

Abstract
Iron-deficient anemia is a prevalent disease among humans. We searched for genes regulated by iron deficiency and its regulated mechanism. cDNA microarrays were performed using Hepa1c1c7 cells treated with 100 μM desferrioxamine (DFO), an iron chelator. Early growth response 1 (Egr1) was upregulated with at least 20-fold increase within 4 h and lasted for 24 h, which was confirmed by qRT-PCR. This activation was not seen by ferric ammonium citrate (FAC). DFO increased the transcriptional activity of Egr1-luc (−604 to +160) and serum response element (SRE)-luc reporters by 2.7-folds. In addition, cycloheximide lowered DFO-induced Egr1 mRNA levels. The upregulation of Egr1 by DFO was accompanied by sustained ERK signals along with phosphorylation of Elk-1. The ERK inhibitor (PD98059) prevented the DFO-induced Egr1 mRNAs. Overexpression of Elk-1 mutant (pElk-1S383A) decreased Egr1 reporter activity. DFO lowered reactive oxygen species (ROS) production and increased caspase 3/7 activity and cell death. DFO-induced iron deficiency upregulates Egr1 in part through transcriptional activation via ERK and Elk-1 signals, which may be important in the regulation of cell death in hepatoma cells. Our study demonstrated that iron depletion controlled the expression of Egr1, which might contribute to decisions about cellular fate in response to iron deficiency.
Electronic supplementary material
The online version of this article (doi:10.1007/s12263-015-0468-0) contains supplementary material, which is available to authorized users.
Keywords: Iron, Deficiency, Egr1, Desferrioxamine, ERK
Introduction
Iron is essential for a range of biological processes including DNA synthesis. Iron is a cofactor for ribonucleotide reductase, oxygen transport, and heme and nonheme enzymatic processes, and is a key component of iron–sulfur proteins and heme proteins used in aerobic respiration (Le and Richardson 2002). Iron deficiency causes anemia in many people worldwide and is the most common micronutrient deficiency disorder (Organization 2008). Although iron is an essential nutrient, excess iron acts as a catalyst in the Fenton reaction to generate free radical species that damage cells. Thus, iron needs to be tightly regulated. The liver has a central role in systemic iron regulation by secreting hepcidin, an iron hormone that acts at the intestine to regulate dietary iron absorption by degrading the iron exporter ferroportin (Ganz and Nemeth 2006, 2012). Physiological factors such as erythropoiesis, hypoxia, inflammation, and hepatic storage affect control of the absorption of dietary iron in the small intestine (Hentze et al. 2004).
Iron deficiency causes increased iron uptake into cells by increasing the expression of the transferrin receptor (TfR) on the plasma membrane. Iron in a complex with transferrin is bound by the TfR and internalized via receptor-mediated endocytosis (Wang and Pantopoulos 2011). In cells, iron is released from the transferrin-TfR complex in endosomes. Some iron is transported transcellularly, exported out of the cell through ferroportin. Some excess iron is stored in cells bound to ferritin, an iron storage protein (Wang and Pantopoulos 2011). TfR, ferritin, and ferroportin are controlled by posttranslational regulation of iron status, which involves iron-responsive elements (IREs) (Wang and Pantopoulos 2011; Casey et al. 1988). In conjunction with the action of iron-responsive proteins (IRPs), IREs are present in either the 5′ untranslated regions of ferritin and ferroportin mRNAs or the 3′ untranslated region of transferrin mRNA blockage or promote translation (Zhang et al. 2009). However, there are limited a number of studies focusing on the transcriptional regulation of iron-responsive genes. The iron storage gene ferritin is regulated transcriptionally (White and Munro 1988; Wu et al. 1999). Previously, global changes in iron-dependent gene expression were investigated in animal tissues including the rat duodenum (Collins et al. 2005) and mouse liver (Kautz et al. 2008). However, limitations to finding genes that respond early to changes in iron conditions include that iron deficiency must be induced over a long period to achieve systemic iron disturbances.
