

Abstract
Expression of the E3 ubiquitin ligase Triad1 is greater in mature granulocytes than in myeloid progenitor cells. HoxA10 actives transcription of the gene encoding Triad1 (ARIH2) during myeloid differentiation, but the contribution of increased Triad1 expression to granulocyte production or function is unknown. Mice with bone marrow-specific disruption of the ARIH2 gene exhibit constitutive inflammation with tissue infiltration by granulocytes and B-cells. In contrast, disruption of the HOXA10 gene in mice neither constitutively activates the innate immune response nor significantly alters steady state granulopoiesis. Our current study explores the impact of HoxA10-induced Triad1 expression on emergency (stress) granulopoiesis. We found that mice with HOXA10 gene disruption exhibited an overwhelming and fatal emergency granulopoiesis response that was characterized by tissue infiltration with granulocytes, but reversed by re-expression of Triad1 in the bone marrow. We determined that HoxA9 repressed ARIH2 transcription in myeloid progenitor cells; antagonizing the effect of HoxA10 on Triad1 expression. And, we found that differentiation-stage-specific ARIH2 transcription was regulated by the tyrosine phosphorylation states of HoxA9 and HoxA10. Our studies demonstrate a previously undescribed role for HoxA10 in terminating emergency granulopoiesis; suggesting an important contribution by Hox proteins to the innate immune response.
Introduction
Granulocytes are produced by two distinct processes; steady state granulopoiesis and emergency (or stress) granulopoiesis. Steady state granulopoiesis is a homeostatic process that replaces cells lost to normal programmed cell death. In contrast, emergency granulopoiesis produces granulocytes in response to infectious or inflammatory stimuli, and contributes to innate immunity. In murine models, steady state granulopoiesis is impaired by disruption of genes encoding G-CSF or GM-CSF, and requires the transcription factors PU.1 and C/EBPα (1-6). Emergency granulopoiesis is also impaired by loss of G-CSF in mice, but is completely abolished by loss of the Interleukin 1 receptor (IL1-R) (2,7).
Emergency granulopoiesis has four stages; release of granulocytes from the bone marrow, expansion of hematopoietic stem cells (HSC) and granulocyte/monocyte progenitor cells, acceleration of differentiation, and termination of the response. CXCR proteins regulate the first step; cells are protected from genotoxic stress during second step by the Fanconi DNA-repair pathway; and the third step requires Stat3 and CEBPβ (8-11). Less is known about termination of emergency granulopoiesis, but dysregulation of this step is implicated in tissue damage during infectious challenge and in auto-inflammatory diseases (12-16).
The hypothesis of this study is that increased expression of Triad1, an E3 ubiquitin ligase, is involved in termination of emergency granulopoiesis. Expression of Triad1 is known to increase during granulopoiesis and impair proliferation of myeloid progenitor cells, but substrates for Triad1 in hematopoietic cells are not defined (17-21). In epithelial cells, Triad1-dependent ubiquitination results in lysosomal degradation versus re-cycling (and sustained signaling) of the receptors for epidermal growth factor and growth hormone (22). In mice, homozygous knockout of the gene encoding Triad1 (ARIH2) is embryonic lethal at ~E16 due to hepatocyte apoptosis (19). ARIH2−/− fetal hematopoietic cells reconstitute hematopoiesis in irradiated wild type mice, but recipients developed constitutive inflammation with tissue infiltration by B-cells and granulocytes, suggesting involvement of Triad1 in immune regulation (19).
We previously identified tandem cis elements in the ARIH2 promoter that are activated by the homeodomain transcription factor, HoxA10 (21). Unlike ARIH2−/− mice, HOXA10−/− mice are viable; unlike mice transplanted with ARIH2−/− bone marrow, HOXA10−/− mice do not exhibit steady state immune activation (23). We previously found that total protein ubiquitination increased during granulocyte differentiation in a HoxA10 and Triad1-dependent manner (21). Increased production of IL1β during emergency granulopoiesis significantly increases expression of G-CSF relative to steady state levels (1,2). Therefore, it was of interest that we found HoxA10-dependent, increased Triad1 expression in murine myeloid progenitor cells stimulated with G-CSF in vitro (21). Also, we found that differentiation of myeloid cell line transfectants with retinoic acid/dimethyl formamide (RA/DMF) augmented activation of the ARIH2 promoter by HoxA10 (21). RA/DMF both differentiates and activates these cells; more closely modeling emergency granulopoiesis rather than steady state (24). These result suggested conditional, rather than constitutive, Triad1 regulation by HoxA10.
HOX genes are found in 4 groups on 4 chromosomes in mouse and man (25). Although the HOX7-11 genes are maximally transcribed in committed progenitor cells, HoxA9 and HoxA10 proteins are present in granulocytes (26-29). Engineered overexpression of HoxA9 or HoxA10 in murine bone marrow induces granulocytosis in vivo that progresses to AML (30-32). However, preservation of steady state granulopoiesis in mice with homozygous knockout of either HOXA9 or HOXA10 suggests that these proteins might be redundant with each other, or other Hox proteins, for this function (23, 33).
Most investigations of HoxA9 and HoxA10 have focused on identifying target genes that are relevant to the roles of these proteins in leukemogenesis. Work in our laboratory also identified target genes for HoxA9 and HoxA10 that are involved in phagocyte functions, including genes encoding gp91phox and p67phox; components of the phagocyte NADPH-oxidase (34-37). HoxA10 represses these genes in myeloid progenitors, but they are activated by HoxA9 during myelopoiesis (34,37). Cytokine-induced phosphorylation of conserved, homeodomain tyrosine residues in HoxA9 and HoxA10 mediates these differentiation-specific effects (34-37). In contrast, we found cooperative, phosphorylation-independent activation of FGF2 transcription by HoxA9 and HoxA10 (27,38). Fgf2 (fibroblast growth factor 2) is involved in expansion of bone marrow progenitor cells, but also primes granulocytes for NADPH-oxidase activity (27,38-40). E selectin is another a phagocyte effector and common HoxA9/HoxA10-target gene (41).
In the current studies, we hypothesize that transcriptional activation of ARIH2 by HoxA10 down-regulates emergency granulopoiesis in a manner that is antagonized by HoxA9. This identifies modulation of protein ubiquitination/degradation as a novel mechanism for regulation of the innate immune response by Hox proteins. We hypothesize that modulation of innate immunity is an important, under explored role for late Hox proteins in normal myelopoiesis.
Methods
Plasmid vectors
Human HoxA10 cDNA was obtained from C. Largman (University of California, San Francisco) (42,43). HoxA9 cDNA was obtained by PCR (12). Y326F/Y343F-HoxA10 (or HD-Y-mutant HoxA10) and Y212F/Y225F-HoxA9 (or HD-Y-mutant HoxA9) we generated by site directed mutagenesis (37,44). Triad1 cDNA was obtained from BA van der Reijden (Radboud University, Netherlands). Triad1, HoxA10 or HoxA9 specific shRNAs and scrambled controls were designed with the Promega website (Promega, Madison, WI) and subcloned into pLKO.1puro vector (from MK Rundell, Northwestern University, Chicago) as described (21). The most efficient three sequences were combined.
The ARIH2 5’ flank was amplified from U937 chromatin by genomic PCR, subcloned into the pGL3-basic reporter vector (Promega) and sequenced to ensure identity with ENSEMBL data base, as described (21). Additional constructs were generated with three copies of the −22 to −48 bp (proximal) or −174 to −198 bp (distal) ARIH2 promoter sequences in pGL3-promoter vector (Promega), as described (21).
Oligonucleotides
Oligonucleotides were synthesized by MWG Biotech (Piedmont, NC). Double stranded oligonucleotides used in electrophoretic mobility shift assays represented −22 to −48 bp (proximal: 5’-TTAAAAATATAAATATAATTCTTTTCA-3’) or −174 to −198 bp (distal: 5’-TCTTGTCAATATAATTATATCATGGA-3’) from the ARIH2 promoter (21). Oligonucleotides with mutations in the Hox binding sites were used in competition studies (mutant proximal; 5’-TTAAAAATCTCCATAGAATTCTTTTCA-3’, mutant distal; 5’-TCTTGTCAAGCGAAGCATATCATGGA-3’) (21). Mutations are underlined.
Myeloid cell lines, culture and assays
The human myelomonocytic cell line U937 (24) was obtained from AS Kraft (Hollings Cancer Center, Medical University of South Carolina, Charleston, SC). Cells were treated for 48 hrs with IL1β for granulocyte differentiation, as described (9,45).
