

Abstract
RAD51 recombinase plays a critical role for cancer cell proliferation and survival. Targeting RAD51 is therefore an attractive strategy for treating difficult-to-treat cancers, e.g. triple negative breast cancers which are often resistant to existing therapeutics. To this end, we have designed, synthesized and evaluated a panel of new RAD51 inhibitors, denoted IBR compounds. Among these compounds, we have identified a novel small molecule RAD51 inhibitor, IBR120, which exhibited a 4.8-fold improved growth inhibition activity in triple negative human breast cancer cell line MBA-MD-468. IBR120 also inhibited the proliferation of a broad spectrum of other cancer cell types. Approximately 10-fold difference between the IC50 values in normal and cancer cells were observed. Moreover, IBR120 was capable of disrupting RAD51 multimerization, impairing homologous recombination repair, and inducing apoptotic cell death. Therefore, these novel RAD51 inhibitors may serve as potential candidates for the development of pharmaceutical strategies against difficult-to-treat cancers.
Keywords: RAD51 inhibitor, indole alkaloid, breast cancer, triple-negative, QSAR, synthesis
1. Introduction
Targeting DNA damage repair pathway as a novel means of treating cancer, especially difficult-to-treat cancers, has attracted much attention in the past a few years. Homologous recombination (HR) is one of the most prominent repair pathways and is required for cancer cells to undergo continuous proliferation under stress. Therefore, various efforts have been taken to inhibit or modulate this pathway, in the hope of rendering cancer cells vulnerable [1–4]. As one of the key players in HR, RAD51 is essential for DNA repair, proliferation and survival. RAD51 protein level is elevated in many cancer cells, contributing to their resistance to chemotherapy and the continuous cell proliferation [5–14]. Therefore, RAD51 inhibition has been pursued by several groups in search of a novel potential treatment for difficult-to-treat-cancers [2, 3, 15].
Recently, our group identified and validated a small molecule synthetic alkaloid, named IBR2 (Figure 1), which showed interesting RAD51 inhibition activities both in vitro and in vivo [15]. RAD51 was rapidly degraded in IBR2-treated cancer cells, and the homologous recombination repair was impaired, subsequently leading to cell death. Therefore, IBR2 represents a novel class of direct and specific RAD51 inhibitors [1–3, 15]. The IC50 values of the original IBR2 were in the range of 12–20 µM for most tested cancer cell lines. Its molecular scaffold contains a chiral center and the structure-activity relationship of this chirality has not been explored in our previous studies. It is well accepted that the interaction between chiral molecules and biological systems can provide more mechanistic insight and important clues for drug discovery and development [16]. Therefore, we sought to develop efficient and stereo-selective synthetic routes such that more potent analogues can be identified [17]. In this report, several types of IBR2 analogues IBR101–124 were designed, synthesized, modeled, and biologically evaluated. These new compounds constitute a focused compound library representing a diversity of modified scaffold or substituents (Figure 1). As a result, a 4.8-fold improved RAD51 inhibitor was identified, which was able to inhibit the growth of triple-negative breast cancer cells and a panel of other malignancies. This finding may provide support for innovative methods and developments in cancer treatment.

2. Results and Discussion
2.1 Synthesis of IBR2 analogues
1,2,3,4-tetrahydroisoquinoline indole alkaloids IBR101–114 were stereoselectively synthesized by the addition of N-Boc-3-bromo-indole 1 to the chiral benzylidene sulfinamide 2 as key steps[17]. 2,3-dihydro-1H–benzo[c]azepine analogues IBR115–116 were stereoselectively synthesized using the bifunctional cinchona alkaloid-thiourea catalyzed addition of unprotected indole 3 to the sulfonyl amide 4 (Scheme 1) [17, 18].

Synthesis of optically pure indazole derivative IBR117 embarked on the reaction of the chiral benzylidene sulfinamide 2 [17] and 3-Bromoindazole 5. The desired indazolylated adduct 6 was obtained in medium yield (43%) and diastereoselectivity (65% dr) (Scheme 2) [19]. Protection of 6 with Boc2O gave compounds 7 and 8 in 66% and 14% yields, respectively. To our delight, the diastereoisomers 7 and 8 could be readily separated by silica gel column chromatography. Then, starting with chiral sulfinamide 8, HCl-mediated removal of the tert-butanesulfinyl group provided the amine product 9 in 90% yield. Subjecting the amine 9 to Pd/C-catalyzed hydrogenation followed by exposure of the resultant alcohol to CbzCl/DIPEA gave the compound 10 in 61% yield over 2 steps. Alcohol 10 was further mesylated to give compound 11 in 91% yield. Once hydrogenation of the mesylate 11 was carried out with Pd/C as catalyst in MeOH, the desired cyclic amine 12 was isolated in 94% yield, which led directly to IBR117 in 61% yield via deprotection with NaOMe/MeOH and subsequent benzylsulfonylation. Utilizing similar reaction procedures, the S configuration indazole derivative IBR118 was also prepared starting from chiral sulfinamide 7 in 37% yield over 5 steps (Scheme 3).


Chiral thiazolylamine derivative IBR119 was synthesized starting from N-Boc-2-amino-5-bromothiazole 17 and chiral benzylidene sulfinamide 2 (Scheme 4). Addition proceeded well by treatment of compound 2 with N-Boc-2-amino-5-lithiothiazole (prepared in situ via reaction of 17 with n-BuLi) at −78 °C, and the desired sulfinamides 18 and 19 were provided in 39% yield with medium diastereoselectivity (59% dr). Diastereoisomers 18 and 19 (1:4 isolated molar ratio) were then successfully separated through silica gel chromatography (isolated yield: 8% and 31%, respectively). The major diastereomer compound 19 was chosen for further modification. tert-Butanesulfinyl group of 19 was removed by treatment with 4M HCl in dioxane and the amine 20 was obtained in almost quantitative yield. Benzylsulfonylation of 20 gave compound 21 in 97% yield, which was subjected to Pd/C-catalyzed hydrogenation to afford the alcohol 22 in 80% yield. Mesylation of 22 by treatment with MsCl/DMAP/DIPEA followed by exposure of resultant mesylate to KHDMS in THF furnished the desired cyclic compound 23. Synthesis of IBR119 was finally accomplished via TFA-mediated removal of Boc protecting group in 23.

Synthesis of isoindolinyl derivative IBR120 started from the indole-derivated compound 24, which was prepared according to our previous report [17, 18]. A direct OsO4-NaIO4 mediated oxidative cleavage reaction on unprotected compound 24 in the presence of 2, 6-lutidine [20] was carried out, followed by reduction with NaBH4 and resulted in alcohol 25 (82%). Alcohol 25 was then treated with mesyl chloride and an appropriate base in DCM to give the corresponding mesylate, which was used as an intermediate for the following cyclisation reaction to form IBR120. Our first attempt on cyclization of alcohol 25 using KHMDS as a base in THF gave a racemic product. Reaction conditions with milder bases were then investigated (Table S1 in Supporting Information). Reactions with Et3N in DCM at r.t. or in dioxane under reflux did not facilitate the conversion. The use of K2CO3 in MeOH gave racemic cyclization product in 42 % yield. Finally, with Hünig's base in acetonitrile, the desired product IBR120 was obtained in 53 % yield with > 95% ee (Scheme 5). Utilizing the similar reaction procedures, the S configuration isomer IBR121 was also prepared starting from the indole-derivated compound 26 (Scheme 6). Chiral HPLC analysis confirmed high enantiomeric purities of IBR120 and IBR121 (See Figure S1-S3 in Supporting Information for representative HPLC profiles).


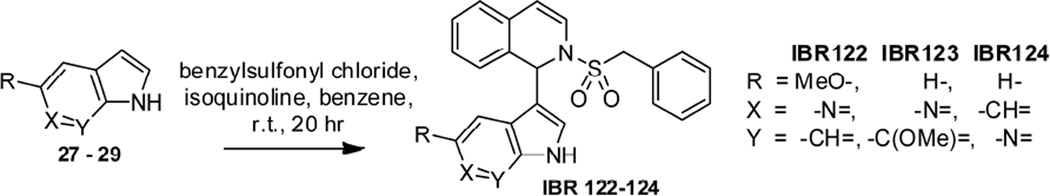
To explore possible effects of other indole bioisosteres on the bioactivity, racemic azaindole derivatives IBR122–124 were synthesized using a facile one-pot synthetic method as described in our previous report [15], starting from azaindoles 27, 28, and 29, respectively (Scheme 7).
2.2 IBR2 Analogues Inhibit Growth of Triple-Negative Human Breast Cancer
Triple-negative breast cancer is often easy to metastasize and difficult to treat using existing therapeutics [21, 22]. As a broad spectrum anti-cancer agent, IBR2 was able to inhibit a number of difficult-to-treat cancers [15]. To test the possibility of inhibiting triple-negative breast cancer, we screened the newly synthesized IBR2 analogues IBR101–124 of their growth inhibition abilities using an XTT assay. As shown in Table 1, most of these synthetic IBR2 analogues can inhibit the growth of triple-negative human breast cancer cell line MBA-MD-468.
Table 1.