IEGs are expressed immediately after various external stimuli including serum growth factor exposure (Johansen and Prywes 1995). Most IEGs are transcription factors that regulate secondary gene expression. IEGs contain serum response elements (SREs) in their promoter regions that are bound by serum response factor (SRF). Mitogenic stimuli activate ERK1/2 MAPK signaling pathways, triggering phosphorylation of SRF coregulators, which are ternary complex factors (TCFs) whose members are ELK-1, SAP1, or Net (Gille et al. 1992). Phosphorylated TCFs form a ternary complex with SRF on SREs to initiate transcription in some IEGs (Yordy and Muise-Helmericks 2000). In addition to growth stimulation, IEG expression is associated with apoptosis (Muthukkumar et al. 1995). Thus, IEG activation results in complicated biological outcomes. Changes in IEG expression in response to iron deficiency have not been studied.
As a starting point to understand how cells respond to iron-deficient conditions, we searched for genes that changed expression early in iron-deficient hepatoma cells using cDNA microarray assays. We found that expression of Egr1 was upregulated upon iron depletion by the iron chelator DFO. Subsequent analysis at the cellular levels revealed that the molecular mechanism of DFO-induced expression of Egr1 appeared to involve activation of ERK1/2 signals and phosphorylation of Elk-1.
Materials and methods
Cell culture and treatments
Mouse hepatoma (Hepa1c1c7) cells were from the Korean Cell Line Bank (Seoul, Korea) and were grown in a 5 % CO2 atmosphere at 37 °C in alpha minimum essential medium (MEM) without nucleosides (Welgene, Daegu, Korea) containing 10 % (v/v) fetal bovine serum (FBS) (Cellgro, Manassas, VA, USA) and 5 % antimycotic–antibiotic (Invitrogen, Carlsbad, CA, USA). Microarray experiments used Hepa1c1c7 cells. Confluent cells were incubated with 100 μM DFO for 4, 8, 12, 16, 20, and 24 h and harvested for cDNA microarray analysis. For other experiments, cells were plated at 5.0 × 105 cells per well in six-well plates on day 0. On day 1, medium was changed to starvation medium of 0.2 % (v/v) FBS in alpha MEM medium. On day 3, cells were treated with ferric ammonium citrate (FAC) or desferrioxamine (DFO) to final concentrations of 3 or 100 μM FAC or 100 μM DFO. After 24 h, cells were collected. When applicable, other additions were 2.5 μg/ml actinomycin D and 100 nM PD98059, all from CalBiochem (San Diego, CA, US) and added 1 h before incubation with DFO.
cDNA microarrays
Reverse transcription using Superscript II reverse transcriptase (Invitrogen) produced amino-modified cDNA from total RNAs after priming with oligo dTs in the presence of aminoallyl dUTP. The resulting cDNA was labeled using Cy3 (0 h) or Cy5 (4, 8, 12, 16, 20, and 24 h) fluorescent dyes (Amersham Biosciences, Piscataway, NJ, USA). Cy3-labeled and Cy5-labeled cDNAs were mixed before hybridization at 65 °C for 16–20 h on cDNA microarrays. Microarrays were generated at the College of Natural Resources Genomics Facility using Brain Mouse Anatomy Project mouse brain UniGene set (Open Biosystems, Huntsville, AL, USA). An ArrayWoRx Biochip Reader (Applied Precision, Issaquah, WA, USA) was used to scan slides to obtain microarray images. Images were quantified using GenePix 3.01 (Molecular Devices, Sunnyvale, CA, USA) to obtain an average hybridization signal intensity for each spot with background of the surrounding area. Spot intensity values were analyzed using an exploratory differential gene expression analysis algorithm (Loguinov et al. 2004). Log2-transformed normalized ratios larger than 1.0 and p values lower than 0.05 were used for subsequent data analysis to identify genes that were candidates for differential expression.