Stable transfectants
U937 cells were transfected by electroporation with a HoxA10 or HoxA9 expression vector or both (or control vector) plus a vector with a neomycin resistance cassette (pSRα) (30 μg total) (37,44). Stable transfectant pools were selected in G418 and aliquots tested for HoxA10 and HoxA9 by real time PCR and Western blot. Other cells were transfected with a lentiviral vector for expression of Triad1, HoxA10 or HoxA9 specific shRNAs (or scrambled controls) (30 μg), selected in puromycin and tested for Triad1, HoxA10 or HoxA9 expression as above (21).
Reporter assays
U937 cells (1.5 × 107) were co-transfected with plasmids containing ARIH2 promoter sequences linked to a Luciferase reporter (or control vector; 10 μg) , and a vector to express shRNAs specific to HoxA9, HoxA10, HoxA9 + HoxA10 (or scrambled control vector; 30 μg) (0.24 V/960 μF) (21). Other cells were co-transfected with a Luciferase reporter vector containing a minimal promoter and the −22 to −48 bp (proximal) or −174 to −198 bp (distal) ARIH2 cis elements (or control vector; 10 μg) (21) and combinations of vectors to overexpress or knockdown HoxA9 or HoxA10 (30 μg). Reporter assays were performed 24 hrs post transfection ± IL1β (to induce granulocyte differentiation) as described (9, 21). Cells were transfected with a β-galactosidase reporter plasmid to control for transfection efficiency (2 μg).
Real time PCR
RNA was isolated with Trizol reagent (Gibco-BRL, Gaithersburg MD). Primers were designed with Applied Biosystems software and real time PCR performed using SYBR green “standard curve” method. Result were normalized to 18S and γ-actin (for mRNA) or input chromatin (for chromatin immuno-precipitation). Primers were described (21).
Chromatin co-immunoprecipitation
Cells were incubated briefly in media supplemented with formaldehyde to generate DNA-protein cross links (46). Lysates were sonicated to generate chromatin fragments with an average size of ~100 bp, chromatin was immuno-precipitated with HoxA10, HoxA9 or control antibody, and amplified by real time PCR, as described (47). HoxA10 antiserum was obtained from Covance Research Products (Richmond, CA) and antibody to HoxA9 from Santa Cruz Biotechnology (Santa Cruz, CA). Each assay used 2 × 106 murine bone marrow cells.
Protein Assays
Western Blots
Cells were lysed by boiling in 2X SDS sample buffer, lysate proteins were separated by SDS-PAGE, transferred to nitrocellulose, and Western blots probed with antibodies to ubiquitin, HoxA9, HoxA10, Triad1 or Gapdh (loading control). Antibodies to Triad1 and Gapdh were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Each experiment was repeated at least 3 times with different lysates. For some studies, cells were pre-treated with the lysosome inhibitor E64 to stabilize ubquitinated proteins (48).
Electrophoretic mobility shift assays
Nuclear proteins were isolated by the method of Dignam (45,49). Oligonucleotides probes were prepared and EMSA performed as described (21,34). Three batches of nuclear proteins were tested in at least 2 independent experiments. Nuclear protein loading was determined in EMSA with a classical CCAAT box probe.
Statistical analysis
Statistical significance was determined by Student’s t-test and ANOVA using SigmaPlot and SigmaStat software. Error bars represent standard error. Statistical significance was a p value of < 0.02.
Murine studies
Ex vivo bone marrow studies
Mononuclear cells were obtained from the femurs of Wt C57/BL6 mice. Lin−Sca1+ cells were separated using the Miltenyi magnetic bead system (according to manufacturer’s instructions; Miltenyi Biotechnology, Auburn, CA). Cells were cultured for 48 hrs in DME media supplemented with 10% fetal calf serum, 1% pen-strep, and murine recombinant GM-CSF (20 ng/ml), Scf (100 ng/ml) and IL-3 (10 ng/ml) (R & D Systems Inc., Minneapolis, MN) (2 × 105 cells per ml). Some CD34+ cells were separated for analysis (using the Miltenyi magnetic bead system); referred to as myeloid progenitor cells in this study. Other cells were differentiated after 24 hrs with G-CSF or IL1β, as described (9,30).
Bone marrow transduction
Retrovirus was generated with Phoenix packaging cells according to manufacturer’s instructions (Stratagene, La Jolla, CA). Lin−Sca1+ bone marrow cells were incubated with retroviral supernatant (~107 pfu/ml) supplemented with polybrene (6 μg/ml) (30). Transgene expression was confirmed by real time PCR. Transduction studies were repeated 3 times with at least 2 batches of retroviruses.
Emergency granulopoiesis assay and analysis of peripheral blood and tissues
Wt or HoxA10−/− mice were injected intraperitoneally (IP) with ovalbumin/alum (referred to as Alum) or saline q4 wks (7 mice per group). Alum was prepared as described and a 0.5 ml volume was injected IP (9). Some Wt mice transplanted with transduced HoxA10−/− bone marrow (6 mice per group; see below) were similarly treated. Wt mice were litter mates of HoxA10−/− mice (HoxA10+/− breeding pairs used due to HoxA10−/− infertility).
Peripheral blood was obtained from the tail vein q2 wks and complete blood counts determined using an automated cell counter. Mice were sacrificed if Hgb was ≤ 6.0, platelets were ≥100,000, or granulocytes ≤100,000. Sternal bone marrow and lung samples were stained using hematoxylin and eosin by the Pathology Core Facility of the Robert H. Lurie Comprehensive Cancer Center. Light microscopy was performed and digital images were captured (40 × magnification). Differential cell counts were obtained from 300 cells (in duplicate) on slides from three different animals.
Bone marrow transplantation
Lin−Sca1+ cells from the bone marrow of HoxA10−/− mice were transduced with a retroviral vector to express Triad1 (Triad1/MSCV) or empty control vector (MSCV), as above. Viable cells were obtained by negative selection for Annexin V using Ab conjugated magnetic beads (Miltenyi Biotech). Lethally irradiated Wt C57 Black 6 mice were injected (retro-orbital) with Lin−Sca1+ cells (2×105 cells).
Approvals
Animal studies were approved by the Animal Care and Use Committees of Northwestern University and Jesse Brown VA Medical Center.
Results
HoxA10−/− mice exhibit an overwhelming and fatal emergency granulopoiesis response
The survival of mice with disruption of the HOXA10 gene is comparable to wild type (Wt) mice in a pathogen free environment, consistent with a lack of constitutive activation of the innate immune response in HoxA10−/− mice (21,23). In the current studies, we investigated the contribution of HoxA10 to emergency granulopoiesis. Emergency granulopoiesis is studied in mice by intraperitoneal (IP) injection with pathogens or an ovalbumin-alum mixture (referred to as Alum in our study) (50,51). Either approach induces IL1β production and an IL1β-dependent increase in G-CSF (51). We chose the Alum-injection method because it permits analysis of multiple episodes of emergency granulopoiesis without causing death in Wt mice (9). We compared Alum-injected HoxA10−/− or Wt mice to matched cohorts injected with saline, as controls for steady state granulopoiesis. Mice were injected every four weeks.
Although 100% of Wt mice survived three cycles of Alum injection, only 20% of HoxA10−/− mice were alive two weeks after the first Alum injection, and no HoxA10−/− mice survived more than a few days after the second injection (Figure 1A). At necropsy, Alum-injected HoxA10−/− mice exhibited enlarged livers and spleens in comparison to saline injected HoxA10−/− mice, or Alum or saline injected Wt mice (~50% increase). Lungs appeared to have areas of consolidation. Wt and HoxA10−/− mice tolerated saline injection without morbidity of mortality.
Figure 1.

