

Abstract
AIM: To investigate changes in advanced glycation end products (AGEs) and their receptor (RAGE) expression in the gastrointestinal (GI) tract in type 2 diabetic rats.
METHODS: Eight inherited type 2 diabetic rats Goto-Kakizak (GK) and ten age-matched normal rats were used in the study. From 18 wk of age, the body weight and blood glucose were measured every week and 2 wk respectively. When the rats reached 32 wk, two-centimeter segments of esophagus, duodenum, jejunum, ileum, and colon were excised and the wet weight was measured. The segments were fixed in 10% formalin, embedded in paraffin and five micron sections were cut. The layer thickness was measured in Hematoxylin and Eosin-stained slides. AGE [N epsilon-(carboxymethyl) lysine and N epsilon-(carboxyethyl)lysine] and RAGE were detected by immunohistochemistry staining and image analysis was done using Sigmascan Pro 4.0 image analysis software.
RESULTS: The blood glucose concentration (mmol/L) at 18 wk age was highest in the GK group (8.88 ± 1.87 vs 6.90 ± 0.43, P < 0.001), a difference that continued to exist until the end of the experiment. The wet weight per unit length (mg/cm) increased in esophagus, jejunum and colon from the normal to the GK group (60.64 ± 9.96 vs 68.56 ± 11.69, P < 0.05 for esophagus; 87.01 ± 9.35 vs 105.29 ± 15.45, P < 0.01 for jejunum; 91.37 ± 7.25 vs 97.28 ± 10.90, P < 0.05 for colon). Histologically, the layer thickness of the GI tract was higher for esophagus, jejunum and colon in the GK group [full thickness (μm): 575.37 ± 69.22 vs 753.20 ± 150.41, P < 0.01 for esophagus; 813.51 ± 44.44 vs 884.81 ± 45.31, P < 0.05 for jejunum; 467.12 ± 65.92 vs 572.26 ± 93.60, P < 0.05 for colon]. In esophagus, the AGE and RAGE mainly distributed in striated muscle cells and squamous epithelial cells. The AGE distribution was much stronger in the GK group compared to the normal group both in the striated muscle layer and mucosa layer (immuno-positive area/ total measuring area %: 4.52 ± 0.89 vs 10.96 ± 1.34, P < 0.01 for muscle; 8.90 ± 2.62 vs 22.45 ± 1.26, P < 0.01 for mucosa). No visible difference was found for RAGE distribution between the two groups. In the intestine AGE and RAGE distributed in epithelial cells of villi and crypt. RAGE was also found in neurons in the myenteric and submucosal plexus. The intensity of AGE staining in mucosa of all segments and RAGE staining in neurons in all segments were strongest in the diabetes group. Significant difference for AGE was found in the epithelial cells of villi and crypt in duodenum (immuno-positive area/total measuring area %: 13.37 ± 3.51 vs 37.48 ± 8.43, P < 0.05 for villi; 0.38 ± 0.12 vs 1.87 ± 0.53, P < 0.05 for crypt) and for RAGE in neurons of all segments (e.g., for jejunum: no staining neurons% 0 vs 0, mild 36.0 ± 5.2 vs 28.7 ± 3.5, moderate 53.2 ± 4.8 vs 55.8 ± 5.4, strong 10.7 ± 1.1 vs 15.4 ± 2.0, P < 0.05). In the colon, RAGE was primarily found in neurons in the myenteric and submucosal plexus. It was stronger in the diabetes group than in the normal group (no staining neurons% 6.2 ± 0.2 vs 0.3 ± 0.04, mild 14.9 ± 2.1 vs 17.6 ± 1.5, moderate 53.1 ± 4.6 vs 44.7 ± 4.4, strong 25.6 ± 18 vs 43.6 ± 4.0, P < 0.05). In the rectum, RAGE was primarily found in the mucosa epithelial cells.
CONCLUSION: The AGE and RAGE expression was up-regulated in the GI tract of GK diabetic rats and may contribute to GI dysfunction in type 2 diabetic patients.
Keywords: Diabetes mellitus, Gastrointestinal complications, Advanced glycation end products, Receptor of advanced glycation end products
Core tip: Changes in advanced glycation end products (AGEs) and their receptor (RAGE) expression in the gastrointestinal (GI) tract in type 2 diabetic rats were studied. The AGE and RAGE were widely distributed in epithelial cells of all segments as well as in striated muscle cells in the esophagus. RAGE also distributed in neurons in all segments. Up-regulated AGE and RAGE expression was found in the GI tract of GK diabetic rats. The altered AGE and RAGE may be a contributing factor for GI dysfunction in type 2 diabetic patients.
INTRODUCTION
Sensory-motor abnormalities are common in the gastrointestinal (GI) tract in diabetes mellitus patients. Symptoms may arise from the entire GI tract. Common complaints are dysphagia, heartburn, abdominal pain, early satiety, nausea, vomiting, constipation, and diarrhea[1-3]. The pathogenesis of such symptoms in diabetes mellitus is complex, multi-factorial with motor dysfunction, glycemic control, autonomic neuropathy, and psychological factors, and is not well understood[4].
Previous studies demonstrated changes in the morphological and biomechanical properties of the GI tract during diabetes, e.g., the wall thickness and stiffness of GI tract increased[5-7]. The structure or deformation changes may alter the relative positions of the mechanosensitive afferents (zero setting of the mechanosensitive afferents). The changes in stress distribution and wall stiffness likely alter the stress in the vicinity of the mechanosensitive afferents. Consequently, the perception and motility of the intestinal tract will change as well. Therefore, the morphological changes and biomechanical remodeling are likely to affect the function of mechanosensitive afferents in the GI wall and further affect the motor and sensory function. Only sparse information, however, is available about the mechanisms for these changes.
Advanced glycation end products (AGEs) are formed in physiological states and gradually increases with age but AGEs formation is accelerated in diabetes[8]. AGEs can lead to changes in structure and function directly in the target protein. They also can bind to their receptor (RAGE), leading to activation of signaling pathways resulting in serial changes[9-11]. AGEs and RAGE play important roles for diabetic complications in the cardiovascular system[12-14] and for retinopathy[15] and nephropathy[16,17]. It was also demonstrated that AGEs and RAGE were associated with diabetic-induced arterial wall stiffening[18-20]. Therefore, they may also play an important role in the diabetic GI tract. In our previous study we demonstrated that AGE [N epsilon-(carboxymethyl)lysine, CML and N epsilon-(carboxyethyl)lysine, CEL] and RAGE were up-regulated in the small intestine and colon of streptozotocin (STZ)-induced diabetic rats[21]. However, to the best of our knowledge, data on the distribution of AGE and RAGE in the GI tract of type-2 diabetes have never been described.
