van't Veer-Korthof E, Gluckman E. Malignant tumors occurring after treatment of aplastic anemia. European Bone Marrow Transplantation-Severe Aplastic Anaemia Working Party. N Engl J Med. 1993;329:1152–1157. doi: 10.1056/NEJM199310143291603. [DOI] [PubMed] [Google Scholar]
- 3.Young NS, Maciejewski JP, Sloand E, Chen G, Zeng W, Risitano A, Miyazato A. The relationship of aplastic anemia and PNH. Int J Hematol. 2002;76(Suppl 2):168–172. doi: 10.1007/BF03165111. [DOI] [PubMed] [Google Scholar]
- 4.Dunn DE, Tanawattanacharoen P, Boccuni P, Nagakura S, Green SW, Kirby MR, Kumar MS, Rosenfeld S, Young NS. Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Ann Intern Med. 1999;131:401–408. doi: 10.7326/0003-4819-131-6-199909210-00002. [DOI] [PubMed] [Google Scholar]
- 5.Katagiri T, Sato-Otsubo A, Kashiwase K, Morishima S, Sato Y, Mori Y, Kato M, Sanada M, Morishima Y, Hosokawa K, Sasaki Y, Ohtake S, Ogawa S, Nakao S. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood. 2011;118:6601–6609. doi: 10.1182/blood-2011-07-365189. [DOI] [PubMed] [Google Scholar]
- 6.Babushok DV, Xie HM, Roth JJ, Perdigones N, Olson TS, Cockroft JD, Gai X, Perin JC, Li Y, Paessler ME, Hakonarson H, Podsakoff GM, Mason PJ, Biegel JA, Bessler M. Single nucleotide polymorphism array analysis of bone marrow failure patients reveals characteristic patterns of genetic changes. Br J Haematol. 2013 doi: 10.1111/bjh.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Afable MG, 2nd, Wlodarski M, Makishima H, Shaik M, Sekeres MA, Tiu RV, Kalaycio M, O'Keefe CL, Maciejewski JP. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011;117:6876–6884. doi: 10.1182/blood-2010-11-314393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulasekararaj AG, Jiang J, Smith AE, Mohamedali AM, Mian S, Gandhi S, Gaken J, Czepulkowski B, Marsh JC, Mufti GJ. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014 doi: 10.1182/blood-2014-05-574889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane AA, Odejide O, Kopp N, Kim S, Yoda A, Erlich R, Wagle N, Abel GA, Rodig SJ, Antin JH, Weinstock DM. Low frequency clonal mutations recoverable by deep sequencing in patients with aplastic anemia. Leukemia. 2013;27:968–971. doi: 10.1038/leu.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heuser M, Schlarmann C, Dobbernack V, Panagiota V, Wiehlmann L, Walter C, Beier F, Ziegler P, Yun H, Kade S, Kirchner A, Huang L, Koenecke C, Eder M, Brummendorf TH, Dugas M, Ganser A, Thol F. Genetic characterization of acquired aplastic anemia by targeted sequencing. Haematologica. 2014;99:e165–e167. doi: 10.3324/haematol.2013.101642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dumitriu B, Feng X, Townsley DM, Ueda Y, Yoshizato T, Calado RT, Yang Y, Wakabayashi Y, Kajigaya S, Ogawa S, Zhu J, Young NS. Telomere attrition and candidate gene mutations preceding monosomy 7 in aplastic anemia. Blood. 2014 doi: 10.1182/blood-2014-10-607572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718–1721. [PubMed] [Google Scholar]
- 13.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008. [Google Scholar]
- 14.American Academy of Pediatrics Council on Child and Adolescent Health: Age limits of pediatrics. Pediatrics. 1988;81:736. [PubMed] [Google Scholar]
- 15.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–1792. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lind C, Ferriola D, Mackiewicz K, Heron S, Rogers M, Slavich L, Walker R, Hsiao T, McLaughlin L, D'Arcy M, Gai X, Goodridge D, Sayer D, Monos D. Next-generation sequencing: the solution for high-resolution, unambiguous human leukocyte antigen typing. Hum Immunol. 2010;71:1033–1042. doi: 10.1016/j.humimm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 19.Du HY, Idol R, Robledo S, Ivanovich J, An P, Londono-Vallejo A, Wilson DB, Mason PJ, Bessler M. Telomerase reverse transcriptase haploinsufficiency and telomere length in individuals with 5p- syndrome. Aging Cell. 2007;6:689–697. doi: 10.1111/j.1474-9726.2007.00324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo V, Lessin SR, Wilson RB, Rennert H, Tozer C, Benoit B, Leonard DG. Detection of clonal T-cell receptor gamma gene rearrangements using fluorescent-based PCR and automated high-resolution capillary electrophoresis. Mol Diagn. 2001;6:169–179. doi: 10.1054/modi.2001.27056. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. 1995;57:289–300. [Google Scholar]
- 23.Shen W, Clemente MJ, Hosono N, Yoshida K, Przychodzen B, Yoshizato T, Shiraishi Y, Miyano S, Ogawa S, Maciejewski JP, Makishima H. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Invest. 2014;124:4529–4538. doi: 10.1172/JCI74747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajala HL, Eldfors S, Kuusanmaki H, van Adrichem AJ, Olson T, Lagstrom S, Andersson EI, Jerez A, Clemente MJ, Yan Y, Zhang D, Awwad A, Ellonen P, Kallioniemi O, Wennerberg K, Porkka K, Maciejewski JP, Loughran TP, Jr, Heckman C, Mustjoki S. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121:4541–4550. doi: 10.1182/blood-2012-12-474577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patton BL, Miller SG, Kennedy MB. Activation of type II calcium/calmodulin-dependent protein kinase by Ca2+/calmodulin is inhibited by autophosphorylation of threonine within the calmodulin-binding domain. J Biol Chem. 1990;265:11204–11212. [PubMed] [Google Scholar]