RNA extraction and quantitative RT-PCR
Cells were collected with TRIzol (Invitrogen) on ice, and total RNA was extracted according to the manufacturer’s protocol. For qRT-PCR, 1 μg of total RNA was used to synthesize cDNA with random hexamer primer and reverse transcription by ImprompII reverse transcriptase (Promega, Madison, WI, USA) according to the manufacturer’s directions. Real-time PCR was performed on a CFX96 sequence detection system (Biorad, Hercules, CA, USA) using EvaGreen qPCR mix plus (Solis BioDyne, Estonia). Expression levels were normalized to the amount of 18S rRNA. Relative mRNA levels were calculated as difference in Ct values among animal groups and expressed as fold change. Primers were Egr1, forward 5′-GAGCGAACAACCCTATGA-3′, reverse 5′-AGGCCACTGACTAGGCTG-3′; TfR, forward 5′-TCCGCTCGTGGAGACTAC-3′, reverse 5′-GAGCCCCAGAAGATATGT-3′; 18SrRNA, forward 5′-TCGAGGCCCTGTAATTGGAAT-3′, reverse 5′-CCCTCCAATGGATCCTCCTTA-3′.
Plasmids and luciferase assays
The luciferase reporter plasmid, pEgr1-luc, containing the human Egr1 promoter region (−604 to +160) was constructed by PCR using primers: forward 5′-AACTCGAGGCTTCCCCAGCCTAGTTCACGCCT-3′ and reverse 5′-TGAAGCTTGAACACTGAGAAGCGTGCAGGCGGC-3′. PCR products were subcloned into XhoI and HindIII sites of the pGL3basic luciferase vector (Promega) and pCGN-Elk-1S383A carrying ELK1 mutant (S383A) was kindly provided by Dr. Ron Prywes (Columbia University, NY, NY) (Lee et al. 2010).
Cells were grown to subconfluency. In each well, a 1:2 ratio of plasmid DNA to transfection reagent (Omics Bio, Taiwan) was used for transfection according to the manufacturer’s instructions. To normalize for transfection efficiency, 500 ng of reporter constructs together with 100 ng of pRLSV40 were added to each well unless otherwise indicated. The overall DNA amount was held constant at 600 ng by addition of empty vector. Approximately 18 h after transfection, transfection medium was changed to complete medium, and cells were stimulated with either DFO or FAC for 18–24 h with or without 1-h pretreatment with PD98059. Cell lysates were harvested and prepared for luciferase assays using the Promega luciferase assay system (Promega). Luciferase activity was measured by a Promega Glomax luminometer (Promega BioSystems Sunnyvale Inc., Sunnyvale, CA, USA). Promoter activity was measured as relative light units (RLU) from firefly luciferase, and RLU values were normalized to Renilla luciferase activities for each well. All experiments were performed at least in triplicate.
Western blots
Cells were washed once with phosphate-buffered saline (PBS) and collected with 500 μl RIPA buffer (50 mM Tris (pH 7.5), 5 mM EDTA, 1 % sodium deoxycholate, 1 % NP-40, and 0.1 % SDS, 400 mM NaCl) with protease inhibitor mixture (Sigma-Aldrich, St. Louis, MO, USA) per well in six-well plates. After 30 min on ice, cells were centrifuged at 13,000 rpm for 20 min at 4 °C. Supernatants were boiled with 5X sample buffer (200 mM Tris (pH 6.8), 3.575 M β-mercaptoethanol, 10 % SDS, 0.05 % bromophenol blue, and 50 % glycerol) for 10 min, and equal amounts of protein lysates were loaded onto SDS-polyacrylamide gels. Mouse anti-pERK (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), rabbit anti-pJNK (Merck Millipore, Darmstadt, Germany), rabbit anti-pElk (Abcam, Cambridge, England), rabbit anti-Elk-1 (Cell Signaling Technology Inc., Beverly, MA, USA), rabbit anti-caspase 3 (Cell Signaling Technology Inc., Beverly, MA, USA), and rabbit anti-β-actin (Merck Millipore, Darmstadt, Germany) were used to detect the proteins.