IBR2 analogues inhibit the growth of the triple-negative human breast cancer cell line MDA-MB-468.
| Cmpd. | IC50 (mM) | Cmpd. | IC50 (mM) | Cmpd. | IC50 (mM) |
|---|---|---|---|---|---|
| IBR2 (rac) | 14.8 | ||||
| IBR101 (R) | 11.7 | IBR109 (R) | 14.8 | IBR117 (R) | 9.5 |
| IBR102 (S) | 13.2 | IBR110 (S) | 16.7 | IBR118 (S) | 14.3 |
| IBR103 (R) | 12.7 | IBR111 (R) | 11.1 | IBR119 (S) | >25 |
| IBR104 (S) | 19.2 | IBR112 (S) | 22.2 | IBR120 (R) | 3.1 |
| IBR105 (R) | 14.2 | IBR113 (R) | 13.6 | IBR121 (S) | >12 |
| IBR106 (S) | 17.2 | IBR114 (S) | 20.7 | IBR122 (rac) | 12.7 |
| IBR107 (R) | 11.2 | IBR115 (R) | 14.7 | IBR123 (rac) | 6.3 |
| IBR108 (S) | 25.7 | IBR116 (S) | >50 | IBR124 (rac) | 6.2 |
We found that the half inhibitory concentrations (IC50) of 1,2,3,4-tetrahydroisoquinoline analogues IBR101, 102, 103, 105, 107, 109, 111, 113, 118, 2,3-dihydro-1H–benzo[c]azepine analogue IBR115, and 1,2-dihydroisoquinoline analogue IBR122 were slightly lower than that of the parental compound IBR2; while 1,2,3,4-tetrahydroisoquinoline analogue IBR117, isoindoline analogue IBR120, and 1,2-dihydroisoquinoline analogues IBR123, 124 were significantly lower than that of IBR2. In all the successfully synthesized enantiomers, the R configuration consistently exhibited better bioactivity than the S configuration. Among all these analogues, IBR120 exhibited a 4.8-fold increase in activity (IC50 = 3.1 µM), followed by IBR124, 123 and 117, with 1.6~2.4-fold increase in activities (IC50 = 6.2, 6.3, 9.5 µM, respecitvely), compared to the parental compound IBR2 (IC50 = 14.8 µM).
2.3 IBR120 inhibits a panel of cancer cell lines growth
We then tested the best analogue IBR120 on the growth inhibition of a panel of cancer cell lines using XTT assay. These cell lines included human Chronic Myelogenous Leukemia cell line K562, human breast cancer cell lines MCF7, MDA-MB-231, MDA-MB-361, MDA-MB-435, MDA-MB-468, Hs578-T, and T47D, human Osteosarcoma cell line U2OS, human Glioblastoma cell line T98G, human Cervical Cancer cell line HeLa, as well as human mammary gland normal epithelial cell line MCF10A (normal control). As shown in Table 2, while essentially nontoxic to the normal cell line MCF10A (IC50 > 30 µM), IBR120 exhibited killing effect in most tested cancer cell lines with IC50 values in low micromolar range (3~5 µM). This approximately 10-fold difference between the IC50 values in normal and cancer cells demonstrates a potential therapeutic window for future drug development.
Table 2.
Inhibition activity of IBR120 on a panel of cancer cell lines.
| Cell line | Cell Type [23] | IC50 (µM) | |
|---|---|---|---|
| K562 | Chronic Myelogenous Leukemia | 3.6 | ± 0.7 |
| MCF7 | Mammary Gland Adenocarcinoma | 3.1 | ± 0.2 |
| MDA-MB-231 | Mammary Gland Adenocarcinoma | 3.5 | ± 0.4 |
| MDA-MB-361 | Mammary Gland Adenocarcinoma | 4.5 | ± 0.7 |
| MDA-MB-468 | Mammary Gland Adenocarcinoma | 3.1 | ± 0.4 |
| MDA-MB-435 | Melanoma / Mammary Gland Ductal Carcinoma | 4.0 | ± 0.7 |
| Hs578-T | Mammary Gland Carcinoma | 4.9 | ± 1.0 |
| T47D | Mammary Gland Ductal Carcinoma | 6.3 | ± 1.5 |
| U2OS | Osteosarcoma | 4.7 | ± 0.5 |
| T98G | Glioblastoma Multiforme | 9.5 | ± 0.9 |
| HeLa | Cervix Adenocarcinoma | 3.6 | ± 0.6 |
| MCF10A | Mammary Gland, Normal epithelial cell line | > 30 | |
2.4 Molecular Docking Model
We have previously shown that the phenylalanine binding site on the RAD51 core domain was responsible for IBR2 binding [15]. Blocking of this site inhibits RAD51 multimerization and leads to deficiencies in DNA repair; while mutation in the center of this site abolishes the binding [15]. ICM software was employed to prepare the structures of RAD51 core domain, and to perform the molecular docking studies with IBR2 analogues. The crystal structure of human RAD51 in complex with BRC4 peptide (PDB No. 1N0W) [24] was chosen for molecular docking studies. Initially a docking box containing all the atoms of RAD51 residues within 5Å distance from the BRC4 peptide in 1N0W was used; and a “blind docking” procedure was performed using IBR2 as probe. Then the docking box was fixed and used for all docking studies with IBR2 analogues; the amino acid residues within the box contain RAD51 residues M158, Y159, I160, F167, P168, L171, S183, V185, L186, D187, N188, V189, A190, Y191, A192, R193, A194, F195, H199, Q202, L203, L204, Y205, Q206, A207, S208, A209, M210, V212, E213, Y216, L219, R247, R250, M251, L252, R254, L255, E258, F259. Molecular docking was then performed following rigid docking protocols; docking scores were collected and conformational analysis was performed based on the results.
Many of the residues in the binding site are hydrophobic and therefore provides hydrophobic surface for interacting with the aromatic ring systems of the IBR compounds. Three potential hydrogen bonding sites were identified between IBR120 and RAD51 residues V189, Y191, and Q206. The distance between the indole nitrogen atom and the backbone carbonyl oxygen of V189 was 3.3 Å, suggesting a hydrogen bond of moderate strength may be involved in the binding. This notion is partly consistent with our previous finding that methylation of the indolyl nitrogen resulted in loss of activity [15]. An alternative explanation for the loss of activity in the N-methylated analogue could be that the methyl group might introduce unfavorable steric interaction with the corresponding amino acid residues on RAD51. Moreover, both of the sulfonyl oxygen atoms seem to be involved in weak hydrogen bonding, which could explain the observed loss of function when changing the sulphonyl into carbonyl group as our previous results showed [15] (Figure 2).

2.5 Quantitative Structure Activity Relationship (QSAR) Analysis
The phenyl moiety of IBR2 is situated in a spatially confined environment on RAD51 protein [15]. In this work, we have modified the indolyl and isoquinoline moieties of IBR2 to further explore the structure-activity relationship. Our new IBR compounds described in this work covered the following structural diversity of interest: (1) different bioisosteres for replacing the indolyl ring, (2) chiral center of IBR2 analogues, and (3) variable sizes of the central ring.
By modifying indole moiety to various bioisosteres, a potentially favorable modification site was identified. We found that compared with IBR2, IBR123 and 124 showed more than 2-fold increase of activity; and both IBR123 and 124 have included a potential hydrogen bond acceptor at the 7-position of the indolyl ring, suggesting further improvement could be made by utilizing this feature. Second, the growth inhibition assay results indicated that, compared with S isomers (average pIC50 = 4.70), the R isomers (average pIC50 = 4.96) are generally more active (Student’s t-test, p = 0.0047) as summarized in Figure 3a. Moreover, increasing the central ring size from six to seven resulted in minimal change of activity of the R isomer (IBR115), but almost complete loss of function in the S isomer (IBR116). Interestingly, decreasing the central ring size from six to five resulted in more than 4-fold increase of activity (IBR120). If we assume that other confounding factors, such as assay variability, solubility differences, or variations in protein binding, remain similar among all the compound structures, these observed effects on activity could be viewed as results of certain molecular recognition driven events. As we have discussed in previous sections, the docked conformation of IBR120 indeed showed several favorable features of binding.

To better understand how these structural factors might have invoked such changes in activity, we decided to further look into the optimal docked conformations of all compounds. We hypothesized that the various sizes of the ring and substitutents could result in differences in the optimal orientations of the indolyl, phenyl, and sulphonyl groups. This orientation can be readily quantified with a dihedral angle in the docking model (C-C-N-S, as shown in Figure 3b, inset). A cluster analysis (k-means, k =2) of this dihedral angle with regard to the IC50’s indicated that the dihedral angles clustered around 75~100 degrees usually correlated with better activity; while the dihedral angles of −60~-110 degrees correlated with poorer activity (Figure 3b). Therefore the combined conformational effect elicited by the ring size and substituents determines the activity of a certain compound, at least in part.