The aims of this study were to investigate the AGE and RAGE distribution in the GI tract in type-2 diabetic rats and to compare those with normal rats. The data obtained may serve as the basis for further studying AGE and RAGE effects on type 2 GI diabetic dysfunctions.
MATERIALS AND METHODS
Reagents
Anti-AGEs mouse monoclonal antibody (6D12), against N(epsilon)-(carboxymethyl)lysine (CML, a major immunological epitope in AGEs) and N epsilon-(carboxyethyl)lysine (CEL) was purchased from COSMO BIO CO.,LTD. Japan. Other substances were rabbit polyclonal antibody against the N-terminal of human RAGE from Cell Applications, INC, United States; LSAB2 System-HRP for rat specimens, proteinase K, citrate buffer (pH = 6.0, 10xconcentrated), bovine serum albumin (BSA) and Mayer haematoxylin from Dako A/S, Denmark; soluble RAGE from Shanghai Yanji Bio; STZ, ethanol, methanol and xylene from Sigma-Aldrich Denmark A/S, Vallensbæk Strand, Denmark. Blood glucose analyzer and test strips were supplied by Hemocue Corporation, Sweden. The slides and cover glasses we used for immunohistochemical staining were Menzel-Glaser products, Germany.
Animal
Approval of the protocol was obtained from the Danish Committee for Animal Experimentation. Eight inherited type 2 diabetic Goto-Kakizak rats (GK group), 12 wk old and weighting about 330 g, were purchased from Taconic Europe DK-8680 Ry, Denmark. Ten age-matched normal rats (same strain as GK rats) served as controls (Normal group). During the breeding, the rats freely drank tap water and ate food except fasting over night before measuring body weight and blood glucose, which were done every week for body weight and every 2 wk for blood glucose from week 18. The rats survived until 32 wk of age.
Sampling
At the termination of the experiments, the rats were anaesthetized with Hypnorm 0.5 mg and Dormicum 0.25 mg per 100 g body weight (Hypnorm: Dormicum: sterile water = 1:1:2; subcutaneous injection). Two-centimeter segments of esophagus, duodenum, jejunum, ileum, and colon were excized. The esophageal segment was taked from the distal end of esophagus; the duodenal segment from 5 cm distal to pylorus sphincter; the jejunal segment from 5 cm distal to the ligament of Treitz; the ileal segment from 5 cm proximal to the ileo-cecal valve; and the colon from the middle part. Residual contents in the lumen were gently cleared using Krebs solution of the following composition (mmol/L): NaCl, 118; KCl, 4.7; NaHCO3, 25; NaH2PO4, 1.0; MgCl2, 1.2; CaCl2-H2O, 2.5; Glucose, 11; ascorbic acid, 0.11 and the wet weight was measured. Thereafter the rats were killed by injecting an overdose anesthetics.
General histological staining
All samples were fixed in 10% phosphate-buffered formalin about 24 h. The specimen were dehydrated in a series of graded ethanol (70%, 96% and 99%) and embedded in paraffin. Five-micron sections were cut perpendicular to the mucosa surface and the paraffin was cleared from the slides with coconut oil (over 15 min, 60 °C). The sections were rehydrated in 99%, 96% and 70% ethanol followed by a 10 min wash in water and stained with hematoxylin and eosin (HE). The layer thickness was measured by the same pathologist in a blinded review and sixteen determinations were made on each specimen and averaged.
Immunohistochemical staining
Tissue pretreatment: The tissue samples for immunohistochemistry were also fixed in 10% phosphate-buffered formalin about 24 h, embedded in paraffin. Five-micron sections were cut perpendicular to the mucosa surface and placed in a water bath at 40 °C. Thereafter, sections were transferred onto pretreated microscopic slides, which electrostatically attracted formalin fixed tissue and binding them to the slides. After drying the slides completely at room temperature, they were treated in an oven at 37 °C overnight to enhance the attachment of tissue to the slides. The sections were deparaffinized two times in xylene, 15 min per time, and rehydrated in 100%, 95%, 90%, 80%, 70%, 60% and 50% ethanol two times respectively, 3 s per time, followed by rinsing for 10 min and washing in 0.01M PBS (pH 7.4).
AGE: After treatment with H2O2 (3% in ethanol, room temperature, 15 min) and proteinase K (100 μg/mL, 100 μL, 37 °C, 20 min), the sections were incubated with 5% BSA-PBS buffer (room temperature, 30 min) for blocking non-specific staining. Afterwards, the sections were incubated with the primary antibody 6D12 [1:100, diluted in 1% bovine serum albumin-Phosphate buffered saline (BSA-PBS)], which has been thoroughly characterized by Ikedas group[22], or normal mouse IgG (250 μg/mL) pre-treated with excessive CML (1:250, diluted in 1% BSA-PBS, negative control) overnight at 4 °C. The sections were then washed and incubated with LINK (biotinylated anti-rabbit and anti-mouse immunoglobulins) and afterwards with STREPTAVIDIN PEROXIDASE (streptavidin conjugated with horseradish peroxidase) at room temperature for 10 min (both are part of reagents of LSAB2 System-HRP, products of Dako Company, Denmark). Then the peroxidase activity was visualized by incubating the sections in substrate working solution containing hydrogen peroxide and 3,3’-diaminobenzidine tetrahydrochloride at room temperature for 5 min. The sections were rinsed for 10 min, counterstained with Mayer Haematoxylin for 1 min, treated in HCl-ethanol for 3 s, dehydrated in 80%, 90%, 95%, 100% ethanol for 3 s, respectively. Then the slides were immersed in xylene for 15 min two times and mounted.
RAGE: The primary antibody against RAGE was produced in rabbits immunized with a synthetic peptide corresponding to a sequence at the N-terminal of human RAGE. Only two amino acids are different from the related rat sequence. For RAGE immunostaining, instead of treating sections with proteinase K, the sections were boiled in 10 mmol/L citrate buffer (pH = 6.0) 18 min for retrieving antigen and using normal rat lung as positive control as the RAGE is highly expressed in the lung[23]. The primary antibody was diluted (1:60) with 1% BSA-PBS and normal rabbit serum (diluted 1:60) pre-treated with excessive soluble RAGE was used as negative control. Other processes were similar to the AGE immunostaining.
Image analysis
To minimize errors, 6 to 10 photographs were randomly taken of different locations of same layer in each slide. After that, images of the different parts such as villus and crypt were saved as individual image files. The region of interest (ROI) was defined using Sigmascan Pro 4.0 image analysis software (Jandel Scientific, Germany). The color due to 3,3’-diaminobenzidene staining was distinguished in the ROI using intensity thresholds. Finally the images were exported as binary images and the area fraction of AGE or RAGE positive staining was calculated by a MATLAB program (MATLAB 6.5, The MathWorks Inc. United States).