- 26.Hanson PI, Schulman H. Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J Biol Chem. 1992;267:17216–17124. [PubMed] [Google Scholar]
- 27.Heuser M, Schlarmann C, Dobbernack D, Panagiota V, Wiehlmann L, Walter C, Beier F, Ziegler P, Yun H, Kade S, Kirchner A, Huang L, Koenecke C, Eder M, Brümmendorf TH, Dugas M, Ganser AFT. Blood. New Orleans, LA: 2013. Genetic Characterization Of Aplastic Anemia Patients By Targeted Sequencing American Society of Hematology; p. 2470. [Google Scholar]
- 28.Mikhailova N, Sessarego M, Fugazza G, Caimo A, De Filippi S, van Lint MT, Bregante S, Valeriani A, Mordini N, Lamparelli T, Gualandi F, Occhini D, Bacigalupo A. Cytogenetic abnormalities in patients with severe aplastic anemia. Haematologica. 1996;81:418–422. [PubMed] [Google Scholar]
- 29.Rioux JD, Goyette P, Vyse TJ, Hammarstrom L, Fernando MM, Green T, De Jager PL, Foisy S, Wang J, de Bakker PI, Leslie S, McVean G, Padyukov L, Alfredsson L, Annese V, Hafler DA, Pan-Hammarstrom Q, Matell R, Sawcer SJ, Compston AD, Cree BA, Mirel DB, Daly MJ, Behrens TW, Klareskog L, Gregersen PK, Oksenberg JR, Hauser SL. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18680–18685. doi: 10.1073/pnas.0909307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osumi T, Miharu M, Saji H, Kusunoki Y, Kojima H, Nakamura J, Shimada H. Nonsense mutation in HLA-B*40:02 in a case with acquired aplastic anaemia: a possible origin of clonal escape from autoimmune insult. Br J Haematol. 2013;162:706–707. doi: 10.1111/bjh.12395. [DOI] [PubMed] [Google Scholar]
- 31.Bessler M, Mason P, Hillmen P, Luzzatto L. Somatic mutations and cellular selection in paroxysmal nocturnal haemoglobinuria. Lancet. 1994;343:951–953. doi: 10.1016/s0140-6736(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 32.Betensky M, Babushok DV, Roth JJ, Mason PJ, Biegel JA, Lind C, Manos D, Bessler M, Olson TS. Clonal Evolution and Clinical Significance of Copy Number Neutral Loss of Heterozygosity of Chromosome Arm 6p in Acquired Aplastic Anemia. American Society of Hematology Annual Meeting. San Francisco. 2014 doi: 10.1016/j.cancergen.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Si J, Collins SJ. Activated Ca2+/calmodulin-dependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res. 2008;68:3733–3742. doi: 10.1158/0008-5472.CAN-07-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Si J, Mueller L, Collins SJ. CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J Clin Invest. 2007;117:1412–1421. doi: 10.1172/JCI30779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubokawa M, Nakamura K, Komagiri Y. Interaction between Calcineurin and Ca/Calmodulin Kinase-II in Modulating Cellular Functions. Enzyme Res. 2011;2011:587359. doi: 10.4061/2011/587359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O'Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL, Hundal J, Cook LL, Swift GW, Reed JP, Alldredge PA, Wylie TN, Walker JR, Watson MA, Heath SE, Shannon WD, Varghese N, Nagarajan R, Payton JE, Baty JD, Kulkarni S, Klco JM, Tomasson MH, Westervelt P, Walter MJ, Graubert TA, DiPersio JF, Ding L, Mardis ER, Wilson RK. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, Hassimi M, Socci N, Bhatt PK, Gonen M, Mason CE, Melnick A, Godley LA, Brennan CW, Abdel-Wahab O, Levine RL. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, Maragh M, Gilliland DG. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 39.Mossner M, Nolte F, Hutter G, Reins J, Klaumunzer M, Nowak V, Oblander J, Ackermann K, Will S, Rohl H, Neumann U, Neumann M, Hopfer O, Baldus CD, Hofmann WK, Nowak D. Skewed X-inactivation patterns in ageing healthy and myelodysplastic haematopoiesis determined by a pyrosequencing based transcriptional clonality assay. J Med Genet. 2013;50:108–117. doi: 10.1136/jmedgenet-2012-101093. [DOI] [PubMed] [Google Scholar]
- 40.Holstege H, Pfeiffer W, Sie D, Hulsman M, Nicholas TJ, Lee CC, Ross T, Lin J, Miller MA, Ylstra B, Meijers-Heijboer H, Brugman MH, Staal FJ, Holstege G, Reinders MJ, Harkins TT, Levy S, Sistermans EA. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res. 2014;24:733–742. doi: 10.1101/gr.162131.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC, Walter MJ, Mardis ER, Dipersio JF, Chen F, Wilson RK, Ley TJ, Ding L. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014 doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landen M, Hoglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, Gronberg H, Hultman CM, McCarroll SA. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med. 2014 doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med. 2014 doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]