We then attempted to build a predicative multivariate linear regression model based on the above observations obtained from all the synthesized chiral and racemic compounds. First, the molecules were divided into a test set (9 compounds) and a training set (16 compounds) using a random number generator to minimize potential bias. We then chose a handful of molecular descriptors (Molecular Weight, LogP, LogS, Polar Surface Area, Molecular Volume, Heat of Formation) to account for the difference in the physicochemical properties of the small molecules, and docking scores as indicators of overall binding capacity with RAD51. And we generated a first predicative model (Multiple R-squared: 0.77, Adjusted R-squared: 0.57, p-value: 0.039, see Figure S4 in Supporting Information). Next, based on the above exploratory analysis, we then included two extra parameters (dihedral angle and chirality) to account for the important conformational preference as we discussed above; and a much improved predicative model was obtained (Multiple R-squared: 0.98, Adjusted R-squared: 0.96, p-value: 0.0001, Figure 3c, Training Set). Using this model, we were able to predict the growth inhibition activity of most compounds in the test tests within 95% confidence interval (Figure 3d, Testing Set).
2.6 IBR120 inhibits RAD51 multimerization
On the RAD51 core domain, a hydrophobic pocket formed between β-strand B3 and α-helix A4 of RAD51 [24] is critical for RAD51 multimerization, which is also responsible for IBR2 competitive binding [15]. Therefore, analogous IBR compounds were expected to inhibit RAD51 multimerization. To demonstrate that IBR120 can indeed do this, we compared the gel filtration profile of RAD51 multimerization in the presence of IBR120 or vehicle (DMSO) alone. In the presence of IBR120, the RAD51 elution profile exhibited a major peak consistent with the molecular weight of a monomer, while in the absence of IBR120, the majority of RAD51 formed multimers (Figure 4), indicating that IBR120, similar to IBR2, can inhibit RAD51 multimerization.

2.7 IBR120 inhibits Homologous Recombination (HR) repair
Inhibition of RAD51 function will lead to failure in HR repair. To test this possibility, we used an I-SceI inducible gene conversion assay that measures the DNA double strand break repair frequency by detecting successful restoration of a fluorescent GFP from the repair substrate DR-GFP [15, 25]. DR-GFP, consisting of two nonfluorescent GFP derivatives, SceGFP and iGFP, was first stably integrated into HeLa cells. Upon I-SceI expression by transient transfection for 24 hours to induce DSBs, cells were treated with DMSO, 10 or 20 µM of IBR120 or IBR2 for another 24 hours and the GFP positive population resulting from successful recombination was measured by flow cytometry. As shown in Figure 5, the HR frequency was significantly reduced after IBR120 treatment in a dose dependent manner. Compared with IBR2 treatment, IBR120 treatment consistently led to greater depression of HR repair activity.

3. Conclusion
In summary, we have developed several stereoselective synthetic routes to synthesize chiral IBR2 analogues. One route featured the addition of N-Boc-3-bromo-indole 1, 3-bromoindazole 5 and N-Boc-2-amino-5-bromothiazole 17 to the benzylidene sulfinamide 2 provided the separable diasteroisomers in medium to good yields. The biological assays and QSAR studies clearly demonstrated that R configuration exhibited improved bioactivity than S configuration, and the 5-membered central ring system proves to be the most active; these effects can be structurally correlated to the fine-tuning of the orientation of the two aromatic groups. As a result, these modifications led to more than 4-fold increase of growth inhibition activity in a panel of cancer cells, providing a significant milestone towards better RAD51 inhibitors.
Our synthetic scheme for most of the described IBR compounds relies on the stereoselective arylation of chiral benzylidene sulfinamide 2, using in situ generated lithium reagent, followed by separation of diastereomers, removal of chiral auxiliary group, and further modifications thereafter. This route calls for a total of 6~10 steps of conversions to obtain the target molecules, in which a chromatographic separation of diastereomers is indispensable. These undesirable hurdles can be overcome in further optimization of the synthetic route. Indeed, in our synthesis of IBR120, we have employed catalytic stereoselective arylation methods to satisfactorily obtain the chiral starting material 24, as previously described [17, 18]. We have then improved the oxidation/reduction [20] and ring closure reactions to achieve moderate to high yields of the desired enantiomers. It is noteworthy that these steps were performed under very mild conditions, and could be telescoped with some adjustments, which is a very attractive feature of this synthetic route. With further optimization, next generation of IBR120 analogues can be synthesized and screened for improved activity.
We have previously shown that IBR2 directly target RAD51 both in vitro and in cells [15]. In this work, we demonstrate that IBR120 can disrupt RAD51 multimer formation in vitro, and inhibit HR repair in cells, leading to growth inhibition and cell death. Our results indicate that IBR120 has similar biochemical and cellular consequences as IBR2, and suggest they may share an identical molecular target. Notably, the central ring structure in IBR120 is only one carbon smaller than that in IBR2, and this structural similarity suggests they might share similar pharmacokinetic and pharmacodynamic properties as well, which awaits further in-depth studies in vivo. With the quantitative structure-activity analysis in this work, further modifications are possible and necessary for this type of molecules to become clinically useful in the future.
IBR120 showed consistent potency in various cancer cell lines with different cancer origins and genotypes, suggesting IBR120 is an effective anti-cancer agent that might be used for various types of tumors. Taken together, we expect further improvement can be achieved based on these findings and may lead to the discovery and development of novel pharmaceutical strategies against difficult-to-treat cancers.
4. Experimental Protocols
4.1 Cell lines and antibodies
Human leukemia cell line K562 and human breast cancer cell line T47D were maintained in RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Human breast cancer cell lines MCF7, MDA-MB-231, MDA-MB-361, MDA-MB-435, MDA-MB468, Hs578-T, human osteosarcoma cell line U20S, human glioblastoma cell line T98G and human cervical adenocarcinoma cell line HeLa were maintained in low glucose Dulbecco Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% FBS and 1% penicillin-streptomycin. Human normal mammary epithelical cell line MCF10A was maintained in DMEM/F12 (50:50) medium (Invitrogen) supplemented with 5% horse serum, 20 ng/mL of epidermal growth factor, 0.5 mg/mL of hydrocortisone, 100 ng/mL of Cholera Toxin, 10 µg/mL of insulin, and 1% penicillin-streptomycin. To establish HeLa cells that stably expressed DR-GFP construct, cells were transfected with DR-GFP plasmid and selected with 2 µg/mL puromycin. Antibody sources were: mouse anti-RAD51 clone 14B4 and mouse anti-p84 (GeneTex), and secondary antibodies conjugated with Horseradish Peroxidase (GeneTex).
4.2 Cell killing assay
Standard XTT assays with a four-day drug treatment procedure were performed to measure the dose dependent cytotoxicity of IBR analogs in cultured cells. In brief, cells were plated on 96-well dishes one day before the drug treatment, followed by drug treatment on day 2 and XTT assay on day 6 after drug addition by using a commercial cell proliferation kit (Roche Scientific) following the instructions. Triplicate sets were measured and compiled for final data presentation.
4.3 Homologous Recombination Assay
As previously described [15], HeLa cells stably expressing DR-GFP were transfected with the I-Sce expression vector pCBASce and treated with compounds or DMSO. Cells were then trypsinized and subjected to flow cytometry. GFP positive cell percentages from three independent experiments were summarized as means±SD.
4.4 Molecular Modeling
The molecular docking was carried out following a previously established protocol [15]. RAD51 coordinates were from PDB (Accession No: 1N0W). RAD51 residues containing atoms within 5Å distance from the BRC4 peptide in 1N0W was designated as binding site for docking, including residues M158, Y159, I160, F167, P168, L171, S183, V185, L186, D187, N188, V189, A190, Y191, A192, R193, A194, F195, H199, Q202, L203, L204, Y205, Q206, A207, S208, A209, M210, V212, E213, Y216, L219, R247, R250, M251, L252, R254, L255, E258, F259. Structures of small molecules were generated and optimized and molecular docking was performed using ICM Pro (Molsoft), following standard procedures as described by the software manual, using default docking parameters at thoroughness = 5. Docked conformations with RMSD < 2Å were considered acceptable and kept for future analysis. Dihedral angles in the lowest energy docked conformation were recorded. Statistic analysis, clustering analysis, and predictive multivariate linear model building was performed using R (Version 3.1.0).
4.5 Multimer Formation Assay
Multimer formation assay was performed following a previously established protocol [15]. A mixture of RAD51 (3.2 µg) and IBR120 (1:10 molar ratio) was incubated for 15 min at 37°C, supplemented with buffer (50mM triethanolamine-HCl [pH7.5], 0.5mM Mg(OAc)2, 1mM DTT, 2mM ATP and 100 µg/ml BSA, total volume 20 µl) and incubated for 15min. The mixture was loaded onto a 2.4 ml Superdex 200 PC 3.2/30 column (Pharmacia) equilibrated with the same buffer as previously described[15]. Fractions (50 µl) were collected and 0.5 µl of each fraction was blotted onto PVDF membrane. RAD51 was detected using anti-RAD51 antibody (mAb 14B4, GeneTex).
4.6 Synthesis of IBR derivatives
4.6.1 Material and Methods
All reagents were used as received from commercial sources, unless specified otherwise, or prepared as described in the literature. Reactions requiring anhydrous conditions were performed in vacuum heat-dried glassware under nitrogen atmosphere. Reaction mixtures were stirred magnetically. DMF, dichloromethane and pyridine were distilled from CaH2. 1H NMR spectra were recorded at either 400 MHz or 500 MHz. 13C NMR spectra were recorded at either 125 MHz or 100 MHz. Chemical shifts (d) are reported in ppm, and coupling constants (J) are in Hz. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet.