HoxA10−/− mice exhibit a fatal emergency granulopoiesis response that is reversed by re-expressing Triad1. Emergency granulopoiesis was studied by injecting mice with Alum versus saline control (IP). Wild type (Wt) mice, HoxA10−/− mice, or Wt mice that were transplanted with HoxA10−/− bone marrow that was transduced with a vector to express Triad1 or MSCV control vector were studied. A. HoxA10−/− mice exhibited excess mortality during emergency granulopoiesis, but this phenotype was reversed by re-expression of Triad1. Mice were followed for survival after injection (indicated by red arrows). B. Re-expression of Triad1 in HoxA10−/− bone marrow normalized the emergency granulopoiesis response in the peripheral blood. Peripheral blood granulocyte counts (PMNs) were determined every 2 weeks. Injections are indicated in red and time points with no surviving mice with an X. Statistically significant differences in circulating PMNs in mice with versus without Alum injection are indicated by *, ***, # or ##. Statistically significant difference between Alum injected Wt versus HoxA10−/− mice is indicated by **. P values< 0.02 were considered statistically significant. C. Inflammatory lung infiltration was observed post Alum injection in mice with HoxA10−/− bone marrow, but not in mice transplanted with Triad1-transduced HoxA10−/− bone marrow. Mice were sacrificed 2 weeks post Alum or saline injection and sternal bone marrow and lung examined histologically. D. Triad1 expression in mice with Triad1-transduced HoxA10−/− bone marrow is comparable to endogenous Triad1 expression in Alum injected Wt mice. Bone marrow was harvested 2 weeks post injection with saline or Alum and Triad1 expression determined by real time PCR. Statistically significant differences in Triad1 mRNA abundance in murine bone marrow with versus without Alum injection are indicated by *, ***, #, ##, or ###. Statistically significant difference Triad1 expression in Wt versus HoxA10−/− Alum injected mice are indicated by **. P values< 0.02 were considered statistically significant.
In the first 24 hours after Alum injection, we found an equivalent increase in circulating granulocytes in Wt or HoxA10−/− mice; representing release from the bone marrow (p>0.2, n=6). Initial granulocytosis resolved by 72 hours in all Alum injected mice (during the first cycle), consistent with transit from circulation to tissues. In Wt mice, circulating granulocytes began to rise again 10 days after Alum injection, were maximal at 14 days, and returned to baseline at 4 weeks (Figure 1B) (9). The increase in circulating granulocytes 14 days post Alum injection was significantly greater in HoxA10−/− mice versus Wt mice (p<0.001, n=6), with highest counts in the most severely affected animals (Figure 1B). In HoxA10−/− mice, the number of circulating granulocytes did not return to baseline prior to the second injection.
Histological examination of sternal bone marrow from Wt mice revealed an increase in mature granulocytes two weeks post Alum injection, as anticipated (Figure 1C). At steady state, HoxA10−/− murine bone marrow was not significantly enriched in granulocytes in comparison to Wt, but two weeks post Alum-injection the percent of granulocytes in HoxA10−/− sternal bone marrow was significantly greater than in Wt mice (44.6% ± 2.9% of nucleated cells for Wt mice versus 60.9% ± 2.6% for HoxA10−/− mice, n=6, p=0.005) (Figure 1C). Two weeks post Alum injection, granulocyte infiltration of the lungs was observed in HoxA10−/− mice, but not Wt (Figure 1C). Pulmonary infiltration was not found in saline injected Wt or HoxA10−/− mice. Granulocyte infiltration of the spleen and liver was also observed in Alum-treated HoxA10−/− mice, but not in Alum injected Wt mice, or saline injected HoxA10−/− or Wt mice.
At steady state, expression of Triad1 mRNA in the bone marrow of HoxA10−/− mice was slightly less than Wt (p=0.02, n=4) and this difference increased significantly two weeks post Alum injection (p=0.001, n=4) (Figure 1D). HoxA10 expression in Wt murine bone marrow was not significantly different two weeks post injection with Alum versus saline (p=0.1, n=4) (Figure 1D). Expression of HoxA10 decreases as steady state granulopoiesis proceeds, but has not been investigated in emergency granulopoiesis. Since HoxA9 shares some HoxA10 target genes, we also investigated HoxA9 expression . We found that HoxA9 mRNA was slightly decreased two weeks post Alum injection in Wt and HoxA10−/− mice. HoxA9 expression was not altered by HoxA10 knockout (Figure 1D).
Re-expression of Triad1 in HoxA10−/− bone marrow normalizes emergency granulopoiesis
To determine if impaired Triad1 expression contributes to the abnormal emergency granulopoiesis phenotype of HoxA10−/− mice, we transduced HoxA10−/− Lin−Sca1+ bone marrow cells with a retroviral vector to re-express Triad1 (Triad1/MSCV) or empty expression vector (MSCV). Transduced bone marrow was transplanted into lethally irradiated Wt mice. Cohorts of mice were injected with Alum or saline and analyzed as above.
We found that mortality, numbers of circulating granulocyte, and granulocyte infiltration of the lungs after Alum injection were not significantly different in HoxA10−/− mice in comparison to Wt mice transplanted with control-vector-transduced HoxA10−/− bone marrow (Figure 1A-C). In contrast, survival of Alum injected mice transplanted with Triad1-transduced HoxA10−/− bone marrow was significantly better than HoxA10−/− mice (Figure 1A). 100% of mice transplanted with Triad1-transduced HoxA10−/− bone marrow survived the first cycle of Alum injection and 80% survived a second cycle. Circulating granulocytes returned to baseline between Alum injections in these mice (Figure 1B).
Pulmonary infiltration with granulocytes was not found post Alum injection in mice transplanted with Triad1-transduced HoxA10−/− bone marrow (Figure 1C). Two weeks after Alum injection, the percent of granulocytes in the bone marrow of control-vector-transduced HoxA10−/− recipients was significantly greater than in recipients of Triad1-transduced HoxA10−/− bone marrow (58.6% ± 3.2% of total nucleated cells for control vector versus 43.8% ± 2.7% with Triad1 vector, p=0.006 n=5) (Figure 1C). We found similar Triad1 expression in the bone marrow of Alum injected Wt mice and mice transplanted with Triad1-transduced HoxA10−/− bone marrow (p=0.6, n=4) (Figure 1D).
HoxA10 and Triad1 influence protein ubiquitination and Fgf-R1 stability during emergency granulopoiesis
We hypothesize that HoxA10-induced expression of Triad1 during emergency granulopoiesis results in ubiquitination and degradation of proteins that facilitate proliferation of myeloid progenitor cells and/or enhance phagocyte function. Fgf-R1 is regulated by ubiquitin-mediated lysosomal degradation, and we investigated involvement of Triad1 in this process. Fgf-R1 was of interest for several reasons. First, Fgf2-binding to Fgf-R1 enhances proliferation of progenitor cells, but also primes mature granulocytes for activation (27,38-40). Second, HoxA10 activates FGF2 transcription, but does not influence Fgf-R1 mRNA (27). Ubiquitination of Fgf-R1 by Triad1 would be a mechanism for HoxA10 to terminate effects of Fgf2.
To pursue this, we first determined if loss of HoxA10 influenced total protein ubiquitination in murine bone marrow cells. For these studies, CD34+ cells were isolated from Wt or HoxA10−/− murine bone marrow after 24 hrs of culture in GM-CSF, IL3 and Scf (referred to as myeloid progenitor conditions in these studies) and analyzed with or without IL1β-induced differentiation (9,30). We found that >70% of cells were Sca1−ckit+CD34+CD38−Gr1− under myeloid progenitor conditions and >80% were CD34−CD38+Gr1+ after treatment with IL1β.
We analyzed cell lysates for protein expression and ubiquitination by Western blot, and for mRNA expression by quantitative real time PCR. We found a significantly greater increase in total protein ubiquitination upon differentiation of Wt myeloid progenitor cells with IL1β in comparison to HoxA10−/− cells (Figure 2A). Differentiation with IL1β significantly increased Triad1 protein (Figure 2A) and mRNA (Figure 2B) in Wt myeloid progenitor cells, but not HoxA10−/− cells, consistent with in vivo results, above. Fgf-R1 protein was relatively less abundant in Wt versus HoxA10−/− cells; with and without IL1β-induced differentiation (Figure 2A). Expression of Fgf-R1 mRNA was not altered by HoxA10 knockout, consistent with our previous studies (not shown) (21).
Figure 2.