Data analysis
According to the image analysis above, the fraction of AGE in mucosa (villi and crypt were analyzed separately in the intestinal segments), muscle layers, and the fraction of RAGE in the mucosa and muscle layer were computed as: Fraction of AGE or RAGE = immuno-positive area/total measuring area. It was difficult to calculate the fraction of RAGE in neurons in the same way. Therefore, the immunoreactivity of RAGE in each neuron was categorized by the stained intensity, i.e., negative, mild, moderate and strong[24].
Statistically analysis
The results were expressed as mean ± SD unless indicated in the text. The differences between the diabetes and normal groups were tested using Student’s t test and Anova. The results were regarded as significant when P < 0.05.
RESULTS
General information
The body weight and blood glucose level of GK group were significantly higher than those of the Normal group during the whole experimental period (Figure 1A and 1B, P < 0.001 and P < 0.01, respectively).
Figure 1.
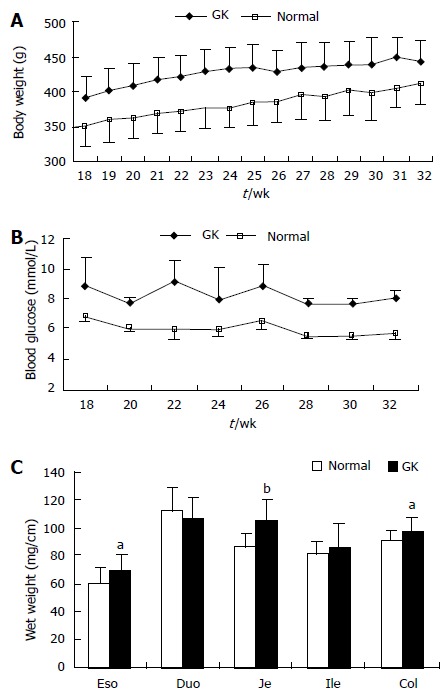
Body weight (A) and the blood glucose level (B) were higher in Goto-Kakizak group than in the normal group (P < 0.001 and P < 0.01). The wet weight per unit length of intestinal and colon segments is shown in Figure 1C (compared with normal group: aP < 0.05, bP < 0.01). Values are mean ± SD, n = 8 for each group. Eso: Esophagus; Duo: Duodenum; Je: Jejunum; Ile: Ileum; Col: Colon; GK: Inherited type 2 diabetic Goto-Kakizak rats.
The wet weights per unit length of esophagus, jejunum and colon segments were highest in the GK group (Figure 1C, P < 0.05 and P < 0.01, respectively). No significant difference were found for duodenum and ileum between the two groups (Figure 1C, P > 0.05).
General histological changes
Compared with the Normal group, the full wall thickness of esophagus, jejunum and colon remarkably increased in the GK group (Figure 2A, P < 0.05 and P < 0.01, respectively). No significant difference was found in duodenum and ileum between two groups. The smooth muscle thickness of esophagus and colon (both circumferential and longitudinal smooth muscle) increased remarkably in GK group. The villous height of jejunum increased in the GK group (Figure 2B-D, P < 0.05 and P < 0.01). No significant difference was found for other layers.
Figure 2.
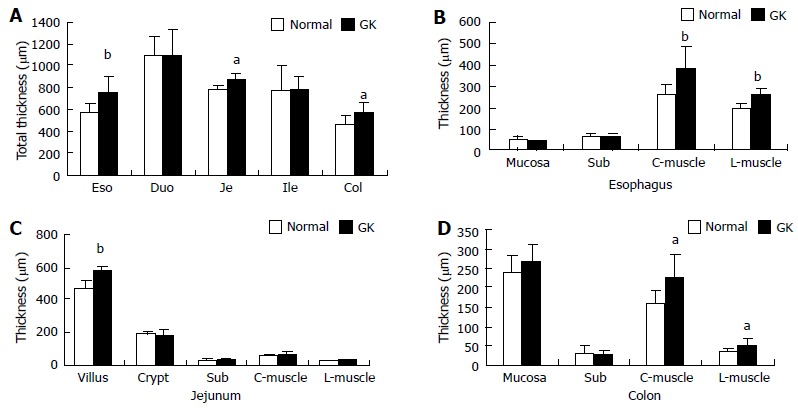
The wall and layer thickness. A: Total wall thickness; B: Layer thickness of esophagus; C: Layer thickness of jejunum; D: Layer thickness of colon. Values are mean ± SD, n = 8 for each group (compared with normal group: aP < 0.05, bP < 0.01). Eso: Esophagus; Duo: Duodenum; Je: Jejunum; Ile: Ileum; Col: Colon; Sub: Submucosa; C-muscle: Circumferential smooth muscle; L-muscle: Longitudinal smooth muscle; GK: Inherited type 2 diabetic Goto-Kakizak rats.
Distribution of AGE
The immune-positive area of AGE was yellow-brown (Figure 3A and B). These colors were not found in the negative control slides (without primary antibody), demonstrating that the stained color was specific for AGE.
Figure 3.
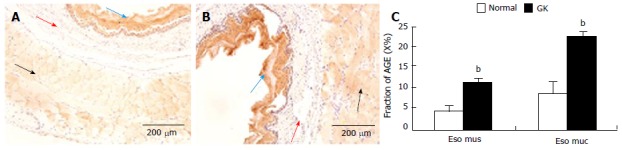
Example of advanced glycation end products immune-staining in esophagus (A, normal; B, diabetic); C: Shows the statistic result of immune-staining intensity in esophagus muscle and mucosa. Values are mean ± SD, n = 8 for each group (compared with normal group: bP < 0.01). The immune-positive area of AGE showed yellow-brown color. Eso mus: Esophageal muscle; Eso muc: Esophageal mucosa; GK: Inherited type 2 diabetic Goto-Kakizak rats; AGE: Advanced glycation end product. Blue arrow: mucosa; Red arrows: Submucosa; Black arrows: Muscle.
In the esophagus, AGE distribution was inhomogeneous and mainly distributed in striated muscle and squamous epithelial cells. Compared with normal group, the intensity of immune-staining for AGE was much stronger in the GK group (Figure 3C, P < 0.01). No visible stained color was found in submucosa layer (Figure 3A and B).
In the intestine, AGE was mainly distributed in the mucosa layer, especially in epithelial cells of villi (Figure 4A-4E) and crypt. No visible stained color was found in submucosa, smooth muscle and ganglia. The crypt epithelial cells in ileum and colon were slightly stained. The distribution of AGE in the epithelial cells was inhomogeneous, the surface part was much stronger than the bottom part in villous epithelial cells but it showed an opposite pattern in the crypt epithelial cells. The intensity of AGE staining of the epithelial cells in villi was stronger than that in crypts (P < 0.01). In the mucosa, the intensity of AGE staining was similar between duodenal and jejunal segments (Figure 4A-C, F, G) but they were stronger than those in colon (Figure 4A, 4C, 4E and 4F). The mucosa of ileum showed the weakest intensity of AGE staining among different intestinal segments (Figure 4D). Compared with the Normal group, the intensity of AGE staining in mucosa of all segments were stronger in the GK group (Figure 4F and G). Significant difference was found in the epithelial cells of villi and crypt in duodenum (Figure 4F and G, P < 0.05).