4.6.2 (R)-2-Methylpropane-2-sulfinic acid [[2-(2-benzyloxy-ethyl)phenyl]-(1H-indazol-3-yl)-methyl]amide (6)
The solution of compound 5 (1.34 g, 6.85 mmol) in THF (40 mL) was cooled to −78 °C and n-BuLi (1.6 M in hexane, 4.28 mL, 6.95 mmol) was added dropwise. The resultant solution was stirred at −78 °C for 5 min. After that, t-BuLi (1.7 M in pentane, 8.06 mL, 13.70 mmol) was added dropwise and the resultant solution was stirred for 15 min at −78 °C. Then, a solution of compound 2[17] (2.35 g, 6.85 mmol) in THF (8 mL) was added dropwise. The mixture was stirred for 1h at −78 °C before saturated aqueous NH4Cl (3 mL) was added to quench the reaction. After the mixture was warmed up to room temperature, the mixture was poured to H2O (200 mL) and extracted with CH2Cl2 (3 × 60 mL). The combined organic phases were dried over anhydrous Na2SO4. The solvent was removed in vacuo and the resultant residue was purified by silica gel chromatography (hexane / EtOAc / NH3 = 100 : 50 : 1) to give compound 6 (1.36 g, 43%) as a yellow foam. 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 8.0 Hz, 0.17H), 7.37–7.33 (m, 1.83H), 7.31–7.23 (m, 8H), 7.17–7.06 (m, 2H), 6.91 (t, J = 8.8 Hz, 0.83H), 6.83 (d, J = 9.0 Hz, 0.17H), 6.78 (t, J = 7.5 Hz, 0.17H), 6.73 (t, J = 8.0 Hz, 0.83H), 6.36 (d, J = 6.5 Hz, 0.83H), 6.24 (d, J = 2.5 Hz, 0.17H), 6.00 (d, J = 6.5 Hz, 1H), 4.48 (s, 1.66H), 4.28–4.22 (m, 0.34H), 3.78–3.69 (m, 1.66H), 3.46–3.41 (m, 0.34H), 3.33–3.23 (m, 1.66H), 3.20–3.07 (m, 0.34H), 1.29 (s, 1.5H), 1.27 (s, 7.5H); 13C NMR (125 MHz, CDCl3) δ 144.9, 144.9, 141.5, 141.5, 139.5, 138.5, 138.4, 138.1, 136.5, 130.9, 130.3, 128.8, 128.5, 128.5, 128.4, 128.0, 127.8, 127.7, 127.7, 127.5, 126.9, 126.8, 126.5, 126.4, 120.9, 120.9, 120.5, 120.5, 120.4, 120.3, 110.7, 110.7, 73.2, 72.9, 71.1, 70.7, 56.9, 56.6, 54.5, 54.4, 33.0, 32.8, 23.4, 23.0; MS (ESI) m/z 484 (M + Na+); HRMS Calcd for C27H31N3O2SNa (M + Na+), 484.2035 Found: 484.2021.
4.6.3 3-[(S)-[2-(2-Benzyloxyethyl)phenyl]-((R)-2-methylpropane-2-sulfinylamino)methyl]indazole-1-carboxylic acid tert-butyl ester (7) and 3-[(R)-[2-(2-Benzyloxyethyl)phenyl]-((R)-2-methyl-propane-2-sulfinylamino)methyl]indazole-1-carboxylic acid tert-butyl ester (8)
To a 0 °C solution of compound 6 (706 mg, 1.53 mmol) in CH2Cl2 (20 mL) was added DMAP (5.0 mg, 0.04 mmol) followed by a solution of Boc2O (350 mg, 1.60 mmol) in CH2Cl2 (2.0 mL). The mixture was warmed up to room temperature and stirred for 1h. Removal of all the solvent in vacuo resulted in a residue, which was purified by silica gel chromatography (hexane / EtOAc = 3 : 1 to 2 : 1) to give compound 8 (less polar, 117 mg, 14%) as an oil and compound 7 (more polar, 565 mg, 66% ) as an oil.
Compound 7: [α]D20 = +7.3 (c 0.9 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.44 (t, J = 8.0Hz, 1H), 7.33–7.19 (m, 8H), 7.15–7.10 (m, 2H), 6.42 (d, J = 3.0 Hz, 1H), 4.54–4.48 (m, 2H), 4.28 (d, J = 3.0 Hz, 1H), 3.86–3.76 (m, 2H), 3.27 (t, J = 6.5 Hz, 2H), 1.70 (s, 9H), 1.25 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 150.9, 149.4, 141.2, 138.4, 138.2, 138.0, 130.8, 128.9, 128.7, 128.7, 128.5, 127.7, 127.6, 127.0, 124.3, 123.4, 121.7, 114.9, 84.7, 73.1, 70.9, 56.5, 54.1, 32.5, 28.4, 23.0; MS (ESI) m/z 584 (M + Na+); HRMS Calcd for C32H39N3O4SNa (M + Na+), 584.2559 Found: 584.2560.
Compound 8: [α]D20 = −112 (c 1.1 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 8.0 Hz, 1H), 7.39 (dd, J = 18.5, 7.5 Hz, 2H), 7.32–7.17 (m, 9H), 7.02–6.96 (m, 2H), 6.26 (d, J = 2.5 Hz, 1H), 4.44–4.36 (m, 2H), 3.69–3.61 (m, 2H), 3.29–3.23 (m, 1H), 3.16–3.10 (m, 1H), 1.71 (s, 9H), 1.25 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 150.8, 149.2, 141.2, 138.5, 138.4, 137.4, 130.8, 130.3, 129.0, 128.7, 128.5, 127.7, 127.6, 127.1, 123.8, 123.6, 121.2, 114.8, 84.8, 73.1, 70.9, 56.1, 53.4, 33.0, 28.4, 22.9; MS (ESI) m/z 584 (M + Na+); HRMS Calcd for C32H39N3O4SNa (M + Na+), 584.2559 Found: 584.2559.
4.6.4 3-[(R)-Amino-[2-(2-benzyloxyethyl)phenyl]methyl]indazole-1-carboxylic acid tert-butyl ester (9)
To a solution of compound 8 (118 mg, 0.21 mmol) in MeOH (2.5 mL) was added 4M HCl solution (in dioxane, 2.5 mL). After the mixture was stirred at room temperature for 30 min, the mixture was diluted with H2O (100 mL) and saturated aqueous NaHCO3 (20 mL) was added. Then, the resultant aqueous solution was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with brine, and then dried over anhydrous Na2SO4. After filtration and removal of the solvent in vacuo, the residue was purified by silica gel column chromatography (CH2Cl2 / MeOH / NH3 = 200 : 10 : 1) to afford compound 9 (87 mg, 90%) as a clear oil. [α]D20 = −85 (c 1.0 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 9.5 Hz, 1H), 7.41 (t, J = 7.3 Hz, 1H), 7.28–7.21 (m, 8H), 7.15–7.10 (m, 2H), 7.03 (t, J = 7.5 Hz, 1H), 5.89 (s, 1H), 4.51 (s, 2H), 3.78 (t, J = 8.0 Hz, 2H), 3.22 (t, J = 6.5 Hz, 2H), 2.98 (br, 2H), 1.73 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 154.0, 149.5, 140.9, 140.4, 138.3, 137.6, 130.6, 128.9, 128.6, 128.2, 128.0, 127.9, 127.8, 127.1, 124.1, 123.4, 121.5, 114.7, 85.0, 73.2, 71.3, 51.1, 33.1, 28.4; MS (ESI) m/z 480 (M + Na+); HRMS Calcd for C28H32N3O3 (M + H+), 458.2444 Found: 458.2424.
4.6.5 3-[(R)-Benzyloxycarbonylamino-[2-(2-hydroxyethyl)phenyl]methyl]indazole-1-carboxylic acid tert-butyl ester (10)
To a stirred solution of 9 (86 mg, 0.19 mmol) in MeOH (8 mL) was added 4M HCl in dioxane (0.4 mL), followed by Pd / C (50 mg, 10% Pd). The mixture was hydrogenated at room temperature under 1 atm for 1h. Then, the mixture was filtrated and the filtrate was neutralized with a NaHCO3 aqueous solution. The resulted mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with brine, and then dried over anhydrous Na2SO4. After filtration and removal of the solvent in vacuo, the residue was solved in CH2Cl2 (3 mL) and the solvent was cooled to 0°C. To this solvent was added DIPEA (40 µL, 0.23 mmol) followed by CbzCl (40 mg, 0.23 mmol) dropwise. The mixture was stirred at 0°C for 20min. Then, all the solvent was removed in vacuo and the residue was purified by silica gel chromatography (hexane / EtOAc = 2 : 1) to give the compound 10 (57 mg, 61%) as a oil. [α]D20 = −95 (c 0.9 CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.4 Hz, 1H), 7.44 (ddd, J = 8.4, 7.2, 4.2 Hz, 1H), 7.33–7.22 (m, 9H), 7.16–7.11 (m, 2H), 6.66 (d, J = 7.6 Hz, 1H), 6.55 (d, J = 6.4 Hz, 1H), 5.14–5.05 (m, 2H), 3.97 (br, 2H), 3.31–3.25 (m, 1H), 3.22–3.16 (m, 1H), 2.77 (br, 1H), 1.72 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 156.1, 150.6, 149.2, 140.9, 138.2, 137.6, 136.4, 130.7, 129.2, 128.7, 128.7, 128.6, 128.3, 128.2, 127.3, 123.8, 123.8, 120.8, 114.9, 85.3, 67.3, 63.8, 50.6, 35.9, 28.4; MS (ESI) m/z 524 (M + Na+); HRMS Calcd for C29H31N3O5Na (M + H+), 524.2161 Found: 524.2156.