HoxA10 influences total protein ubiquitination and Fgf-R1 protein stability in a Triad1-dependent manner. Bone marrow from wild type (Wt) or HoxA10−/− mice was assayed under myeloid progenitor conditions (GM-CSF, IL3 and Scf followed by CD34+ separation) or after IL1β-induced differentiation. Some cells were transduced with a Triad1 expression vector or MSCV control vector. A. Loss of HoxA10 decreases IL1β-induced total protein ubiquitination and increases Fgf-R1 protein, but this is reversed by re-expression of Triad1. Cell lysates were analyzed by Western blots probed with antibodies to total ubiquinated (Ub) protein, Triad1, Fgf-R1 or Gapdh (as a loading control). B. HoxA10-knockout prevents increased Triad1 mRNA expression during IL1β-induced differentiation. These cells were also analyzed for Triad1 mRNA expression by real time PCR. Statistically significant differences in Triad1 mRNA abundance with versus without Triad1 vector are indicated by *. Statistically significant difference after IL1β differentiation is indicated by **, and in Wt versus HoxA10−/− cells by ***. P values< 0.02 were considered statistically significant C. Triad1 overexpression does not alter HoxA9 or HoxA10 expression, and HoxA10-knockout does not alter HoxA9 expression. Cells were analyzed for HoxA9 or HoxA10 mRNA by real time PCR. Statistically significant differences in HoxA10 mRNA abundance with HoxA10-knockout versus Wt cells are indicated by * or **. P values< 0.02 were considered statistically significant D. HoxA10-knockout decreases Fgf-R1 ubiquitination in a Triad1-dependent manner. The cells described above were treated with a lysosomal stabilizer (E64) prior to harvesting. Cell lysates were immuno-precipitated with an anti-ubiquitin antibody followed by Western blotting with antibody to Fgf-R1. Western blots of total cell lysates were probed with antibody to Gapdh as a loading control. Lysates that were immuno-precipitated with an irrelevant, control antibody demonstrated no immuno-reactive Fgf-R1 on Western blot (not shown).
We next investigated the role of Triad1 in the decrease in total protein ubiquitination, and increase in Fgf-R1 protein, in HoxA10−/− bone marrow. For these studies, we transduced Lin−Sca1+ bone marrow mononuclear cells with a retroviral vector to express Triad1 or with control MSCV vector. Cells were analyzed as above. We found that re-expression of Triad1 in HoxA10−/− cells increased total protein ubiquitination and decreased Fgf-R1 protein (Figure 2A). In control experiments, we demonstrated that expression of HoxA9 and HoxA10 mRNA were not altered by Triad1 re-expression (Figure 2C).
To determine if the observed increase in Fgf-R1 protein in HoxA10−/− cells was due to decreased ubiquitination, some of the cells (described above) were treated with a lysosomal stabilizer (E64) prior to harvesting, and lysate proteins were immuno-precipitated with an anti-ubiquitin antibody followed by Western blot for Fgf-R1. We found increased Fgf-R1 ubiquitination during IL1β-differentiation of Wt cells, but not HoxA10−/− cells. Fgf-R1 ubiquitination was greater in Wt versus HoxA10−/− cells, but was increased by re-expressing Triad1 in HoxA10−/− cells (Figure 2D).
HoxA9 and HoxA10 regulate ARIH2 transcription during emergency granulopoiesis
Mechanisms that regulate ARIH2 transcription during myelopoiesis are undefined. We hypothesized that activation of ARIH2 by HoxA10 during emergency granulopoiesis might be antagonized by HoxA9. This would be the opposite of the mechanism for regulation of phagocyte effector genes, which are repressed by HoxA10 in myeloid progenitor cells, but activated by HoxA9 during myelopoiesis (34-37). We investigated this hypothesis using ARIH2 promoter/reporter constructs designed around Hox-DNA-binding consensus sequences (previously described) (21,52). U937 myeloid cells were co-transfected with these constructs and vectors to express shRNAs specific to HoxA9, HoxA10 or both, or control scrambled shRNA. Transfectants were assayed for reporter activity with or without IL1β-induced differentiation (21,24). We found that knockdown of HoxA9 significantly increased activity of constructs with 198 bp of ARIH2 promoter (p<0.001, n=6; similar results with 333 bp and 629 bp constructs), but not constructs with 167 bp or less (p>0.6, n=6) (Figure 3A). Differentiation significantly decreased this effect of HoxA9-knockdown (p<0.01, n=6 for percent repression by HoxA9 with versus without differentiation). Knockdown of HoxA10 decreased activity of the ARIH2 promoter constructs in a manner that was consistent with known HoxA10-binding cis elements between −32 to −41 bp and −182 to −191 bp (Figure 3B) (21). Knockdown of HoxA9 + HoxA10 increased activity of 198 bp construct in untreated transfectants (p<0.01, n=6), but decreased activity in differentiated transfectants (p<0.001, n=6). Control experiments confirmed equivalent knockdown of HoxA9 and HoxA10 (Figure 3C).
Figure 3.

HoxA9 and HoxA10 regulate ARIH2 transcription during emergency granulopoiesis. A. HoxA9 represses a ARIH2 cis element between −198 and −167 bp, but this effect is decreased by differentiation with IL1β. U937 cells were co-transfected with promoter-reporter gene constructs with truncations of the ARIH2 5’ flank and vectors to knockdown HoxA9, HoxA10 or both. Statistically significant differences in reporter activity with versus without HoxA10 specific shRNA are indicated by * or #, with versus without HoxA9 shRNA by ** or ##, and with versus without both shRNAs by *** or ###. Statistically significant difference in reporter activity with versus without IL1β is indicated by &. P values< 0.02 were considered statistically significant B. Schematic identifying the ARIH2 cis elements. Human sequences are in black, murine in blue and conserved sequences in gray. Hox-consensus sequences are in red and the HoxA10-binding cis elements are underlined. Truncations used in reporter assays are indicated by an arrows. C. Control studies demonstrate equivalent shRNA knockdown of HoxA9 or HoxA10. Hox protein expression was determined by real time PCR or Western blot (inset). Statistically significant differences in HoxA9 or HoxA10 mRNA with versus without knockdown of HoxA9 or HoxA10 are indicated by * or **, respectively. P values< 0.02 were considered statistically significant.
To further investigate repression of ARIH2 by HoxA9, we co-transfected U937 cells with a reporter construct that included three copies of the proximal or distal cis element linked to a minimal promoter plus vectors to knockdown or overexpress various combinations of HoxA9 and HoxA10 (or relevant control vectors). Transfectants were analyzed for reporter activity with or without IL1β-differentiation. HoxA9-knockdown did not alter activity of the proximal ARIH2 cis element (not shown). However, distal cis element activity was increased by expression of HoxA9-specific shRNA (p<0.001, n=6) and decreased by HoxA10-knockdown (p<0.01, n=6) (Figure 4A). Simultaneously knocking down HoxA9 and overexpressing HoxA10 activated the distal cis element in comparison to overexpressing HoxA10 alone (p<0.0001, n=6) (Figure 4A). And, HoxA10-knockdown in HoxA9-overexpressing transfectants further decreased cis element activity in comparison to overexpressing HoxA9 alone (p<0.01, n=6) (Figure 4A).
Figure 4.

HoxA9 and HoxA10 are antagonists for ARIH2 transcription during emergency granulopoiesis. A. HoxA9 antagonizes activation of the distal ARIH2 cis element by HoxA10 in untreated, but not in differentiating, transfectants. U937 cells were co-transfected with a reporter construct with 3 copies of the distal ARIH2 cis element linked to a minimal promoter and vectors to overexpress and/or knockdown HoxA9 and HoxA10. Statistically significant differences in reporter activity with versus without overexpression of HoxA10 are indicated by * or #, and with versus without overexpression of HoxA9 by ** or ##. Statistically significant difference reporter activity in HoxA9 + HoxA10 overexpressing cells with versus without IL1β is indicated by ***. Statistically significant differences in HoxA10 overexpressing cells with versus without HoxA9 shRNA are indicated by ### or &, and in HoxA9 overexpressing cells with versus without HoxA10 shRNA by & or @. Statistically significant differences in reporter activity with versus without expression of HoxA10 specific shRNA are indicated by @@ or ^, and with versus without HoxA9 specific shRNA by @@@ or ^^. P values< 0.02 were considered statistically significant. B. Blocking tyrosine phosphorylation decreases ARIH2 activation by HoxA10 and increases ARIH2 repression by HoxA9. U937 cells were co-transfected with the distal ARIH2 cis element reporter construct and vectors to express HoxA9 or HoxA10, forms of these proteins with mutation of tyrosine residues in the homeodomain (HD-Y-mut), or Wt Hox proteins + constitutively active Shp2 (E76K). Statistically significant differences in reporter activity in cells overexpressing Wt versus HD-Y-Mut HoxA9, or Wt versus HD-Y-Mut HoxA10 are indicated by * or *** (respectively) in undifferentiated transfectants, and by ## or & in IL1β differentiated transfectants. Statistically significant differences in reporter activity in HoxA9 or HoxA10 overexpressing cells with versus without E76K-Shp2 expression are indicated by ** or # (respectively) in undifferentiated transfectants, and by ### or && in IL1β differentiated transfectants. P values< 0.02 were considered statistically significant.