Figure 4.
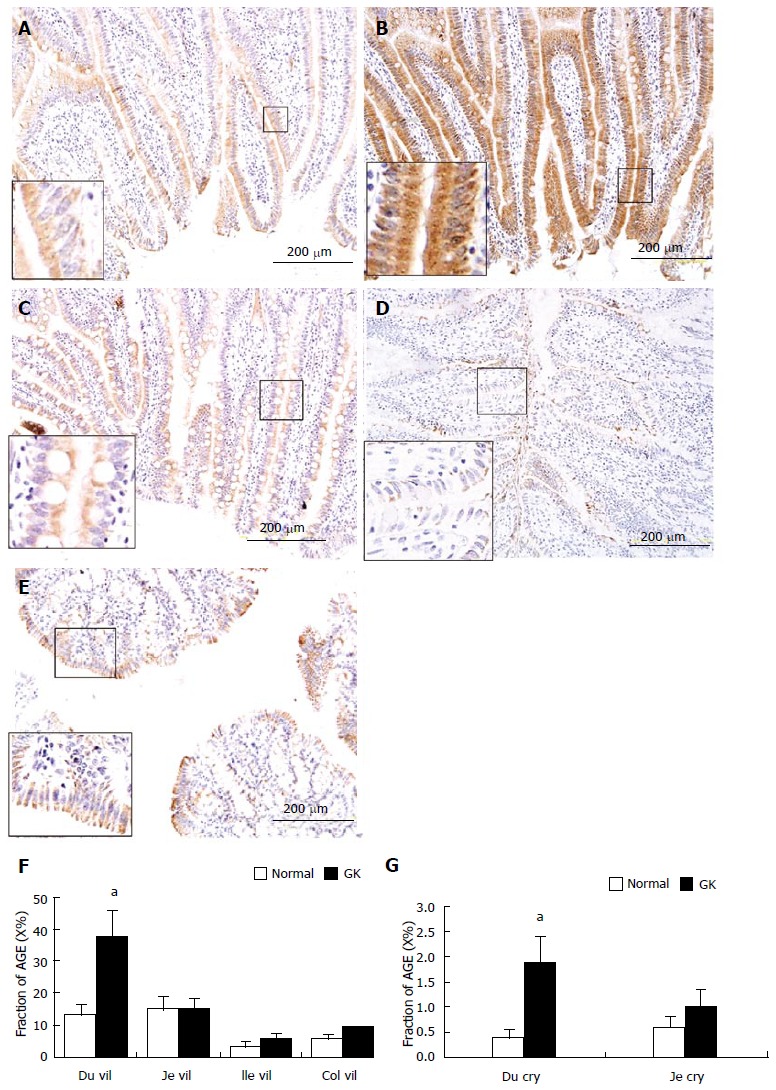
Example of advanced glycation end products immune-staining in villi of duodenum (A, normal; B, diabetic), jejunum (C), ileum (D) and mucosa in colon (E). The immune-positive area of AGE showed yellow-brown color; 4F and 4G: Show the statistical result of immune-staining intensity in villous epithelial cells of duodenum, jejunum, ileum and colon as well as in crypt epithelial cells of duodenum and jejunum. As shown in the magnification area (big frame vs small frame), the AGE distribution in epithelial cells was inhomogeneous, the surface part was much stronger than bottom part. Values are mean ± SD, n = for each group (compared with normal group: aP < 0.05). Du vil: Duodenum villi; Je vil: Jejunum villi; Ile vil: Ileum villi; Col mu: Colon mucosa; Du cry: Duodenum crypt; GK: Inherited type 2 diabetic Goto-Kakizak rats; AGE: Advanced glycation end products.
Distribution of RAGE
The immune-positive area of RAGE also showed yellow-brown color (Figure 5) that was not found in the negative control slides (without primary antibody). Therefore, the stained color was specific for RAGE.
Figure 5.
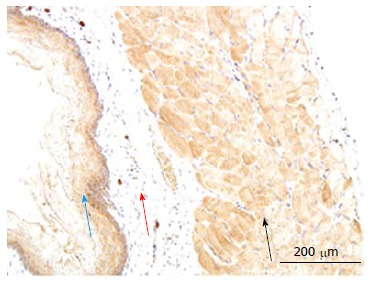
Example of receptor of advanced glycation end products immune-staining in normal esophagus. The immune-positive area of RAGE showed yellow-brown color. RAGE: Receptor of advanced glycation end products. Blue arrow: mucosa; Red arrows: Submucosa; Black arrows: Muscle.
In esophagus, the immune-positive staining for RAGE was mainly observed in the striated muscle cells and mucosa squamous epithelial cells. The RAGE distribution was inhomogeneous in the striated muscle layer and graduated decreased from bottom to surface. No visible stained color was found in the submucosa layer (Figure 5). The intensity of RAGE staining did not differ between Normal and GK group both in the striated muscle cells and mucosa squamous epithelial cells (P > 0.05).
In the small intestine, the immune-positive staining for RAGE was observed in the epithelial cells of villi (Figure 6A, C, E) and crypts (Figure 6B, D, F), and in neurons in the myenteric and submucosal plexus (Figure 6G). The RAGE was homogeneously distributed in the cells, as shown in villus and crypt epithelial cells, but the intensity of immune-staining was much stronger in villous epithelial cells than in crypt epithelial cells. The strongest staining color occurred in duodenum and the weakest in ileum among the three segments (duodenum > jejunum > ileum, Figure 6A-F). In neurons, RAGE distributed both in cytoplasm and cell membrane (Figure 6G). Compared with the Normal group, the intensity of immune-staining for RAGE increased in the neurons for the three segments (Figure 6G and H) in the GK group (P < 0.05), but no significant difference was found in other cells between two groups.
Figure 6.