4.6.6 3-[(R)-Benzyloxycarbonylamino-[2-(2-methanesulfonyloxyethyl)phenyl]methyl]indazole-1-carboxylic acid tert-butyl ester (11)
The compound 10 (57 mg, 0.11 mmol) was dissolved in CH2Cl2 (3 mL) and DIPEA (40 µL, 0.23 mmol) were added. Then, MsCl (13 µL, 0.17 mmol) was added dropwise. The mixture was stirred at room temperature for 20 min. After that, the solvent was removed in vacuo and the residue was purified by silica gel column chromatography (hexane / EtOAc = 2 : 1) to afford 11 (60 mg, 91%) as a white foam. [α]D20 = −88 (c 0.8 CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1H), 7.47 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 7.34–7.26 (m, 9H), 7.22–7.15 (m, 2H), 6.54 (d, J = 7.2 Hz, 1H), 6.41 (d, J = 6.8 Hz, 1H), 5.15–5.07 (m, 2H), 5.48–4.43 (m, 2H), 3.39 (s, 2H), 2.84 (s, 3H), 1.73 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.9, 150.1, 149.1, 140.9, 138.2, 136.4, 134.6, 131.0, 129.4, 129.2, 128.7, 128.7, 128.3, 128.2, 128.0, 124.0, 123.6, 120.7, 115.0, 85.5, 70.2, 67.3, 50.8, 37.3, 32.2, 28.4; MS (ESI) m/z 602 (M + Na+); HRMS Calcd for C30H33N3O7SNa (M + Na+), 602.1937 Found: 602.1948.
4.6.7 (R)-3-(1,2,3,4-Tetrahydro-isoquinolin-1-yl)indazole-1-carboxylic acid tert-butyl ester (12)
To a solution of compound 11 (60 mg, 0.10 mmol) in MeOH (4 mL) was added Pd / C (55 mg, 10% Pd). The mixture was hydrogenated at room temperature under 1 atm for 1 h. Then, the solvent was removed in vacuo and the residue was purified by silica gel column chromatography (CH2Cl2 / MeOH / NH3 = 150 : 10 : 1) to afford 12 (34 mg, 94%) as a foam. [α]D20 = +106 (c 1.0 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.06 (d, J = 9.0 Hz, 1H), 7.41 (ddd, J = 7.5, 7.5, 0.8 Hz, 1H), 7.22–7.15 (m, 3H), 7.08 (dd, J = 7.8, 7.5 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 5.68 (s, 1H), 3.41–3.36 (m, 1H), 3.24–3.16 (m, 2H), 2.91–2.86 (m, 1H), 2.21 (br, 1H), 1.75 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 153.9, 149.6, 141.1, 135.4, 135.3, 129.4, 128.8, 127.8, 127.0, 126.2, 124.2, 123.5, 122.5, 114.8, 85.2, 56.4, 43.3, 30.0, 28.5; MS (ESI) m/z 372 (M + Na+); HRMS Calcd for C21H23N3O2Na (M + Na+), 372.1688 Found: 372.1687.
4.6.8 (R)-1-(1H-Indazol-3-yl)-2-phenylmethanesulfonyl-1,2,3,4-tetrahydro-isoquinoline (IBR117)
To a solution of compound 12 (34 mg, 0.097 mmol) in MeOH (1 mL) was added a solution of NaOMe (2.7 mg, 0.05 mmol) in MeOH (0.1 mL). The resultant mixture was stirred at room temperature for 3h. CH2Cl2 (20 mL) and H2O (50 mL) were added to dilute the mixture and the aqueous phase was extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were dried over anhydrous Na2SO4. Removal of all the solvent in vacuo gave a residue, which was dissolved in CH2Cl2 (1 mL). The resultant solution was cooled to 0°C and DIPEA (22 µL, 0.13 mmol) was added. Then, a solution of BnSO2Cl (20 mg, 0.10 mmol) in CH2Cl2 (0.25 mL) was added dropwise. The mixture was stirred at 0°C for 20min. Removal of all the solvent gave a residue, which was purified by silica gel chromatography (hexane / EtOAc = 2 : 1) to afford compound IBR117 (24 mg, 61%) as a white foam. [α]D20 = +93 (c 0.6 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 10.43 (br, 1H), 7.56 (dd, J = 8.3, 3.5 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.26–7.10 (m, 5H), 7.07 (t, J = 7.5 Hz, 2H), 6.97 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 7.5 Hz, 2H), 6.63 (s, 1H), 4.07–3.97 (m, 2H), 3.52 (dd, J = 14.0, 6.5 Hz, 1H), 3.28 (ddd, J = 12.5, 12.0, 3.5 Hz, 1H), 3.02 (ddd, J = 17.0, 11.5, 6.0 Hz, 1H), 2.77 (dd, J = 17.0, 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 146.7, 141.3, 134.1, 133.9, 130.7, 129.5, 128.8, 128.5, 128.5, 128.2, 127.6, 127.4, 126.6, 122.1, 121.8, 121.0, 110.2, 59.4, 53.2, 40.8, 29.1; MS (ESI) m/z 426 (M + Na+); HRMS Calcd for C23H22N3O2S (M + H+), 404.1433 Found: 404.1430.
4.6.9 3-[(S)-Amino-[2-(2-benzyloxyethyl)phenyl]methyl] indazole-1-carboxylic acid tert-butyl ester (13)
Compound 13 (203 mg, 91%) was prepared as a clear oil using the same conditions as described for the compound 9. [α]D20 = +85 (c 1.0 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 8.5 Hz, 1H), 7.41 (t, J = 9.5 Hz, 1H), 7.30–7.20 (m, 8H), 7.16–7.10 (m, 2H), 7.03 (t, J = 7.5 Hz, 1H), 5.89 (s, 1H), 4.51 (s, 2H), 3.78 (t, J = 7.3 Hz, 2H), 3.22 (t, J = 7.0 Hz, 2H), 2.99 (br, 2H), 1.73 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 154.0, 149.5, 140.9, 140.4, 138.3, 137.6, 130.6, 128.9, 128.5, 128.2, 128.0, 127.9, 127.8, 127.1, 124.1, 123.4, 121.5, 114.7, 85.0, 73.2, 71.3, 51.1, 33.1, 28.4; MS (ESI) m/z 458 (M + H+); HRMS Calcd for C28H32N3O3 (M + H+), 458.2444 Found: 458.2435.
4.6.10 3-[(S)-Benzyloxycarbonylamino-[2-(2-hydroxyethyl)phenyl]methyl]indazole-1-carboxylic acid tert-butyl ester (14)
Compound 14 (159 mg, 71%) was prepared as a white foam using the same conditions as described for the compound 10. [α]D20 = +99 (c 0.9 CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.4 Hz, 1H), 7.44 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 7.33–7.22 (m, 9H), 7.16–7.11 (m, 2H), 6.65 (d, J = 7.2 Hz, 1H), 6.55 (d, J = 6.4 Hz, 1H), 5.14–5.05 (m, 2H), 3.96 (br, 2H), 3.31–3.25 (m, 1H), 3.22–3.15 (m, 1H), 2.77 (br, 1H), 1.72 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 156.1, 150.6, 149.2, 140.9, 138.2, 137.6, 136.4, 130.7, 129.2, 128.7, 128.7, 128.6, 128.3, 128.2, 127.3, 123.8, 123.8, 120.8, 114.9, 85.3, 67.3, 63.8, 50.7, 35.9, 28.4; MS (ESI) m/z 524 (M + Na+); HRMS Calcd for C29H31N3O5Na (M + Na+), 524.2161 Found: 524.2169.
4.6.11 3-[(S)-Benzyloxycarbonylamino-[2-(2-methanesulfonyloxyethyl)phenyl]methyl]indazole-1-carboxylic acid tert-butyl ester (15)
Compound 15 (161 mg, 88%) was prepared as a white foam using the same conditions as described for the compound 11. [α]D20 = +86 (c 1.2 CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1H), 7.47 (ddd, J = 8.4, 7.2, 0.8 Hz, 1H), 7.34–7.26 (m, 9H), 7.22–7.15 (m, 2H), 6.54 (d, J = 7.2 Hz, 1H), 6.41 (d, J = 5.6 Hz, 1H), 5.15–5.07 (m, 2H), 5.57–4.43 (m, 2H), 3.39 (s, 2H), 2.84 (s, 3H), 1.73 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.9, 150.1, 149.2, 140.9, 138.2, 136.4, 134.6, 131.0, 129.4, 129.2, 128.7, 128.7, 128.3, 128.2, 128.0, 124.0, 123.6, 120.7, 115.0, 85.4, 70.1, 67.3, 50.8, 37.3, 32.2, 28.4; MS (ESI) m/z 602 (M + Na+); HRMS Calcd for C30H33N3O7SNa (M + Na+), 602.1937 Found: 602.1941.