Cell viability-MTT assay
Cells were plated at 2 × 104 cells per well into 96-well plates and treated with 0, 50, 100, 500, or 1000 μM DFO and incubated for 24, 48, or 72 h. After removing medium, cells were incubated with 1 mg/ml 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT, Sigma-Aldrich) solution for 4 h. MTT solution was removed carefully, and 200 µl dimethyl sulfoxide (DMSO, Sigma-Aldrich) was added to dissolve purple formazan crystals. Plate was covered with tinfoil and agitated on orbital shaker at 200 rpm for 10 min. Absorbance was measured by (Tecan Infinite 200 PRO, Tecan Australia Pty Ltd, Grodig, Austria) at 570 nm.
ROS assays
After incubation of cells with FAC 3 or 100 μM or DFO 100 μM for 24 h, medium was removed and cells were washed with PBS buffer three times and incubated at 37 °C for 30 min with 20 μM 2′,7′-dichlorofluorescin diacetate (DCFH-DA, Sigma-Aldrich) dissolved in cell culture medium. After removal of medium, cells were washed three times with PBS buffer and incubated with 200 µl PBS buffer for 15 min. Fluorescence from cells was measured by Tecan Infinite 200 PRO with excitation filter 485 nm and emission filter 530 nm.
Cell apoptosis (caspase 3/7 cleavage) assay
For detection of caspase 3 and caspase 7 activations, Caspase 3/7-Glo Assay kit (Promega, Medison, WI, USA) was used according to the manufacturer’s instructions. Cells were plated with a concentration of 1.0 × 104 cells per well into 96-well plates and treated DFO 100 μM for 24 h. After medium was removed, the cells were incubated with 50 µl of the substrate of caspase 3/7 diluted in lysis buffer at room temperature for 1 h. The luminescence signal was detected on a Glomax 20/20 Luminometer (Promega, Medison, WI, USA). The experiment was performed in quadruplicate.
Statistical analyses
SPSS was used to perform statistical analyses (Statistical Package for the Social Science; SPSS Inc., Chicago, IL, USA). All data are presented as mean ± SE. Statistical significance for differences among experimental groups was tested by one-way analysis of variance (ANOVA) followed by Duncan’s multiple comparisons test or Student’s t test. p < 0.05 was used as the criterion for significance.
Results
cDNA microarray assays revealed an increase in Egr1 mRNA levels in DFO-treated cells
We performed cDNA microarray time course experiments using DFO-treated Hepa1c1c cells to identify differential expression profiles of 105 genes over 24 h of treatment (Supplemental Table 1). Among the genes with an expression change detected within 4 h, only one gene was identifiable: Egr1 (Table 1). Egr1 expression levels remained increased at 24 h (Table 1). An increase in mRNA levels of Atf3, Map2k3, Txnip, Pex7, Fam162a, and Hbegf was detected at 8 h. Dusp1 mRNAs was elevated at 12 h and Cts1 at 20 h. Several genes were decreased at 4 h after DFO treatment: Lpl, Ddx54, FlnaA, and Arrb2. However, this reduction in mRNA levels was not consistently detected over 24 h (Supplemental Table 1). A reduction in Atp1a1, G6pdx, C1s, Flna, Maoa, Ly6a, and Cbx1 levels was detected three out of four times in 24 h samples but was not consistent over time. Egr1 mRNAs alone were significantly increased as early as 4 h after DFO treatment with continuous elevation over time. Thus, Egr1 might have been a direct target of DFO.
Table 1.