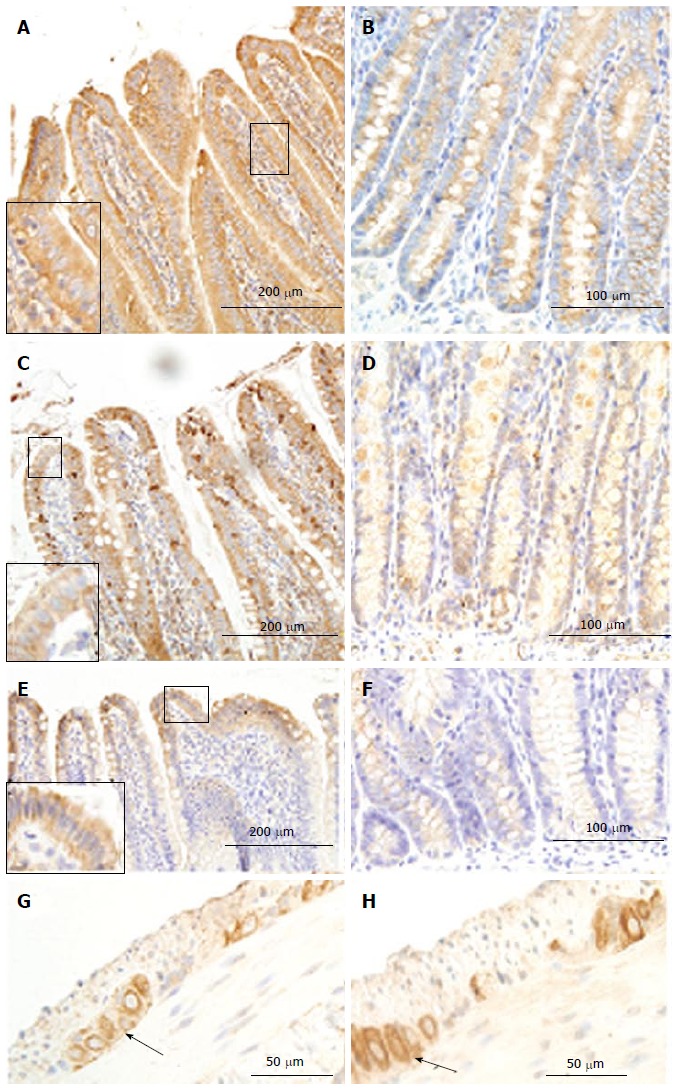
Receptor of advanced glycation end products immune-staining in villi (A, C, E) and crypt (B, D, F) of duodenum (A, B), jejunum (C, D) and ileum(E, F) as well as in ileum ganglia (arrowhead; G: Normal group; H: GK group). As shown in the magnification area (big frame vs small frame), the RAGE homogeneously distributed in the epithelia cells. The intensity of immune-staining in ganglia was stronger in the diabetic group (H) than in the normal group (G) (arrowhead). RAGE: Receptor of advanced glycation end products; GK: Inherited type 2 diabetic Goto-Kakizak rats.
In the colon, the immune-positive staining for RAGE was observed in neurons of the myenteric (Figure 7A) and submucosal plexus. It was stronger in the GK group than in Normal group (P < 0.05) (Figure 7A and B). Furthermore, a mild positive staining was also observed in mucosa epithelial cells both in the GK and Normal groups (Figure 7C).
Figure 7.
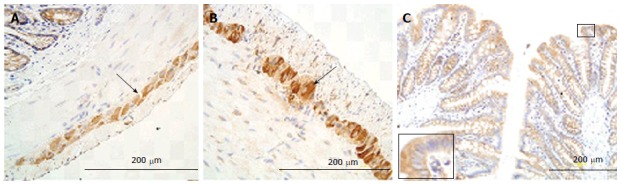
Receptor of advanced glycation end products immune-staining in colon ganglia (arrow; A: Normal; B: Diabetic) and mucosa (C). The intensity of immune-staining in ganglia was stronger in the GK group (B) than in the normal group (A) (arrow). Big frame area vs small frame area. RAGE: Receptor of advanced glycation end products; GK: Inherited type 2 diabetic Goto-Kakizak rats.
DISCUSSION
Our previous study showed that the expression of AGE and RAGE was up-regulated in the small intestine and colon of STZ-induced type 1 diabetic rats[21]. STZ rats have high blood glucose but formation of free radicals and STZ cytotoxicity plus its direct effects on AGE formation and RAGE expression in GI tract may be questioned. Using the present model, the confounding effect of STZ can be avoided. The major discovery was that the intensity of AGE immune-staining was significantly increased in striated muscle and mucosa layer of esophagus, and in epithelial cells located in intestinal villi and crypts in the GK group compared to normal rats. RAGE was significantly increased in myenteric and submucosa plexus neurons of all intestinal segments in the GK group.
The distribution of AGE and RAGE in normal GI tract
Ling et al[25] reported the existence of four kinds of AGEs in stomach and small intestinal epithelial cells in normal rats. Our previous study[21] showed homogenous AGE distribution in the cytoplasm of smooth muscle cells, epithelial cells, and neurons of the myenteric and submucosal plexus in the layers of colon and small intestine. Furthermore, homogeneous distribution of RAGE was found in epithelial cells and neurons. The present study confirmed the distribution of both compounds in the colon and small intestine as reported from our previous study. Furthermore, we also found that the AGE and RAGE distributed in the striated muscle and squamous epithelial cells of esophagus and also in the stomach (unpublished data). The present study together with our previous study[21] is the first reports of the localization of AGE and RAGE in the whole rat GI tract. This provides a basis for further comparison study of the distribution of AGE and RAGE on GI tract with diseases, such as diabetes.
AGE and RAGE changes in GI tract of GK diabetic rats
RAGE and AGE distribution in the GI tract of GK rats was similar to that in normal controls. However, compared with the normal controls, the level of AGE and RAGE at some GI locations was increased in GK rats. However, compared with our previous study[21], the intensity of AGE and RAGE immune-staining were not so strong in the present study. It is well known that the accumulation and production of AGEs and expression of RAGE are associated with blood glucose level[26]. The blood glucose level is much lower in GK diabetic rats compared to STZ-induced diabetic rats. This is one plausible explanation for the weaker increasing AGE and RAGE in the GK type-2 diabetic rat model.
Histomorphological and biomechanical GI remodeling occurred during the development of diabetes[27]. For example the esophagus and colon were morphologically and biomechanically remodeled during the development of diabetes[6,7]. Abnormal levels of AGE and RAGE found in the present study may be associated with the GI remodeling in the GK diabetic rats. In the present study we found that the mucosa of small intestine and muscle layer of all segments proliferated, accordingly the AGE expression is up-regulated in the mucosa in all segments and esophageal muscle layer in the GK diabetic rats. However, the intensity of immune-staining in muscle had no apparent increase in muscle layer of intestine in GK group despite the fact that the muscle layer showed hyperplasia. Therefore it is speculated that in addition to the affection of AGE, other factors may affect the hyperplasia of GI muscle in GK diabetic rats, such as glucagon-like peptide 2[28].