To determine if phosphorylation of conserved, tyrosine residues in the homeodomains HoxA9 and HoxA10 influenced ARIH2 promoter activity, we co-transfected U937 cells with the distal cis element/reporter construct and vectors to overexpress tyrosine-mutant forms of HoxA9 (referred to as HD Y-mut-HoxA9) or HoxA10 (HD Y-mut-HoxA10) (36,37). As another method to prevent phosphorylation, we co-transfected other cells with vectors to overexpress HoxA9 or HoxA10 plus a constitutively active form of Shp2 (E76K). HoxA9 and HoxA10 are substrates for Shp2 only in myeloid progenitor cells, but E76K-Shp2 de-phosphorylates these proteins throughout IL1β-induced myelopoiesis (30,36,37). For these studies, we used an amount of E76K-Shp2 was used that had a minimal effect on the ARIH2 cis element alone (determined by dose titration studies; not shown).
We found that distal cis element activation was less efficient in transfectants overexpressing HD Y-mut-HoxA10 or HoxA10 + E76K-Shp2 compared to transfectants with Wt HoxA10 alone, with or without IL1β-induced differentiation (p<0.0001, n=6) (Figure 4B). Conversely, overexpression of either HD Y-mut-HoxA9 or HoxA9 + E76K-Shp2 more efficiently repressed the distal cis element than overexpressed Wt HoxA9 alone (p<0.01, n=6) (Figure 4B). We previously determined that Wt and HD Y-mutant proteins have comparable expression and stability in U937 cells (30,36,37).
HoxA9 and HoxA10 interact with the ARIH2 promoter during emergency granulopoiesis
To investigate interaction between HoxA9 or HoxA10 and the ARIH2 promoter during emergency granulopoiesis, we performed chromatin immuno-precipitation assays. For these studies, Wt murine bone marrow mononuclear cells were cultured under myeloid progenitor conditions (see above) and analyzed with or without differentiation with G-CSF or IL1β (9,30). We found that either cytokine significantly decreased in vivo interaction of HoxA9 with the distal ARIH2 cis element, but increased binding of HoxA10 (p<0.001, n=3) (Figure 5A).
Figure 5.

Emergency granulopoiesis alters in vivo HoxA9 and HoxA10 binding to the ARIH2 promoter. A. Differentiation of murine myeloid progenitor cells with IL1β or G-CSF increases in vivo HoxA10 binding and decreases HoxA9 binding to the distal ARIH2 cis element. Murine bone marrow cells were assayed under myeloid progenitor conditions, or after G-CSF or IL1β-induced differentiation. Chromatin was co-immuno-precipitated with antibody to HoxA9, HoxA10 or irrelevant control antibody and analyzed by real time PCR with primers flanking the distal ARIH2 cis element. Statistically significant differences in co-precipitation of cis element DNA with HoxA9 or HoxA10 in cells with versus without G-CSF or IL1β induced differentiation are indicated by *, **, ***, or #. P values< 0.02 were considered statistically significant. B. Constitutive activation of Shp2-PTP increases HoxA9 binding to the distal ARIH2 cis element, but decreases binding of HoxA10. Bone marrow cells from Wt mice were transduced with a retroviral vector to express E76K-Shp2 or with MSCV control vector. Cells were analyzed by chromatin immuno-precipitation with antibodies to HoxA9 or HoxA10, as described above. Statistically significant differences in co-precipitation of cis element DNA with HoxA9 or HoxA10 in cells with versus without E76K-Shp2-expression are indicated by *, **, ***, or #. P values< 0.02 were considered statistically significant. C. Constitutive activation of Shp2 blocks IL1β-induced tyrosine phosphorylation of HoxA9 or HoxA10. Lysates from cells described above were subjected to immuno-precipitation with an anti-phospho tyrosine antibody (or irrelevant control antibody) followed by Western blots with antibodies to total HoxA9 or HoxA10 protein. D. Control studies demonstrate Shp2 overexpression in E76K-Shp2 transduced cells, but no change in expression of HoxA9 or HoxA10. Western blots of non-immuno-precipitated proteins from the experiment described above were probed with antibody to Shp2, HoxA9, HoxA10 or Gapdh (as a loading control).
Based on the reporter assays in the previous section, we also investigated the role of tyrosine phosphorylation on binding of HoxA9 or HoxA10 to the ARIH2 cis element. For these studies, Lin−Sca1+ murine bone marrow mononuclear cells were transduced with a retroviral to express constitutively active Shp2 (E76K) or with empty control vector. Cells were cultured under myeloid progenitor conditions, with or without IL1β-induced differentiation, and chromatin immuno-precipitation assays were performed, as above. We found that constitutive activation of Shp2 significantly decreased HoxA10 binding to the ARIH2 promoter in IL1β-differentiated cells, but increased HoxA9 binding under these conditions (for both experiments p<0.01, n=4) (Figure 5B). In control experiments, lysate proteins from these cells were also analyzed for tyrosine phosphorylation of HoxA9 and HoxA10 by immuno-precipitation and Western blot. IL1β increased tyrosine phosphorylation of both HoxA9 and HoxA10, but this was blocked in cells expressing E76K-Shp2 (Figure 5C). Other control experiments verified that Shp2 was overexpressed in E76K-Shp2 transduced cells, but expression of total HoxA9 or HoxA10 protein was not altered (Figure 5D).
We also performed in vitro assays as an additional approach to evaluate this interaction. For these studies, U937 nuclear proteins were pre-incubated with HoxA9, HoxA10 or irrelevant control antibody, and analyzed by electrophoretic mobility shift assay. We previously demonstrated that the distal ARIH2 cis element probe generates a low mobility, HoxA10-containing complex in such assays (21). In the current studies, we found that HoxA9 antibody disrupted the complex most efficiently in assays with untreated cells, and HoxA10 antibody was most efficient with differentiated cells (Figure 6A). In binding assays with unlabeled oligonucleotide competitors, we found cross competitive binding specificities between the ARIH2 cis element and HoxA9/A10 binding cis elements from several previously described HoxA9/HoxA10 target genes, but not irrelevant oligonucleotide competitors (Figure 6B).
Figure 6.

Differentiation of U937 cells decreases in vitro HoxA9 binding to the distal ARIH2 cis element. EMSA were performed with a radiolabeled probe representing the cis element and U937 nuclear proteins. Some binding assays were incubated with; A. Antibodies to HoxA9 or HoxA10 or, B. Competitor oligonucleotides (200 fold molar excess) representing other Hox-binding cis elements (with or without Hox-binding-site mutations). The arrow represents free probe.
HoxA9 influences Triad1-expression and total protein ubiquitination
We first investigated the influence of IL1β on expression of HoxA9, HoxA10 and their common target genes in primary murine bone marrow cells. We found that expression of HoxA9 and HoxA10 increased in IL1β-treated cells at early time points, but decreased by 48 hrs (Figure 7A). We also found an IL1β-induced increase in expression of other Hox-target-genes involved in the innate immunity, including β3 integrin, Fgf2 and gp91phox (Figure 7A) (27,36,47).
Figure 7.

HoxA9 and HoxA10 regulate Triad1 expression during IL1β-induced differentiation of murine myeloid progenitor cells. A. Expression of HoxA9, HoxA10 and target genes that are relevant to innate immunity increases during differentiation with IL1β. Murine bone marrow cells were cultured under myeloid progenitor conditions with or without differentiation with IL1β. Expression of mRNA for HoxA9, HoxA10, β3 integrin, Fgf2, gp91phox, or Triad1 was determined by real time PCR. Statistically significant differences in mRNA expression of these genes in IL1β-differentiated cells versus control cells are indicated by *, **, ***, #, ##, or ###, respectively. P values< 0.02 were considered statistically significant. B. Knockdown of HoxA9 increases total protein ubiquitination in a Triad1-dependent manner. Bone marrow derived myeloid progenitor cells were transduced with retroviral vectors to express shRNAs specific to HoxA9, Triad1, both, or control (scrambled) shRNAs and analyzed with or without IL1β-differentiation. Western blots of lysate proteins were probed with antibodies to total ubiquitinated proteins, Triad1, or Gapdh as a loading control. C. Knockdown of HoxA9 increases Triad1 mRNA expression. These cells were also analyzed by real time PCR for mRNA abundance of Triad1 and HoxA9. Statistically significant differences in Triad1 expression with versus without HoxA9 knockdown are indicated by * or #, and with versus without knockdown of Triad1 by ** or ##. Statistically significant differences in Triad1 expression in cells with Triad1 knockdown with versus without HoxA9 shRNA are indicated by *** or ###. Statistically significant differences in HoxA9 mRNA with versus without HoxA9 knockdown are indicated by & or &&. P values< 0.02 were considered statistically significant.