Genes significantly upregulated in iron-deficient mouse Hepa1c1c7 cells
| Fold change relative to 0 h after DFO treatment | Clone ID | Gene name | Description | UniGene | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 h | 8 h | 12 h | 16 h | 20 h | 24 h(1) | 24 h(2) | 24 h(3) | 24 h(4) | ||||
| 4.92 | 9.58 | 7.78 | 8.69 | 7.78 | 4.41 | 3.41 | 8.40 | IMAGE:608153 | Egr1 | Early growth response 1 | Mm.181959 | |
| 3.53 | 8.82 | 11.63 | 12.82 | 9.06 | 13.18 | 5.35 | 2.83 | 5.35 | IMAGE:775996 | Umod | Uromodulin | Mm.10826 |
| 7.57 | 18.38 | 8.69 | 23.59 | 20.68 | 16.22 | 2.07 | 8.06 | IMAGE:576356 | Egr1 | Early growth response 1 | Mm.181959 | |
| 7.41 | 10.20 | 7.31 | 4.59 | 6.15 | 7.01 | IMAGE:317759 | Egr1 | Early growth response 1 | Mm.181959 | |||
| 17.15 | 8.88 | 15.24 | 11.47 | 7.16 | 4.32 | 8.34 | IMAGE:617677 | Atf3 | Activating transcription factor 3 | Mm.2706 | ||
| 10.13 | 5.46 | 9.71 | 6.68 | 10.34 | 6.32 | 3.05 | IMAGE:585108 | Map2k3 | Mitogen-activated protein kinase kinase 3 | Mm.18494 | ||
| 3.97 | 8.06 | 10.13 | 12.13 | 15.78 | 2.85 | 3.58 | IMAGE:871172 | Txnip | Thioredoxin interacting protein | Mm.410189 | ||
| 5.06 | 2.81 | 3.12 | 2.16 | 3.51 | 7.73 | 4.82 | 3.12 | IMAGE:458895 | Pex7 | Peroxisome biogenesis factor 7 | Mm.338363 | |
| 2.19 | 3.68 | 4.29 | 3.68 | 3.86 | 3.27 | 2.25 | 2.39 | IMAGE:870961 | Fam162a | Family with sequence similarity 162, member A | Mm.212991 | |
| 3.41 | 3.25 | 2.91 | 2.22 | 5.03 | 2.25 | IMAGE:870831 | Transcribed locus, strongly similar to NP_033441.1 triosephosphate isomerase 1 | Mm.439915 | ||||
| 2.48 | 2.62 | 2.28 | 2.17 | 3.23 | 2.53 | 3.01 | IMAGE:871120 | Hbegf | Heparin-binding EGF-like growth factor | Mm.289681 | ||
| 6.15 | 6.32 | 8.46 | 4.56 | 9.13 | 3.05 | IMAGE:MDB0085 | ||||||
| 4.79 | 3.81 | 2.93 | 4.82 | 6.32 | IMAGE:582081 | Dusp1 | Dual specificity phosphatase 1 | Mm.239041 | ||||
| 1.89 | 2.07 | 4.08 | 3.12 | 4.17 | IMAGE:317700 | Ctsl | Cathepsin L | Mm.930 | ||||
DFO upregulates Egr1 partially via transcriptional activation
To confirm the microarray results on Egr1, real-time RT-PCR was performed using Hepa1c1c7 cells treated with either 3 or 100 μM FAC or 100 μM DFO to mimic iron-sufficient, iron-overload, or iron-deficient conditions. At 24 h DFO, Egr1 was significantly increased, and this change was not observed in cells treated with 3 or 100 μM FAC (Fig. 1a). An increase in TfR1 mRNA levels after DFO treatment indicated that cells were depleted for iron (Fig. 1b). Time course experiments showed an increase in Egr1 mRNA as early as 6 h and progressively up to 24 h after DFO incubation (Fig. 1c). When the concentration of DFO was increased from 10 to 100 μM, Egr1 mRNA levels increased dose dependently (Fig. 1d). In order to investigate whether the effects of DFO on Egr1 gene is via transcriptional regulation, the cells were pretreated with actinomycin D or transfected with a reporter construct containing Egr1 promoter (−604 to +160) prior to the exposure of DFO. Actinomycin D pretreatment completely abolished the response of the cells to 100 μM DFO (Fig. 1e). DFO increased the luciferase activities of reporter constructs containing human promoter regions of Egr1 (pEgr1-luc) by 2.7-folds compared with FACs (Fig. 1f). These data suggested that DFO activated transcription of Egr1, but its induction levels were not sufficient enough to explain the full induction of Egr1 gene expression. Overall, our results demonstrated that DFO specifically increases Egr1 transcript abundance in a time- and dose-dependent manner, but its induction appeared to be partly through activation of transcription because DFO modestly increased the transcriptional activity of Egr1 gene.