In the present experiment no direct evidence showed how tissue growth was affected by AGEs and RAGE. From studies in other organs, it is known that AGE through AGE-RAGE-mediated ROS generation activating angiotensin II-tissue growth factor beta (TGF-β)-Smad signaling can increase renal interstitial fibroblasts mitogenesis and type I collagen production[29], mesangial cell hypertrophy and fibronectin synthesis[9], through AGE-RAGE interaction can cause epithelial myofibroblast transdifferentiation[30] and vascular smooth muscle proliferation[31], and through galectin-3 induce smooth muscle proliferation[32]. Therefore, it is feasible that GI tissue proliferation at least in part may be induced by AGE accumulation through the same pathways. Further studies must explore mechanisms for AGE- and RAGE-induced GI tissue growth and the association with the biomechanical remodelling in diabetes.
AGE and RAGE accumulation impact on GI dysfunction
It is well documented that small intestinal epithelial cells are important for digestion and absorption. Many kinds of enzymes located in the enterocytes lining the intestinal villi brush border are involved in digestion[33]. Furthermore, the small intestinal mucosa is important for absorptive function[33]. We demonstrated a stronger intensity of AGE immune-staining in the epithelial cells of intestinal crypts and villi in the diabetes group compared to the normal group. Digestive enzyme activity and cell membrane properties may potentially be affected by AGE accumulation. It is also known that non-enzymatic glycation and oxidative stress are important for changes of brush border membrane fluidity[34]. Digestion and intestinal transport processes occur at the brush border membrane. Changes in fluidity as well as in the membrane composition can alter the enzyme activity in the villi brush border membrane[35,36]. Multiple cellular signaling cascades can be activated by binding of AGEs to RAGE[37]. The increased AGE linking with RAGE may change epithelial cell function. Mechanisms linking AGEs/RAGE compounds to intestinal mucosa function in diabetics need more work.
Numerous studies demonstrated abnormal GI motility in diabetics [27,38]. Diabetic autonomic neuropathy is considered important in the pathogenesis of sensory-motor disordered function in diabetic patients[39-41]. AGEs and RAGE likely are key players in development of diabetic neuropathy[23]. Synergistic action of AGEs and endogenous nitric oxide can lead to neuronal apoptosis in vitro[42]. The neuronal AGE formation and accumulation may account for the development of GI neuropathy, primarily as a direct effect on structural and functional proteins, alternatively by activating RAGE indirectly[43]. GI nerves express nitric oxide synthase (nNOS), which generates a key transmitter nitric oxide in the regulation of GI motility[44]. Korenagas group demonstrated that AGEs inhibit via RAGE nNOS expression in vitro[45]. The expression and function of neuronal nitric oxide synthase decreased in the stomach of spontaneously diabetic BB-rats[41] and also decreased in duodenum of STZ-induced diabetic rats, which can be prevented by aminoguanidine (a drug that prevents AGE formation) and ALT-711 (AGE cross-link breaker)[8]. In the present study direct evidence was provided that RAGE is localized in myenteric and submucosal plexus neurons in the esophagus and intestine. In addition, DM enhanced RAGE intensity and therefore, AGE-RAGE interaction is likely of importance for GI diabetic autonomic neuropathy. We also demonstrated that the intensity muscle tissue AGE immune-staining was strongest in the diabetic esophagus. The accumulation of AGEs in muscle may alter the architecture and contractile proteins of smooth muscle[46], resulting in the alteration of muscle contraction propertiess.
AGE was mainly distributed in striated muscle and squamous epithelial cells in esophagus; in small intestinal epithelial cells of crypt and villi and in epithelial cells in colon and rectum. RAGE was mainly distributed in striated muscle and squamous epithelial in esophagus; in epithelial cells in intestine mucosa and neurons in ganglia. High AGE density was found in striated muscle and mucosa layers in esophagus, and villus, crypt in the GK rat small intestine, and the expression of RAGE in the intestine increased in ganglia of GK rats. Increased expression of AGE and RAGE likely contributes to GI disorders associated with DM.
ACKNOWLEDGMENTS
Technicians Torben Madsen, Ole Sørensen, and Jens Sørensen are thanked for handling the animals.
COMMENTS
Background
In this previous study the authors demonstrated that advanced glycation end products (AGEs) and their receptor (RAGE) were up-regulated in the small intestine and colon of streptozotocin-induced diabetic rats. However, to the best of our knowledge, data on the distribution of AGE and RAGE in the gastrointestinal (GI) tract of type 2 diabetes have never been described.
Research frontiers
Previous studies demonstrated the morphological and biomechanical properties of the GI tract were remodeled during diabetes. However the mechanisms for these changes are not well understood. Therefore, investigation on the distribution of AGE and RAGE in the GI tract of type-2 diabetes is important for understanding the mechanism of GI remodeling in diabetes.
Innovations and breakthroughs
At present study the authors demonstrated that the AGE and RAGE expression was up-regulated in the GI tract of GK diabetic rats. The increased AGE and RAGE levels may contribute to diabetic GI dysfunction in type 2 diabetic patients.
Applications
The most common type diabetes is type 2 diabetes; therefore it is important to understand the expression of AGE and RAGE in the GI tissues in type 2 diabetes. Knowing the over-expression of AGE and RAGE in the diabetic GI tract may in somehow direct the treatment in the patients suffering from type 2 diabetes.
Terminology
AGEs are formed by non-enzymatic attachment of sugars to the amino groups of various proteins through a series of complex intermediary reactions. Diabetic hyperglycemia accelerates the accumulation of AGEs in the tissues. RAGE is a 55kD transmembrane receptor of the immunoglobulin super family, which binds AGEs. AGEs contribute to diabetic complications through receptor-dependent and -independent pathways.
Peer-review
This is an interesting study. In future, more data should provide to further demonstrate the relationship between increasing AGE and morphological or functional changes in diabetes.
Footnotes
Supported by Karen Elise Jensens Foundation.
Ethics approval: Ethics of the study was approved by the Danish Committee for Animal Experimentation. The license number is 2008/561-1530.
Institutional animal care and use committee: All procedures involving animals were reviewed and approved by the Danish Committee for Animal Experimentation. The license number is 2008/561-1530. Animals in poor clinical condition were euthanized and excluded from the study. The rats were euthanized with CO2 inspiration during the anesthesia. The animals were acclimatized to laboratory conditions (22 °C, 12 h/12 h light/dark, 50% humidity, ad libitum access to food and water) for 2 wk prior to experimentation. The animal protocol was designed to minimize pain or discomfort to the animals.
Conflict-of-interest: We declare that we have no proprietary, financial, professional or other personal interest of any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitle “Advanced Glycation End-Product expression is upregulated in the Gastrointestinal Tract of Type 2 Diabetic Rats”.