Based on our studies of the ARIH2 promoter, we hypothesized that Triad1 expression and total protein ubiquitination are inversely related to HoxA9 expression level. To investigate this, we transduced Wt Lin−Sca1+ murine bone marrow cells with vectors to express HoxA9 specific shRNAs or scrambled shRNA control vectors. Transduced cells were analyzed by Western blot and real time PCR under myeloid progenitor conditions (as defined above) or after differentiation with IL1β. We found that HoxA9 knockdown increased total protein ubiquitination (Figure 7B) and expression of Triad1 protein (Figure 7B) and mRNA (Figure 7C), consistent with our hypothesis. Control studies verified knockdown of HoxA9.
To investigate the influence of Triad1 on HoxA9 related changes in protein ubiquitination, we also transduced Lin−Sca1+ bone marrow cells with retroviral vectors to express Triad1 specific shRNAs, with or without HoxA9 shRNAs. Cells were analyzed as above. We found that blocking Triad1 expression decreased total protein ubiquitination in cells with HoxA9 knockdown (Figure 7B). Control studies demonstrated decreased expression of Triad1 protein (Figure 7B) and mRNA (Figure 7C) with these shRNA vectors.
HoxA9 and Triad1 influence cell proliferation
In previous studies, we found that the HoxA10-induced Triad1 expression antagonized the net pro-proliferative effect of overexpressing HoxA10 in myeloid progenitor cells (21). Based on the results in the current work, we hypothesize that repression of ARIH2 by HoxA9 facilitates cytokine-induced proliferation in HoxA9 overexpressing cells. To investigate this, we transduced Lin−Sca1+ murine bone marrow cells with retroviral vectors to express HoxA9, Triad or both (or control vectors). Cells were analyzed for proliferation in response to a dose titration of GM-CSF. We found that GM-CSF induced significantly more proliferation in HoxA9-overexpressing cells in comparison to control cells at most doses (p<0.01, n=6) (Figure 8A). We also found that re-expression of Triad1 decreased GM-CSF-hypersensitivity in HoxA9 overexpressing cells (Figure 8A). In control studies, we found that HoxA9 overexpression significantly decreased Triad1 expression, with or without IL1β-induced differentiation (p<0.001, n=4) (Figure 8B). These studies also overexpression of Triad1 and that overexpressed Triad1 did not influence HoxA9 mRNA abundance.
Figure 8.

Regulation of Triad1 by HoxA9 influences cell proliferation. A. Triad1 reverses HoxA9-induced cytokine hypersensitivity. Murine myeloid progenitor cells were transduced with retroviral vectors to express HoxA9, Triad1, both, or control vector and analyzed for proliferation in response to a dose titration of GM-CSF. Statistically significant differences in cell proliferation at a given cytokine dose (as determined by 3H-thymidine uptake) are indicated by * or **. B. Overexpression of HoxA9 decreases Triad1 expression. Murine bone marrow cells described above were analyzed for Triad1 and HoxA9 expression by real time PCR. Statistically significant differences in Triad1 mRNA abundance with versus without HoxA9 overexpression are indicated by *, **, ***, or #. Statistically significant difference in HoxA9 mRNA abundance with versus without HoxA9 overexpression are indicated by ##. P values< 0.02 were considered statistically significant
Discussion
In this work, we found that mice with knockout of HoxA10 in the bone marrow have a fatal emergency granulopoiesis response; a phenotype this is rescued by re-expression of Triad1. Inflammatory infiltrates and granulocytosis are not observed at steady state in HoxA10−/− mice, but constitutive inflammation is a characteristic of mice with Triad1−/− bone marrow (19,23). Therefore, our studies suggest that conditional regulation of ARIH2 by HoxA10 contributes to terminating the innate immune response. Hox proteins were not previously known to modulate emergency granulopoiesis (or any other process) by facilitating ubiquitin-mediated protein degradation. We found that expression of Triad1 and transcription of ARIH2 during emergency granulopoiesis was dependent on tyrosine phosphorylation of HoxA10. And, that non-tyrosine phosphorylated HoxA9 antagonized HoxA10 by repressing ARIH2 in myeloid progenitor cells. Therefore, these Hox proteins cooperate to regulate the innate immune response in addition to performing their well described proto-oncogene functions during leukemogenesis.
Mice with Triad1−/− bone marrow exhibit constitutive infiltration of tissues with granulocyte and B-cells, suggesting that Triad1 controls inflammatory processes (19). We find only slightly decreased Triad1 expression in the bone marrow of HoxA10−/− mice at steady state, but an absence of normal induction of Triad1 expression during emergency granulopoiesis. Although HoxA10-induced Triad1 expression is important for termination of emergency granulopoiesis, our studies do not exclude a role for Triad1 in the steady state process. Conversely, steady state granulopoiesis is relatively normal in HoxA10−/− mice, suggesting that regulation of emergency granulopoiesis may be a major, but previously unknown, function of HoxA10.
Total protein ubiquitination increases in a HoxA10 and Triad1 dependent manner in differentiating myeloid cells. Many proteins involved in innate immunity are metabolized by ubiquitination mediated degradation, including Fgf-R1, gp91phox and intermediates downstream from the IL1-R (29,53,54). HoxA9 protein may also be regulated by ubiquitination (55). Such proteins may be direct substrates for Triad1 or for another ligase that is regulated by Triad1 (56). Previous studies indicated that HoxA10 inhibits granulocyte function by repressing phagocyte effector genes in myeloid progenitor cells, including genes encoding the rate limiting NADPH-oxidase proteins and secondary granule proteins (34-36,57,58). Activation of ARIH2 is a novel immune modulatory mechanism for HoxA10, involving induction of protein degradation. Conversely, HoxA9 has been shown to activate genes encoding NADPH-oxidase proteins and E selectin in differentiating phagocytes (37,41). ARIH2-repression may enhance pro-inflammatory effects of HoxA9 by stabilizing these or other inflammatory mediator proteins.
Although HoxA9 and HoxA10 mRNA is maximally expressed in myeloid progenitors, HoxA9 and HoxA10 proteins are relatively stable and found in granulocytes (25,26). Consistent with this, we found that phosphorylation of conserved tyrosine residues in the homeodomains HoxA9 and HoxA10 during IL1β, G-CSF or M-CSF induced differentiation of myeloid progenitor cells influences expression of some target genes (34-37). HoxA10-phosphorylation decreased affinity for cis elements in the NADPH-oxidase genes, but increases the affinity for ARIH2 (34-37). Conversely, HoxA9-phosphorylation increased affinity for NADPH-oxidase genes, but decreases affinity for ARIH2 (34-37). Differences between HoxA9 and HoxA10 in the impact of homeodomain phosphorylation on target gene regulation may reflect the influence of non-conserved domains outside of the homeodomain. For example, we previously defined activation and repression domains in HoxA10 that are not present in HoxA9 (47, 59).
In contrast to these genes, HoxA9 and HoxA10 cooperate to activate the FGF2 gene in a manner that is not influenced by tyrosine phosphorylation (27,38). Instead, our current studies suggest that HoxA10 regulates the effects of Fgf2 via Triad1-induced ubiquitination and degradation of Fgf-R1. This may terminate the effect of Fgf2 on progenitor expansion and phagocyte function, and down-regulate emergency granulopoiesis (Figure 9).
Figure 9.