Fig. 1.
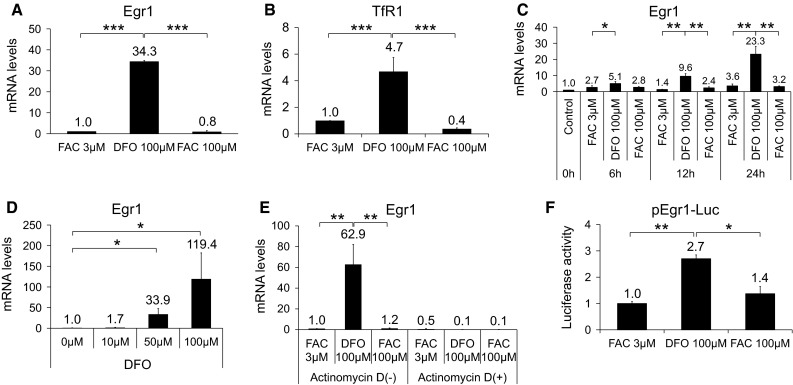
Effects of DFO on Egr1 mRNA and transcriptional activity. Egr1 (a) and TfR1 (b) mRNA in cells treated with 3 or 100 μM FAC or 100 μM DFO for 24 h. Changes in Egr1 transcripts over 24 h after DFO addition (c). Changes in Egr1 mRNA with DFO at 0, 10, 50, 100 μM for 24 h (d). Egr1 mRNA in presence or absence of actinomycin d (2.5 μg/ml) for 1 h before addition of 3 or 100 μM FAC or 100 μM DFO (e). Relative luciferase activity of Egr1-luc from −604 to +160 for cells incubated with 3 or 100 μM FAC or 100 μM DFO (f). Except Fig. 1c, all the treatments were done for 24 h. Data are expressed as degree of induction over control and are mean ± SE. Student’s t test was performed between the groups. *p < 0.05, **p < 0.01, ***p < 0.001. All experiments were performed at least three independent times
DFO-induced Egr1 expression requires the activation of ERK signaling pathway and Elk-1 phosphorylation
DFO induced phosphorylation of ERK but not JNK (Fig. 2a, b). ERK activation indicated by phosphorylated ERK gradually increased to 24 h after DFO treatment (Fig. 2b). To confirm the necessity of ERK activation for expression of Egr1 and c-fos genes, we used the specific ERK inhibitor PD098059. Pretreatment of cells with PD098059 attenuated the DFO-induced increase in Egr1 transcripts from 70.6-fold to 7.7-fold (Fig. 2c). Egr1 promoter contains SRE in its proximal region where Elk-1 binds (McMahon and Monroe 1995). We investigated whether DFO-mediated Egr1 induction is via SRE in the promoter region. When SRE-luc transcriptional activity was examined, DFO-treated cells showed about two-fold induction, which indicated that the effects of DFO on transcriptional activation of Egr1 ranging −604 to +160 was likely to be via SRE (Fig. 2d). DFO treatment increased phosphorylation of Elk-1 (Fig. 2e). In addition, transfection with a dominant-negative mutant ELK1 (pElk-1S383A) diminished the luciferase activity of a construct containing Egr1 promoter regions (pEgr1-luc) after DFO treatment (Fig. 2f). These results might indicate that DFO-induced activation of Egr1 promoter harboring SRE involved activation of the ERK1/2 and Elk-1 signals.
Fig. 2.