Data sharing: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 7, 2014
First decision: October 28, 2014
Article in press: March 18, 2015
P- Reviewer: Nishio K, Tamemoto H S- Editor: Tian YL L- Editor: A E- Editor: Wu HL
References
- 1.Mjörnheim AC, Finizia C, Blohmé G, Attvall S, Lundell L, Ruth M. Gastrointestinal symptoms in type 1 diabetic patients, as compared to a general population. A questionnaire-based study. Digestion. 2003;68:102–108. doi: 10.1159/000074523. [DOI] [PubMed] [Google Scholar]
- 2.Oh JH, Choi MG, Kang MI, Lee KM, Kim JI, Kim BW, Lee DS, Kim SS, Choi H, Han SW, et al. The prevalence of gastrointestinal symptoms in patients with non-insulin dependent diabetes mellitus. Korean J Intern Med. 2009;24:309–317. doi: 10.3904/kjim.2009.24.4.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim JH, Park HS, Ko SY, Hong SN, Sung IK, Shim CS, Song KH, Kim DL, Kim SK, Oh J. Diabetic factors associated with gastrointestinal symptoms in patients with type 2 diabetes. World J Gastroenterol. 2010;16:1782–1787. doi: 10.3748/wjg.v16.i14.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horowitz M, Samsom M. Gastrointestinal function in diabetes mellitus. Chichester, England: John Wiley & Sons, Ltd; 2004. pp. 1–349. [Google Scholar]
- 5.Zhao J, Yang J, Gregersen H. Biomechanical and morphometric intestinal remodelling during experimental diabetes in rats. Diabetologia. 2003;46:1688–1697. doi: 10.1007/s00125-003-1233-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhao J, Liao D, Gregersen H. Biomechanical and histomorphometric esophageal remodeling in type 2 diabetic GK rats. J Diabetes Complications. 2007;21:34–40. doi: 10.1016/j.jdiacomp.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Zhao J, Nakaguchi T, Gregersen H. Biomechanical and histomorphometric colon remodelling in STZ-induced diabetic rats. Dig Dis Sci. 2009;54:1636–1642. doi: 10.1007/s10620-008-0540-3. [DOI] [PubMed] [Google Scholar]
- 8.Jeyabal PV, Kumar R, Gangula PR, Micci MA, Pasricha PJ. Inhibitors of advanced glycation end-products prevent loss of enteric neuronal nitric oxide synthase in diabetic rats. Neurogastroenterol Motil. 2008;20:253–261. doi: 10.1111/j.1365-2982.2007.01018.x. [DOI] [PubMed] [Google Scholar]
- 9.Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, Motomiya Y, Bucala R, Iida S, Tamaki K, et al. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004;66:2137–2147. doi: 10.1111/j.1523-1755.2004.66004.x. [DOI] [PubMed] [Google Scholar]
- 10.Sajithlal G, Huttunen H, Rauvala H, Munch G. Receptor for advanced glycation end products plays a more important role in cellular survival than in neurite outgrowth during retinoic acid-induced differentiation of neuroblastoma cells. J Biol Chem. 2002;277:6888–6897. doi: 10.1074/jbc.M107627200. [DOI] [PubMed] [Google Scholar]
- 11.Reber F, Geffarth R, Kasper M, Reichenbach A, Schleicher ED, Siegner A, Funk RH. Graded sensitiveness of the various retinal neuron populations on the glyoxal-mediated formation of advanced glycation end products and ways of protection. Graefes Arch Clin Exp Ophthalmol. 2003;241:213–225. doi: 10.1007/s00417-002-0528-1. [DOI] [PubMed] [Google Scholar]
- 12.Grossin N, Wautier MP, Wautier JL. Red blood cell adhesion in diabetes mellitus is mediated by advanced glycation end product receptor and is modulated by nitric oxide. Biorheology. 2009;46:63–72. doi: 10.3233/BIR-2009-0519. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen JM, Kristiansen SB, Nørregaard R, Andersen CL, Denner L, Nielsen TT, Flyvbjerg A, Bøtker HE. Blockage of receptor for advanced glycation end products prevents development of cardiac dysfunction in db/db type 2 diabetic mice. Eur J Heart Fail. 2009;11:638–647. doi: 10.1093/eurjhf/hfp070. [DOI] [PubMed] [Google Scholar]
- 14.Tekabe Y, Luma J, Einstein AJ, Sedlar M, Li Q, Schmidt AM, Johnson LL. A novel monoclonal antibody for RAGE-directed imaging identifies accelerated atherosclerosis in diabetes. J Nucl Med. 2010;51:92–97. doi: 10.2967/jnumed.109.064659. [DOI] [PubMed] [Google Scholar]
- 15.Sugiyama T, Okuno T, Fukuhara M, Oku H, Ikeda T, Obayashi H, Ohta M, Fukui M, Hasegawa G, Nakamura N. Angiotensin II receptor blocker inhibits abnormal accumulation of advanced glycation end products and retinal damage in a rat model of type 2 diabetes. Exp Eye Res. 2007;85:406–412. doi: 10.1016/j.exer.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Fukami K, Yamagishi S, Ueda S, Okuda S. Role of AGEs in diabetic nephropathy. Curr Pharm Des. 2008;14:946–952. doi: 10.2174/138161208784139710. [DOI] [PubMed] [Google Scholar]
- 17.Matsui T, Nishino Y, Maeda S, Takeuchi M, Yamagishi S. Irbesartan inhibits advanced glycation end product (AGE)-induced up-regulation of vascular cell adhesion molecule-1 (VCAM-1) mRNA levels in glomerular endothelial cells. Microvasc Res. 2011;81:269–273. doi: 10.1016/j.mvr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Monnier VM, Glomb M, Elgawish A, Sell DR. The mechanism of collagen cross-linking in diabetes: a puzzle nearing resolution. Diabetes. 1996;45 Suppl 3:S67–S72. doi: 10.2337/diab.45.3.s67. [DOI] [PubMed] [Google Scholar]
- 19.Sims TJ, Rasmussen LM, Oxlund H, Bailey AJ. The role of glycation cross-links in diabetic vascular stiffening. Diabetologia. 1996;39:946–951. doi: 10.1007/BF00403914. [DOI] [PubMed] [Google Scholar]
- 20.Wolffenbuttel BH, Boulanger CM, Crijns FR, Huijberts MS, Poitevin P, Swennen GN, Vasan S, Egan JJ, Ulrich P, Cerami A, et al. Breakers of advanced glycation end products restore large artery properties in experimental diabetes. Proc Natl Acad Sci USA. 1998;95:4630–4634. doi: 10.1073/pnas.95.8.4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen P, Zhao J, Gregersen H. Up-regulated expression of advanced glycation end-products and their receptor in the small intestine and colon of diabetic rats. Dig Dis Sci. 2012;57:48–57. doi: 10.1007/s10620-011-1951-0. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda K, Higashi T, Sano H, Jinnouchi Y, Yoshida M, Araki T, Ueda S, Horiuchi S. N (epsilon)-(carboxymethyl)lysine protein adduct is a major immunological epitope in proteins modified with advanced glycation end products of the Maillard reaction. Biochemistry. 1996;35:8075–8083. doi: 10.1021/bi9530550. [DOI] [PubMed] [Google Scholar]