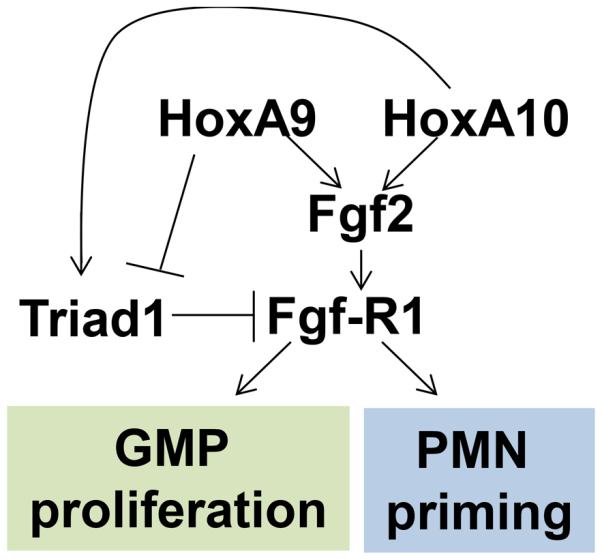
Schematic representation of regulatory network involving HoxA10, HoxA9, Triad1 and Fgf2. Regulation of Triad1 influences progenitor proliferation and phagocyte function via Fgf2 and Fgf-R1.
The physiology of emergency granulopoiesis is relevant to human auto-inflammatory joint diseases (12-16). IL1β-activated events drive these conditions, and IL1-receptor blockade is effective in some patients. Emergency granulopoiesis physiology is also found in G-CSF-treated patients with severe congenital neutropenia and may contribute to the risk of progression to acute myeloid leukemia in these individuals (60). Identification of HoxA10-induced Triad1 expression as a component of the process that down-regulates emergency granulopoiesis suggests possible therapeutic targets for these conditions.
Footnotes
These studies were supported by NIH HL87717, NIH DK098812, a VA Merit Review, the Mander Foundation and the Director’s Fund of the Robert H. Lurie Comprehensive Cancer Center to EAE
References
- 1.Lord BI, Molineux G, Pojda Z, Souza LM, Mermod JJ, Dexter TM. Myeloid cell kinetics in mice treated with recombinant interleukin-3, G-CSF, or GM-CSF in vivo. Blood. 1991;77:2154–9. [PubMed] [Google Scholar]
- 2.Panopoulos AD, Watchwich SS. Granulocyte colony stimulating factor: molecular mechanisms of activation during steady state and emergency hematopoiesis. Cytokine. 2007;42:277–88. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieschke GJ, Grail D, Kodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking G-CSF have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency and impaired neutrophil mobilization. Blood. 1994;84(6):1737–46. [PubMed] [Google Scholar]
- 4.Zhan Y, Lieschke GJ, Grail D, Dunn AR, Cheers C. Essential roles for GM-CSF and G-CSF in the sustained hematopoietic response of Listeria monocytogenes infected mice. Blood. 1998;91:863–9. [PubMed] [Google Scholar]
- 5.Dakic A, Metcalf D, Di Rago L, Mifsud S, Wu L, Nutt SL. PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. 2005;201:1487–502. doi: 10.1084/jem.20050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skokowa J, Welte K. Dysregulation of myeloid-specific transcription factors in congenital neutropenia. Ann N Y Acad Sci. 2009;1176:94–100. doi: 10.1111/j.1749-6632.2009.04963.x. [DOI] [PubMed] [Google Scholar]
- 7.Ueda Y, Cain DW, Kuraoka M, Kondo M, Kelso G. IL1RI dependent hematopoietic stem cell proliferation is necessary for inflammatory granulopoiesis and reactive neutrophilia. J. Immunol. 2009;182:6477–84. doi: 10.4049/jimmunol.0803961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423–31. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu L, Huang W, Hjort EE, Eklund EA. Increased Fanconi C Expression Contributes to the Emergency Granulopoiesis Response. J. Clin. Invest. 2013;123:3952–66. doi: 10.1172/JCI69032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, Tenen DG. C/EBPbeta is required for 'emergency' granulopoiesis. Nat Immunol. 2006;7:732–9. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 11.Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS. Stat3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–90. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 2010;49:1618–31. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- 13.Popa-Nita O, Naccache PH. Crystal-induced neutrophil activation. Immunol Cell Biol. 2010;88:32–40. doi: 10.1038/icb.2009.98. [DOI] [PubMed] [Google Scholar]
- 14.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 15.Emery P, Sebba A, Huizinga TW. Biologic and oral disease-modifying antirheumatic drug monotherapy in rheumatoid arthritis. Ann Rheum Dis. 2013;72:1897–904. doi: 10.1136/annrheumdis-2013-203485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleishmann RM. Safety of anakinra, a recombinant interleukin-1 receptor antagonist, in patients with rheumatoid arthritis and comparison to anti-TNF-alpha agents. Clin Exp Rheumatol. 2002;20:S35–41. [PubMed] [Google Scholar]
- 17.Van Der Reijden BA, Erpelinck-Verschueren CAJ, Lowenberg B, Jansen JH. TRIADs: a new class of proteins with a cysteine-rich signature. Protein Science. 1999;8:1557–61. doi: 10.1110/ps.8.7.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marteijn JAF, van Emst L, Erpelinck-Verschueren CAJ, Nikoloski G, Menke A, de Witte T, Löwenberg B, Jansen JH, van der Reijden BA. The E3 ubiquitin-protein ligase Triad1 inhibits clonogenic growth of primary myeloid progenitor cells. Blood. 2005;106:4114–23. doi: 10.1182/blood-2005-04-1450. [DOI] [PubMed] [Google Scholar]
- 19.Lin AE, Ebert G, Ow Y, Preston SP, Toe JG, Cooney JP, Scott HW, Sasaki M, Saibil SD, Dissanayake D, Kim RH, Wakeham A, You-Ten A, Shahinian A, Duncan G, Silvester J, Ohashi PS, Mak TW, Pellegrini M. ARIH2 is essential for embryogenesis, and its hematopoietic deficiency causes lethal activation of the immune system. Nat Immunol. 2013;14:27–33. doi: 10.1038/ni.2478. [DOI] [PubMed] [Google Scholar]
- 20.Marteijn JA, van der Meer LT, Smit JJ, Noordermeer SM, Wissink W, Jansen P, Swarts HG, Hibbert RG, de Witte T, Sixma TK, Jansen JH, van der Reijden BA. The ubiquitin ligase Triad1 inhibits myelopoiesis through UbcH7 and Ubc13 interacting domains. Leukemia. 2009;23:14809. doi: 10.1038/leu.2009.57. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Bei L, Shah CA, Horvath E, Eklund EA. HoxA10 influences protein ubiquitination by activating transcription of ARIH2; the gene encoding Triad1. J. Biol. Chem. 2011;286:16832–45. doi: 10.1074/jbc.M110.213975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassink G, Slotman J, Oorschot V, Van Der Reijden BA, Monteferrario D, Noordermeer SM, Van Kerkhof P, Klumperman J, Strous GJ. Identification of the ubiquitin ligase Triad1 as a regulator of endosomal transport. Biol Open. 2012;1:607–14. doi: 10.1242/bio.2012778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satokata I, Benson G, Maas R. Sexually dimorphic sterility phenotypes in Hoxa10-deficient mice. Nature. 1995;374:460–3. doi: 10.1038/374460a0. [DOI] [PubMed] [Google Scholar]
- 24.Larrick JW, Fischer DG, Anderson SJ, Koren HS. Characterization of a human macrophage-like cell line stimulated in vitro. J. Immunol. 1980;125:6–12. [PubMed] [Google Scholar]
- 25.Acampora D, D'Esposito M, Faiella A, Pannese M, Migliaccio E, Morelli F, Stornaiuolo A, Nigro V, Simeone A, Boncinelli E. The human HOX gene family. Nucleic Acids Res. 1989;17:10385–10400. doi: 10.1093/nar/17.24.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid ES, Largman C, Lawrence HJ, Humphries RK. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl. Acad. Sci. USA. 1994;9:12223–7. doi: 10.1073/pnas.91.25.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah CA, Bei L, Wang H, Platanias LC, Eklund EA. HoxA10 regulates transcription of the gene encoding Fibroblast Growth Factor 2 (FGF2) in myeloid cells. J Biol Chem. 2012;287:18230–48. doi: 10.1074/jbc.M111.328401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah CA, Wang H, Bei L, Platanias LC, Eklund EA. HoxA10 regulates transcription of the gene encoding transforming growth factor beta 2 in myeloid cells. J. Biol. Chem. 2011;286:3161–76. doi: 10.1074/jbc.M110.183251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B, Yang FC, Clapp DW, Chun KT. Enforced expression of CUL4A interferes with granulocytic differentiation and exit from the cell cycle. Blood. 2003;101:1769–76. doi: 10.1182/blood-2002-05-1517. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Lindsey S, Konieczna I, Eklund EA. Constitutively Active SHP2 Cooperates with HoxA10-overexpression to Induce Acute Myeloid Leukemia. J. Biol. Chem. 2009;284:2549–67. doi: 10.1074/jbc.M804704200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Lansdorp PM, Lawrence HJ, Largman C, Humphries RK. Overexpression of HoxA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol. Cell. Biol. 1995;17:495–505. doi: 10.1128/mcb.17.1.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. HoxA9 transforms primary bone marrow cells through collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17:3714–25. doi: 10.1093/emboj/17.13.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence HJ, Helgason CD, Sauvageau G, Fong S, Izon DJ, Humphries RK, Largman C. Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood. 1997;89:1922–30. [PubMed] [Google Scholar]