4.6.12 (S)-3-(1,2,3,4-Tetrahydro-isoquinolin-1-yl)indazole-1-carboxylic acid tert-butyl ester (16)
Compound 16 (95 mg, 97%) was prepared as a white foam using the same conditions as described for the compound 12. [α]D20 = −108 (c 0.7 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.06 (d, J = 8.5 Hz, 1H), 7.41 (ddd, J = 7.5, 7.3, 0.8 Hz, 1H), 7.22–7.15 (m, 3H), 7.07 (t, J = 7.5 Hz, 1H), 6.99 (t, J = 4.0 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 5.68 (s, 1H), 3.41–3.36 (m, 1H), 3.24–3.16 (m, 2H), 2.91–2.86 (m, 1H), 2.21 (br, 1H), 1.75 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 153.9, 149.5, 141.1, 135.4, 135.3, 129.4, 128.8, 127.7, 127.0, 126.1, 124.2, 123.5, 122.5, 114.8, 85.2, 56.4, 43.3, 29.9, 28.5; MS (ESI) m/z 350 (M + H+); HRMS Calcd for C21H24N3O2 (M + H+), 350.1869 Found: 350.1863.
4.6.13 (S)-1-(1H-Indazol-3-yl)-2-phenylmethanesulfonyl-1,2,3,4-tetrahydro-isoquinoline (IBR118)
IBR118 (73 mg, 67%) was prepared as white foam using the same conditions as described for the compound IBR117. [α]D20 = −90 (c 0.8 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 10.43 (br, 1H), 7.55 (dd, J = 8.0, 3.0 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.40 (t, J = 7.0 Hz, 1H), 7.26–7.10 (m, 5H), 7.06 (t, J = 7.0 Hz, 2H), 6.97 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 7.5 Hz, 2H), 6.63 (s, 1H), 4.07–3.97 (m, 2H), 3.53 (dd, J = 12.5, 6.5 Hz, 1H), 3.28 (ddd, J = 12.5, 12.0, 3.5 Hz, 1H), 3.02 (ddd, J = 18.0, 11.5, 6.0 Hz, 1H), 2.77 (dd, J = 17.0, 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 146.7, 141.3, 134.1, 140.0, 130.7, 129.5, 128.8, 128.5, 128.5, 128.2, 127.6, 127.4, 126.6, 122.1, 121.8, 121.0, 110.2, 59.4, 53.2, 40.8, 29.1; MS (ESI) m/z 426 (M + Na+); HRMS Calcd for C23H21N3O2SNa (M + Na+), 426.1252 Found: 426.1252.
4.6.14 [5-[(R)-[2-(2-Benzyloxyethyl)phenyl]-((R)-2-methylpropane-2-sulfinylamino)methyl]thiazol-2-yl]carbamic acid tert-butyl ester (18) and [5-[(S)-[2-(2-Benzyloxyethyl)phenyl]-((R)-2-methylpropane-2-sulfinylamino)methyl] thiazol-2-yl]carbamic acid tert-butyl ester (19)
A solution of compound 17 (390 mg, 1.40 mmol) in THF (8 mL) was cooled to −78 °C and n-BuLi (2.86 M, 1.22 mL, 3.50 mmol) was added slowly. The resultant mixture was stirred at −78 °C for 20min. After that, a solution of compound 2 (480 mg, 1.40 mmol) in THF (4 mL) was added slowly. The mixture was stirred at −78 °C for 20min. Saturated aqueous NH4Cl (5 mL) was added to quench the reaction and the mixture was warmed up to room temperature. H2O (50 mL) was added and the mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were dried over anhydrous Na2SO4. Removal of the solvent in vacuo resulted in a residue, which was purified by silica gel chromatography (hexane / EtOAc / NH3 = 100 / 200 / 0.6 to 100 / 200 / 3) to give compound 19 (less polar, 235 mg, 31%) as a foam and compound 18 (more polar, 60 mg, 7.9%) as a foam.
Compound 18: [α]D20 = −55.8 (c 0.88 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 11.73 (s, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.30–7.18 (m, 8H), 7.15 (s, 1H), 6.12 (d, J = 2.5 Hz, 1H), 4.53–4.46 (m, 2H), 3.81 (d, J = 2.5 Hz, 1H), 3.73–3.68 (m, 1H), 3.66–3.61 (m, 1H), 3.08–2.95 (m, 2H), 1.49 (s, 9H), 1.22 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 162.3, 153.0, 138.5, 138.5, 137.0, 135.1, 133.3, 130.8, 128.5, 128.3, 127.8, 127.8, 127.6, 127.2, 82.4, 73.2, 70.7, 56.2, 51.9, 33.2, 28.4, 22.9; MS (ESI) m/z 566 (M + Na+); HRMS Calcd for C28H37N3O4S2Na (M + Na+), 566.2123 Found: 566.2122.
Compound 19: [α]D20 = +3.5 (c 0.83 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 7.0 Hz, 1H), 7.29–7.18 (m, 9H), 7.11 (s, 1H), 6.06 (s, 1H), 4.49 (s, 2H), 4.00 (br, 1H), 3.68–3.60 (m, 2H), 3.02 (t, J = 6.5 Hz, 2H), 1.47 (s, 9H), 1.24 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 162.6, 152.8, 139.6, 138.3, 136.7, 132.8, 130.7, 128.6, 128.5, 127.8, 127.7, 127.5, 127.1, 82.5, 73.2, 70.6, 56.3, 52.9, 32.9, 28.4, 23.0; MS (ESI) m/z 566 (M + Na+); HRMS Calcd for C28H37N3O4S2Na (M + Na+), 566.2123 Found: 566.2128.
4.6.15 (5-[(S)-Amino-[2-(2-benzyloxyethyl)phenyl]methyl]thiazol-2-yl)carbamic acid tert-butyl ester (20)
A solution of compound 19 (219 mg, 0.40 mmol) in MeOH (5 mL) was added 4 M HCl in dioxane (5 mL). The mixture was stirred at room temperature for 30 min. After that, the mixture was poured to an aqueous NaHCO3 (2.0 g in 50 mL H2O). The mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were dried over anhydrous Na2SO4. Removal of the solvent in vacuo resulted in a residue, which was purified by silica gel chromatography (CH2Cl2 / MeOH / NH3 = 200 / 20 / 1) to afford compound 20 (178 mg, quant.) as an oil. [α]D20 = +49.3 (c 0.94 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 7.5 Hz, 1H), 7.31–7.17 (m, 8H), 6.90 (s, 1H), 5.59 (s, 1H), 4.47 (s, 2H), 3.67–3.59 (m, 3H), 3.07–3.01 (m, 1H), 2.94–2.88 (m, 1H), 2.26 (br, 2H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 161.6, 153.0, 142.4, 138.3, 136.6, 136.2, 133.7, 130.3, 128.6, 127.8, 127.8, 127.8, 127.3, 126.6, 81.9, 73.3, 71.2, 49.7, 33.1, 28.4; MS (ESI) m/z 462 (M + Na+); HRMS Calcd for C24H29N3O3SNa (M + Na+), 462.1827 Found: 462.1828.
4.6.16 (5-[(S)-[2-(2-Benzyloxyethyl)phenyl]phenylmethanesulfonylamino-methyl]thiazol-2-yl)carbamic acid tert-butyl ester (21)
A solution of compound 20 (178 mg, 0.40 mmol) in CH2Cl2 (8 mL) was added DIPEA (116 µL, 0.65 mmol) and DMAP (8.0 mg, 0.066 mmol) at 0 °C. Then, a solution of BnSO2Cl (130 mg, 0.68 mmol) was added dropwise. The mixture was warmed up to room temperature and stirred for 20min. Removal of the solvent in vacuo resulted in a residue, which was purified by silica gel chromatography (hexane / EtOAc = 2 / 1) to give compound 21 (232 mg, 97%) as a yellow oil. [α]D20 = −33.6 (c 1.26 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 12.06 (s, 1H), 7.35 (td, J = 7.5, 1.5 Hz, 1H), 7.28–7.22 (m, 7H), 7.19–7.14 (m, 4H), 7.05 (s, 1H), 7.00 (d, J = 7.0 Hz, 2H), 6.77 (br, 1H), 5.84 (d, J = 8.0 Hz, 1H), 4.20–4.11 (m, 2H), 3.99 (s, 2H), 3.64 (dt, J = 9.0, 4.5 Hz, 1H), 3.48 (ddd, J = 9.5, 9.5, 4.5 Hz, 1H), 2.91 (ddd, J = 14.5, 9.3, 4.5 Hz, 1H), 2.67 (dt, J = 14.5, 5.0 Hz, 1H), 1.46 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 162.2, 153.0, 138.7, 138.3, 137.5, 135.1, 133.8, 132.8, 131.5, 131.1, 129.2, 129.1, 128.9, 128.7, 128.6, 128.2, 128.0, 127.3, 82.2, 73.1, 70.7, 59.9, 55.3, 33.1, 28.4; MS (ESI) m/z 616 (M + Na+); HRMS Calcd for C31H35N3O5S2Na (M + Na+), 616.1916 Found: 616.1909.