- 23.Wada R, Yagihashi S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann N Y Acad Sci. 2005;1043:598–604. doi: 10.1196/annals.1338.067. [DOI] [PubMed] [Google Scholar]
- 24.Nazratun N, Mahmood AA, Kuppusamy UR, Ahmad TS, Tan SY. Diabetes mellitus exacerbates advanced glycation end product accumulation in the veins of end-stage renal failure patients. Vasc Med. 2006;11:245–250. doi: 10.1177/1358863x06072202. [DOI] [PubMed] [Google Scholar]
- 25.Ling X, Nagai R, Sakashita N, Takeya M, Horiuchi S, Takahashi K. Immunohistochemical distribution and quantitative biochemical detection of advanced glycation end products in fetal to adult rats and in rats with streptozotocin-induced diabetes. Lab Invest. 2001;81:845–861. doi: 10.1038/labinvest.3780294. [DOI] [PubMed] [Google Scholar]
- 26.Hegab Z, Gibbons S, Neyses L, Mamas MA. Role of advanced glycation end products in cardiovascular disease. World J Cardiol. 2012;4:90–102. doi: 10.4330/wjc.v4.i4.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Frøkjaer JB, Drewes AM, Ejskjaer N. Upper gastrointestinal sensory-motor dysfunction in diabetes mellitus. World J Gastroenterol. 2006;12:2846–2857. doi: 10.3748/wjg.v12.i18.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer KD, Dhanvantari S, Drucker DJ, Brubaker PL. Intestinal growth is associated with elevated levels of glucagon-like peptide 2 in diabetic rats. Am J Physiol. 1997;273:E815–E820. doi: 10.1152/ajpendo.1997.273.4.E815. [DOI] [PubMed] [Google Scholar]
- 29.Lee CI, Guh JY, Chen HC, Hung WC, Yang YL, Chuang LY. Advanced glycation end-product-induced mitogenesis and collagen production are dependent on angiotensin II and connective tissue growth factor in NRK-49F cells. J Cell Biochem. 2005;95:281–292. doi: 10.1002/jcb.20380. [DOI] [PubMed] [Google Scholar]
- 30.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang R, Kudo M, Yokoyama M, Asano G. Roles of advanced glycation endproducts (AGE) and receptor for AGE on vascular smooth muscle cell growth. J Nippon Med Sch. 2001;68:472–481. doi: 10.1272/jnms.68.472. [DOI] [PubMed] [Google Scholar]
- 32.Seki N, Hashimoto N, Sano H, Horiuchi S, Yagui K, Makino H, Saito Y. Mechanisms involved in the stimulatory effect of advanced glycation end products on growth of rat aortic smooth muscle cells. Metabolism. 2003;52:1558–1563. doi: 10.1016/j.metabol.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 33.Guyton AC, Hall JE. Textbook of medical physiology. 10th ed. USA: W.B. Saunders Company; 2000. pp. 754–763. [Google Scholar]
- 34.Bhor VM, Sivakami S. Regional variations in intestinal brush border membrane fluidity and function during diabetes and the role of oxidative stress and non-enzymatic glycation. Mol Cell Biochem. 2003;252:125–132. doi: 10.1023/a:1025599126840. [DOI] [PubMed] [Google Scholar]
- 35.Olsen WA, Korsmo H. The intestinal brush border membrane in diabetes. Studies of sucrase-isomaltase metabolism in rats with streptozotocin diabetes. J Clin Invest. 1977;60:181–188. doi: 10.1172/JCI108755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keelan M, Walker K, Thomson AB. Intestinal brush border membrane marker enzymes, lipid composition and villus morphology: effect of fasting and diabetes mellitus in rats. Comp Biochem Physiol A Comp Physiol. 1985;82:83–89. doi: 10.1016/0300-9629(85)90708-x. [DOI] [PubMed] [Google Scholar]
- 37.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 2005;83:876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 38.Yamada K, Hosokawa M, Fujimoto S, Nagashima K, Fukuda K, Fujiwara H, Ogawa E, Fujita Y, Ueda N, Matsuyama F, et al. The spontaneously diabetic Torii rat with gastroenteropathy. Diabetes Res Clin Pract. 2007;75:127–134. doi: 10.1016/j.diabres.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 39.Phillips LK, Rayner CK, Jones KL, Horowitz M. An update on autonomic neuropathy affecting the gastrointestinal tract. Curr Diab Rep. 2006;6:417–423. doi: 10.1007/s11892-006-0073-0. [DOI] [PubMed] [Google Scholar]
- 40.Iwasaki H, Kajimura M, Osawa S, Kanaoka S, Furuta T, Ikuma M, Hishida A. A deficiency of gastric interstitial cells of Cajal accompanied by decreased expression of neuronal nitric oxide synthase and substance P in patients with type 2 diabetes mellitus. J Gastroenterol. 2006;41:1076–1087. doi: 10.1007/s00535-006-1909-8. [DOI] [PubMed] [Google Scholar]
- 41.Zandecki M, Vanden Berghe P, Depoortere I, Geboes K, Peeters T, Janssens J, Tack J. Characterization of myenteric neuropathy in the jejunum of spontaneously diabetic BB-rats. Neurogastroenterol Motil. 2008;20:818–828. doi: 10.1111/j.1365-2982.2008.01091.x. [DOI] [PubMed] [Google Scholar]
- 42.Cellek S, Qu W, Schmidt AM, Moncada S. Synergistic action of advanced glycation end products and endogenous nitric oxide leads to neuronal apoptosis in vitro: a new insight into selective nitrergic neuropathy in diabetes. Diabetologia. 2004;47:331–339. doi: 10.1007/s00125-003-1298-y. [DOI] [PubMed] [Google Scholar]
- 43.Toth C, Martinez J, Zochodne DW. RAGE, diabetes, and the nervous system. Curr Mol Med. 2007;7:766–776. doi: 10.2174/156652407783220705. [DOI] [PubMed] [Google Scholar]
- 44.Russo A, Fraser R, Adachi K, Horowitz M, Boeckxstaens G. Evidence that nitric oxide mechanisms regulate small intestinal motility in humans. Gut. 1999;44:72–76. doi: 10.1136/gut.44.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korenaga K, Micci MA, Taglialatela G, Pasricha PJ. Suppression of nNOS expression in rat enteric neurones by the receptor for advanced glycation end-products. Neurogastroenterol Motil. 2006;18:392–400. doi: 10.1111/j.1365-2982.2006.00774.x. [DOI] [PubMed] [Google Scholar]
- 46.Orasanu G, Plutzky J. The pathologic continuum of diabetic vascular disease. J Am Coll Cardiol. 2009;53:S35–S42. doi: 10.1016/j.jacc.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]