- 34.Eklund EA, Jalava A, Kakar R. Tyrosine phosphorylation decreases HoxA10 DNA-binding and transcriptional repression during IFNγ differentiation in myeloid cell lines. J Biol Chem. 2000;275:20117–26. doi: 10.1074/jbc.M907915199. [DOI] [PubMed] [Google Scholar]
- 35.Lindsey S, Zhu CL, Lu YF, Eklund EA. HoxA10 represses transcription of the gene encoding p67PHOX in phagocytic cells. J Immunol. 2005;175:5269–5279. doi: 10.4049/jimmunol.175.8.5269. [DOI] [PubMed] [Google Scholar]
- 36.Lindsay S, Huang W, Wang H, Horvath E, Zhu C, Eklund EA. Activation of SHP2 increases HoxA10-induced repression of genes encoding gp91PHOX and p67PHOX. J Biol Chem. 2007;282:2237–49. doi: 10.1074/jbc.M608642200. [DOI] [PubMed] [Google Scholar]
- 37.Bei L, Lu YF, Eklund EA. HoxA9 activates transcription of the gene encoding gp91PHOX during myeloid differentiation. J. Biol. Chem. 2005;280:12359–70. doi: 10.1074/jbc.M408138200. [DOI] [PubMed] [Google Scholar]
- 38.Shah CA, Bei L, Wang H, Platanias LC, Eklund EA. The leukemia-associated Mll-Ell oncoprotein induces Fibroblast growth factor 2 (Fgf2)-dependent cytokine hypersensitivity in myeloid progenitor cells. J. Biol. Chem. 2013;288:32490–505. doi: 10.1074/jbc.M113.496109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gabbianelli M, Sargiacomo M, Pelosi E, Testa U, Isacchi G, Peschle C. "Pure" human hematopoietic progenitors: permissive action of basic fibroblast growth factor. Science. 1990;249:1561–4. doi: 10.1126/science.2218497. [DOI] [PubMed] [Google Scholar]
- 40.Takagi S, Takahashi K, Katsura Y, Matsuoka T, Ohsaka A. Basic fibroblast growth factor modulates the surface expression of effector cell molecules and primes respiratory burst activity in human neutrophils. Acta Haematol. 2000;103:78–83. doi: 10.1159/000041024. [DOI] [PubMed] [Google Scholar]
- 41.Bandyopadhyay ,S, Ashraf MZ, Daher P, Howe PH, DiCorleto PE. HOXA9 participates in the transcriptional activation of E-selectin in endothelial cells. Mol Cell Biol. 2007;27:4207–16. doi: 10.1128/MCB.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowney P, Corral J, Detmer K, LeBeau MM, Deaven L, Lawrence HJ, Largman C. A human Hox 1 homeobox gene exhibits myeloid-specific expression of alternative transcripts in human hematopoietic cells. Nucl. Acids Res. 1991;19:3443–3449. doi: 10.1093/nar/19.12.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takebe Y, Seiki M, Fujisajwa J, Hoy P, Yokota K, Arai K, Yoshida M, Arai N. SR alpha promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol. Cell. Biol. 1988;8:466–72. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eklund EA, Goldenberg I, Lu YF, Andrejic J, Kakar R. SHP1 protein-tyrosine phosphatase regulates HoxA10 DNA binding and transcriptional repression activity in undifferentiated myeloid cells. J. Biol. Chem. 2002;277:36878–88. doi: 10.1074/jbc.M203917200. [DOI] [PubMed] [Google Scholar]
- 45.Eklund EA, Jalava A, Kakar R. PU.1, interferon regulatory factor 1, and interferon consensus sequence-binding protein cooperate to increase gp91(phox) expression. J. Biol. Chem. 1998;273:13957–65. doi: 10.1074/jbc.273.22.13957. [DOI] [PubMed] [Google Scholar]
- 46.Oberley MJ, Farnham PJ. Probing chromatin immunoprecipitates with CpG-island microarrays to identify genomic sites occupied by DNA-binding proteins. Methods in Enzymol. 2003;71:577–96. doi: 10.1016/S0076-6879(03)71043-X. [DOI] [PubMed] [Google Scholar]
- 47.Bei L, Lu YF, Bellis SL, Zhou W, Horvath E, Eklund EA. Identification of a HoxA10 activation domain necessary for transcription of the Beta 3 integrin gene during myeloid differentiation. J. Biol. Chem. 2007;282:16846–59. doi: 10.1074/jbc.M609744200. [DOI] [PubMed] [Google Scholar]
- 48.Grinde B. Selective inhibition of lysosomal protein degradation by thiol proteinase inhibitors E-64, Ep-459 and Ep-457 in rat hepatocytes. Biochim Biophys Acta. 1982;701:328–33. doi: 10.1016/0167-4838(82)90235-7. [DOI] [PubMed] [Google Scholar]
- 49.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from mammalian nuclei. Nucl. Acids Res. 1983;11:1475–9. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basu S, Hodgson G, Zhang HH, Katz M, Quilici C, Dunn AR. Emergency granulopoiesis in G-CSF deficient mice in response to Candida albicans infection. Blood. 2000;95:3725–33. [PubMed] [Google Scholar]
- 51.Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, Bergen IM, Castillo R, Lambrecht BN, Tschopp J. Cutting Edge: Alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol. 2008;181:3755–9. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 52.Loots G, Ovcharenko I, Pachter L, Dubchak I, Rubin EM. rVISTA for comparative sequence-based discovery of transcription factor binding sites. Genome. Res. 2002;12:832–9. doi: 10.1101/gr.225502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haugsten EM, Malecki J, Bjørklund SM, Olsnes S, Wesche J. Ubiquitination of fibroblast growth factor receptor1 is required for intracellular sorting but not for endocytosis. Mol Biol Cell. 2008;19:3390–403. doi: 10.1091/mbc.E07-12-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen R, Li M, Zhang Y, Zhou Q, Shu HB. The E3 ubiquitin ligase MARCH8 negatively regulates IL-1β-induced NF-κB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2012;109:14128–33. doi: 10.1073/pnas.1205246109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Morrone G, Zhang J, Chen X, Lu X, Ma L, Moore M, Zhou P. CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. EMBO J. 2003;22:6057–67. doi: 10.1093/emboj/cdg577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohh M, Kim WY, Moslehi JJ, Chen Y, Chau V, Read MA, Kaelin WG., Jr An intact NEDD8 pathway is required for Cullin-dependent ubiquitylation in mammalian cells. EMBO Rep. 2002;3:177–82. doi: 10.1093/embo-reports/kvf028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khanna-Gupta A, Zibello T, Kolla S, Neufeld EJ, Berliner N. CCAAT displacement protein (CDP/cut) recognizes a silencer element within the lactoferrin gene promoter. Blood. 1997;90:2784–95. [PubMed] [Google Scholar]
- 58.Khanna-Gupta A, Zibello T, Sun H, Lekstrom-Himes J, Berliner N. C/EBP epsilon mediates myeloid differentiation and is regulated by the CCAAT displacement protein (CDP/cut) Proc Natl Acad Sci U S A. 2001;98:8000–5. doi: 10.1073/pnas.141229598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu Y, Goldenberg I, Bei L, Andrejic J, Eklund EA. HoxA10 represses gene transcription in undifferentiated myeloid cells by interaction with Histone deacetylase 2. J. Biol. Chem. 2003;278:47792–802. doi: 10.1074/jbc.M305885200. [DOI] [PubMed] [Google Scholar]
- 60.Beekman RI, Touw IP. G-CSF and its receptor in myeloid malignancy. Blood. 2010;115:5131–6. doi: 10.1182/blood-2010-01-234120. [DOI] [PubMed] [Google Scholar]