4.6.17 (5-[(S)-[2-(2-Hydroxyethyl)phenyl]phenylmethanesulfonylamino-methyl]thiazol-2-yl)-carbamic acid tert-butyl ester (22)
To a solution of compound 21 (225 mg, 0.38 mmol) in MeOH (10 mL) was added Pd / C (10% Pd, 198 mg) and 4 M HCl in dioxane (0.5 mL). The, the mixture was hydrogenated (1atm) for 5h. The mixture was subjected to filtration and the filtrate was poured to H2O (50 ml). Saturated aqueous NaHCO3 (10 mL) was added and the mixture was extracted with CH2Cl2 (3 × 25 mL). The combined organic phases were dried over anhydrous Na2SO4. Removal of the solvent in vacuo resulted in a residue, which was purified by silica gel chromatography (CH2Cl2 / MeOH = 15 / 1) to afford compound 22 (152 mg, 80%) as a foam. [α]D20 = −41 (c 0.58 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.37 (t, J = 7.0 Hz, 2H), 7.32–7.24 (m, 4H), 7.20 (t, J = 7.5 Hz, 2H), 7.03 (d, J = 7.5 Hz, 2H), 6.97 (s, 1H), 6.50 (br, 1H), 5.96 (d, J = 8.5 Hz, 1H), 4.21 (d, J = 14.0 Hz, 1H), 4.07 (d, J = 13.5 Hz, 1H), 3.80 (dt, J = 9.5, 5.0 Hz, 1H), 3.71–3.65 (m, 1H), 2.81 (ddd, J = 14.0, 9.3, 5.5 Hz, 1H), 2.68 (dt, J = 14.5, 5.0 Hz, 1H), 1.46 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 162.0, 152.8, 139.1, 137.9, 135.3, 133.3, 131.6, 131.1, 129.3, 129.1, 128.7, 128.6, 127.5; 82.5, 64.1, 60.0, 54.7, 35.4, 28.4; MS (ESI) m/z 526 (M + Na+); HRMS Calcd for C24H29N3O5S2Na (M + Na+), 526.1447 Found: 526.1442.
4.6.18 [5-((S)-2-Phenylmethanesulfonyl-1,2,3,4-tetrahydroisoquinolin-1-yl)thiazol-2-yl]carbamic acid tert-butyl ester (23)
To a solution of compound 22 (152 mg, 0.30 mmol) in CH2Cl2 (5 mL) was added DMAP (3.0 mg, 0.025 mmol) and DIPEA (63 µL, 0.36 mmol) at 0 °C. Then, MsCl (80 µL, 1.03 mmol) was added dropwise. The mixture was stirred at room temperature for 20 min. After that, the solvent was removed in vacuo to afford a residue, which was purified by gel chromatography (CH2Cl2 / MeOH = 30 / 1) to give a foam (168 mg). The yielded foam (168 mg) was dissolved in THF (10 mL) and the solution was cooled to 0 °C. To this solution was added KHMDS (0.5 M in toluene, 1.15 mL, 0.58 mmol) and the mixture was stirred at 0 °C for 5 min. The reaction was quenched with aqueous saturated NH4Cl (5 mL) and H2O (30 mL) was added. The mixture was extracted with CH2Cl2 (3 × 25 mL) and the combined organic phases were dried over anhydrous Na2SO4. The solvent was removed in vacuo and the resultant residue was purified by silica gel chromatography (hexane / EtOAc = 2 / 1) to give compound 23 (118 mg, 81%) as a foam. [α]D20 = −50.7 (c 0.63 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 11.92 (s, 1H), 7.27–7.24 (m, 1H), 7.21–7.17 (m, 3H), 7.14–7.09 (m, 5H), 6.97 (d, J = 7.0 Hz, 1H), 6.08 (s, 1H), 4.13–4.08 (m, 2H), 3.63 (dd, J = 14.0, 6.0 Hz, 1H), 3.29 (ddd, J = 15.5, 10.5, 3.0 Hz, 1H), 2.82 (ddd, J = 16.5, 12.0, 5.0 Hz, 1H), 2.67 (dt, J = 15.0, 3.0 Hz, 1H), 1.47 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 162.5, 152.9, 136.8, 134.0, 133.5, 132.4, 130.9, 129.5, 128.8, 128.7, 128.7, 128.2, 127.9, 126.6, 82.4, 59.6, 53.4, 39.9, 29.9, 28.5; MS (ESI) m/z 508 (M + Na+); HRMS Calcd for C24H27N3O4S2Na (M + Na+), 508.1341 Found: 508.1344.
4.6.19 5-((S)-2-Phenylmethanesulfonyl-1,2,3,4-tetrahydro-isoquinolin-1-yl)thiazol-2-ylamine (IBR119)
To a solution of compound 23 (106 mg, 0.22 mmol) in CH2Cl2 (10 mL) was added TFA (1 mL) at 0 °C. The mixture was warmed up to room temperature and stirred for 5h. CH2Cl2 (20 mL) and H2O (30 mL) were added to the reaction mixture. The reaction was quenched with aqueous saturated NH4Cl (5 mL) and H2O (30 mL) was added. After saturated aqueous NaHCO3 (10 ml) was added, the mixture was extracted with CH2Cl2 (3 × 20 mL) and the combined organic phases were dried over anhydrous Na2SO4. The solvent was removed in vacuo and the resultant residue was purified by silica gel chromatography (hexane / EtOAc / NH3 = 1 : 1 : 0 to 1 : 2 : 0.005 ) to give compound IBR119 (73 mg, 87%) as a foam. [α]D20 = −62.9 (c 0.43 CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.19–7.26 (m, 1H), 7.24–7.19 (m, 3H), 7.16–7.10 (m, 4H), 6.98 (d, J = 8.0 Hz, 1H), 6.61 (s, 1H), 5.93 (s, 1H), 5.07 (br, 2H), 4.19–4.10 (m, 2H), 3.62 (dd, J = 14.3, 6.0 Hz, 1H), 3.22 (ddd, J = 15.5, 10.5, 3.5 Hz, 1H), 2.78 (ddd, J = 17.8, 11.3, 5.0 Hz, 1H), 2.66 (dd, J = 16.5, 1.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 169.1, 138.9, 133.8, 133.6, 130.9, 129.4, 129.3, 128.8, 128.7, 128.7, 128.3, 127.9, 126.5, 59.8, 53.7, 39.6, 29.9; MS (ESI) m/z 386 (M + H+); HRMS Calcd for C19H20N3O2S2 (M + H+), 386.0997 Found: 386.0996.
4.6.20 (R)-N-((2-(hydroxymethyl)phenyl)(1H-indol-3-yl)methyl)-1-phenylmethanesulfonamide (25)
To a solution of compound 24 [17] (176 mg, 0.44 mmol) in dioxane-water (3:1, 8 mL) were added 2, 6-lutidine (0.101 mL, 0.88 mmol), OsO4 (2.5% in tert-butanol, 89 µL, 8.8 µmol) and NaIO4 (371.4 mg, 1.75 mmol). The reaction was stirred at r.t and monitored by TLC. After the reaction was complete, water (10 mL) and CH2Cl2 (20 mL) were added. The organic layer was separated, and the water layer was extracted by CH2Cl2 (10 mL × 3). The combined organic layers were washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was absorbed onto SiO2, and eluted with EtOAc/hexane (1:3) to afford the aldehyde intermediate as white foam (158 mg). This foam was dissolved in THF-MeOH (1:1, 4 mL) at 0°C and NaBH4 (44 mg, 1.17 mmol) in THF (1 mL) was added. The reaction was stirred for 10 min at room temperature and saturated aqueous NH4Cl (3 mL) was added to quench the reaction. H2O (30 mL) was added and the mixture was extracted with Et2O (3 × 20 mL). The combined organic phases were dried with anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was absorbed onto silica gel and eluted with hexane / EtOAc (1 : 2) to give compound 25 (146 mg, 82%) as a foam. [α]D20 = + 70.2 (c 1.06 CH2Cl2); 1H NMR (400 MHz, CD2Cl2) δ 8.26 (s, 1H), 7.62 (d, J = 7.5 Hz, 1H), 7.36–7.52 (m, 6H), 7.15–7.31 (m, 4H), 7.02–7.04 (d, J = 7.5 Hz, 2 H), 6.94 (s, 1H), 6.27 (d, J = 7.5 Hz, 1H), 5.76 (d, J = 7.8 Hz, 1H), 4.47–4.57 (m, 2H), 4.13 (d, J = 13.8 Hz, 1H), 4.02 (d, J = 13.8 Hz, 1H), 1.80 (t, J = 6.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 140.1, 138.2, 136.9, 130.9, 130.7, 129.1, 129.0, 128.9, 128.7, 128.6, 128.6, 128.6, 125.5, 123.8, 122.6, 120.1, 119.6, 116.0, 111.6, 63.3, 60.1, 53.8; MS (ESI) m/z 429 (M + Na+); HRMS Calcd for C23H22N2O3SNa (M + Na+), 429.1249 Found:429.1230.
4.6.21 (R)-3-(2-(benzylsulfonyl)isoindolin-1-yl)-1H-indole (IBR120)
To a stirred solution of compound 25 (28 mg, 0.069 mmol) in CH2Cl2 (2.0 mL) was added Et3N (9.6 µL, 0.069 mmol) at 0°C. Then, MsCl (7.1 µL, 0.092 mmol) was added dropwise. The mixture was warmed up to room temperature and stirred for 1h. The reaction solution was diluted with CH2Cl2 (5 ml) and washed with ice water. Organic phases were dried with anhydrous Na2SO4 and removed under vacuum. The residue was then dissolved in dry MeCN (2 mL) and to this solution was added DIPEA dropwise at 0°C. The reaction was allowed warm up to r.t and monitored by TLC. After the reaction was completed, saturated aqueous NH4Cl (1 mL) was added and the mixture was extracted with with CH2Cl2 (3 × 5 mL). The combined organic phases were dried over anhydrous Na2SO4. Removal of the solvent in vacuo resulted in a residue, which was purified by silica gel chromatography (EtOAc / hexane = 1 : 3) to give compound IBR120 (14.2 mg, 53 %) as a white solid. [α]D20 = +89.5 (c 0.44 CH2Cl2); 1H NMR (400 MHz, CD2Cl2) δ 8.40 (s, 1H), 7.37–7.42 (m, 2H), 7.31–7.37 (t, J = 7.3 Hz, 1H), 7.21–7.30 (m, 3H), 7.13–7.19 (m, 3H), 7.03–7.07 (d, J = 7.6 Hz, 1H), 6.85–6.97 (m, 4H), 6.41 (d, , J = 1.8 Hz, 1H), 4.86 (d, J = 13.3 Hz, 1H), 4.38–4.42 (dd, J = 2.8, 13.6 Hz, 1H), 3.75 (d, J = 13.6 Hz, 1H), 3.64 (d, J = 13.7 Hz, 1H); 13C NMR (125MHz, CD2Cl2) δ 140.7, 137.4, 136.5, 131.2, 129.7, 128.8, 128.7, 128.5, 128.4, 125.9, 125.7, 124.0, 122.9, 122.8, 120.4, 120.1, 116.0, 112.1, 63.2, 59.5, 54.2; MS (ESI) m/z 411 (M + Na+); HRMS Calcd for C23H20N2O2SNa (M + Na+), 411.1143 Found: 411.1134.
4.6.22 (S)-3-(2-(benzylsulfonyl)isoindolin-1-yl)-1H-indole (IBR121)
IBR121 (13.6 mg, 51 %) was prepared as white solid using the same conditions as described for the compound IBR120. [α]D20 = - 90.1 (c 0.75 CH2Cl2); 1H NMR (400 MHz, CD2Cl2) δ 8.37 (s, 1H), 7.39–7.42 (m, 2H), 7.32–7.35 (m, 1H), 7.22–7.28 (m, 3H), 7.13–7.17 (m, 3H), 7.03–7.05 (d, J = 7.7 Hz, 1H), 6.85–6.96 (m, 4H), 6.41 (s, 1H), 4.85 (d, J = 13.3 Hz, 1H), 4.37 (d, J = 13.8 Hz, 1H), 3.74 (d, J = 13.5 Hz, 1H), 3.63 (d, J = 13.6 Hz, 1H); 13C NMR (125MHz, CD2Cl2) δ 140.7, 137.3, 136.4, 131.2, 129.7, 128.7, 128.7, 128.4, 128.4, 125.7, 124.0, 122.9, 122.8, 120.4, 120.1, 116.0, 112.1, 63.1, 59.5, 54.2; MS (ESI) m/z 411 (M + Na+); HRMS Calcd for C23H20N2O2SNa (M + Na+), 411.1143 Found: 411.1150.
Supplementary Material
Highlights.
A class of novel indole alkaloid derivatives were synthesized.
These compounds efficiently inhibit the growth of a panel of cancer cell lines.
IBR120 shows 5-fold increase of activity on a triple-negative breast cancer cell line.
IBR120 disrupts RAD51 multimerization, impairs homologous recombination, induces apoptosis.
Structure activity relationship and molecular modeling studies were included.
Acknowledgement
This work is supported by NIH grant to W.-H. Lee (CA107568). We thank the UCI Molecular Modeling Facility for technical support.
Abbreviations
- DMAP
4,4-dimethylaminopyridine
- XTT
sodium 3’-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro-) benzene sulfonate
- IBR
RAD51 inhibitor
Appendix A. Supplementary data
Supporting information related to this article can be found online. Supporting information contains: (A) Cyclization Conditions; (B) Raw dataset for QSAR analysis; (C) Chiral HPLC analysis; (D) Initial Predictive Multivariate Linear Model; (E) NMR spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions:
HC, XLQ, and JZ designed, synthesized, and characterized all compounds under the guidance of ARC; XEG and CMH performed XTT assay and HR assay; JZ performed the gel filtration assay, QSAR analysis, and molecular modeling. JZ and WHL wrote the paper with inputs from all althors.
Reference
- 1.Carvalho JF, Kanaar R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opinion on Therapeutic Targets. 2014;18:427–458. doi: 10.1517/14728222.2014.882900. [DOI] [PubMed] [Google Scholar]
- 2.Guo XE, Ngo B, Modrek AS, Lee W-H. Targeting Tumor Suppressor Networks for Cancer Therapeutics. Current Drug Targets. 2014;15:2–16. doi: 10.2174/1389450114666140106095151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward A, Khanna KK, Wiegmans AP. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treatment Reviews. 2014;41:35–45. doi: 10.1016/j.ctrv.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 5.Flygare J, Fält S, Ottervald J, Castro J, Dackland Å-L, Hellgren D, Wennborg A. Effects of HsRad51 Overexpression on Cell Proliferation, Cell Cycle Progression, and Apoptosis. Exp. Cell Res. 2001;268:61–69. doi: 10.1006/excr.2001.5265. [DOI] [PubMed] [Google Scholar]
- 6.Chen C-F, Chen P-L, Zhong Q, Sharp ZD, Lee W-H. Expression of BRC Repeats in Breast Cancer Cells Disrupts the BRCA2-Rad51 Complex and Leads to Radiation Hypersensitivity and Loss of G2/M Checkpoint Control. J. Biol. Chem. 1999;274:32931–32935. doi: 10.1074/jbc.274.46.32931. [DOI] [PubMed] [Google Scholar]
- 7.Chen P-L, Chen C-F, Chen Y, Xiao J, Sharp ZD, Lee W-H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA. 1998;95:5287–5292. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair. 2008;7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, Luttges J, Kalthoff H, Sturtzbecher H-W. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–2795. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- 10.Qiao G-B, Wu Y-L, Yang X-N, Zhong W-Z, Xie D, Guan X-Y, Fischer D, Kolberg H-C, Kruger S, Stuerzbecher H-W. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br. J. Cancer. 2005;93:137–143. doi: 10.1038/sj.bjc.6602665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated Levels of Rad51 Recombination Protein in Tumor Cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- 12.Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23:546–553. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- 13.Robu ME, Inman RB, Cox MM. RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. USA. 2001;98:8211–8218. doi: 10.1073/pnas.131022698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, Skorski T. BCR/ABL Regulates Mammalian RecA Homologs, Resulting in Drug Resistance. Mol. Cell. 2001;8:795–806. doi: 10.1016/s1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- 15.Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G, Qiu X-L, Chen C-F, Hu C-M, Goldblatt E, Bhatia R, Chamberlin AR, Chen P-L, Lee W-H. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Molecular Medicine. 2013;5:1–13. doi: 10.1002/emmm.201201760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy IK, Mehvar R. Chirality in Drug Design and Development. 1 edn. CRC Press; 2004. [Google Scholar]
- 17.Qiu X-L, Zhu J, Wu G, Lee W-H, Chamberlin AR. Stereoselective synthesis of chiral IBR2 analogues. J. Org. Chem. 2009;74:2018–2027. doi: 10.1021/jo802607f. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y-Q, Song J, Hong R, Li H, Deng L. Asymmetric Friedel-Crafts Reaction of Indoles with Imines by an Organic Catalyst. J. Am. Chem. Soc. 2006;128:8156–8157. doi: 10.1021/ja062700v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welch WM, Hanau CE, Whalen WM. A novel synthesis of 3-substituted indazole derivatives. Synthesis. 1992:937–939. [Google Scholar]
- 20.Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Improved Procedure for the Oxidative Cleavage of Olefins by OsO4/NaIO4 . Org. Lett. 2004;6:3217–3219. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- 21.Hudis CA, Gianni L. Triple-Negative Breast Cancer: An Unmet Medical Need. The Oncologist. 2011;16:1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 22.Peddi PF, Ellis MJ, Ma C. Molecular Basis of Triple Negative Breast Cancer and Implications for Therapy. International Journal of Breast Cancer. 2012;2012:1–7. doi: 10.1155/2012/217185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.ATCC Cell Lines - ATCC: The Global Bioresource Center. http://www.atcc.org.
- 24.Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 25.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.