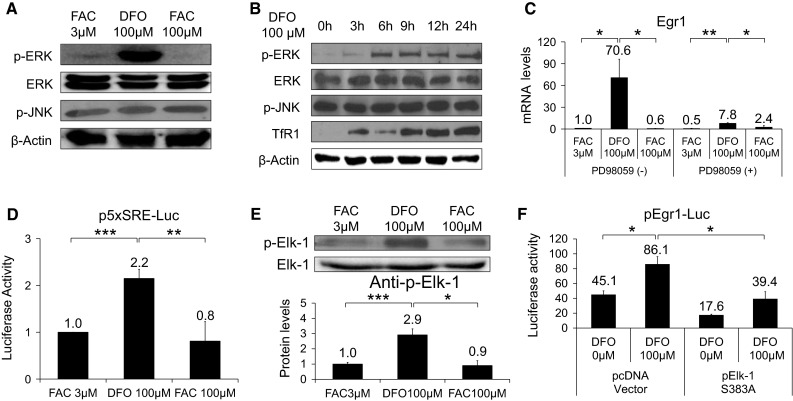
Effects of DFO on phosphorylation of ERK and Elk-1. Phosphorylated protein levels of ERK (p-ERK) and JNK (p-JNK) (a) and Elk (p-Elk) (e) in cells treated with 3 or 100 μM FAC or 100 μM DFO. Time course changes in p-ERK, ERK, p-JNK and TfR1 in cells incubated with 100 μM DFO (b). Egr1 mRNA from cells pretreated with or without PD98059 in the presence of 3 or 100 μM FAC or 100 μM DFO (c). Cells were transfected with 500 ng of p5X SRE reporter and 100 ng of pRLSV40. Next day transfection medium was changed with complete medium. The cells were stimulated with 3 or 100 μM FAC or 100 μM DFO for 24 h. Afterward, the cells were lysed and measured for luciferase activity (d). Cells were transfected with 250 ng of pEgr1-luc, 50 ng of pRLSV40 and 100 ng of pElk-1 S383A or empty vector. In following day, the cells were treated with vehicle or 100 μM DFO for 24 h. Relative luciferase activities in lysed cells were measured (f). Data are expressed as degree of induction over control and are mean ± SE. Student’s t test was performed between the groups. *p < 0.05, **p < 0.01, ***p < 0.001. All experiments were performed at least three independent times
DFO decreased cell viability and ROS production
The effects of DFO on cell viability were examined by MTT assays. As the concentration of DFO increased, cell viability decreased at 24 h (Fig. 3a). After 24 h with 50 μM DFO, cell viability was not significantly reduced but longer than 24-h incubation significantly lowered cell viability. DFO at higher than 50 μM resulted in a significant reduction in cell viability as early as 24 h after treatment began. In order to investigate whether decrease in cell viability was due to apoptosis, caspase 3/7 activation was determined in cells either untreated or treated with 100 μM DFO (Fig. 3b). There was a significant induction of caspase 3/7 activity upon DFO treatment. This was further confirmed by the elevated protein levels of cleaved caspase 3 (Fig. 3c). These data indicated that DFO induced caspase-mediated apoptotic cell death. When intracellular ROS levels were measured, 100 μM DFO treatment resulted in small but significant reductions in ROS levels at 24 h compared with the control, 3 or 100 μM FAC (Fig. 3b). Our data suggested that DFO increased apoptotic cell death and decreased ROS levels modestly.
Fig. 3.

Effects of DFO on cell viability, ROS levels and caspase 3/7 activation. a Viability of cells treated with DFO 0, 50, 100, 500, or 100 μM for 24, 48 or 72 h measured by MTT assay. b ROS levels detected by incubation with DCFH-DA (b) and the protein levels of cleaved form of caspase 3 (c) in cells treated with FAC 3 or 100 μM or DFO 100 μM for 24 h. Levels of caspase 3/7 activation in cells either untreated or treated with 100 μM DFO, which were measured using caspase 3/7-Glo assay kit (d). Values not sharing the same letter were significantly different (p < 0.05) by ANOVA. Student’s t test was performed between the groups. *p < 0.05, ***p < 0.001. All experiments were performed at least three independent times
Discussion
Although many genes are affected by iron deficiency, genes that respond early to iron deficiency in the liver have not been previously elucidated. We used microarray assays and real-time RT-PCR to identify Egr1 as an gene that responded early to iron depletion. Significant upregulation of Egr1 in a hepatoma cell line was detected as early as 4 h and until 24 h after iron depletion by DFO. Activation of ERK1/2 and Elk-1 signals was necessary for the DFO-induced expression of Egr1.
Egr1 is an IEG (Gashler and Sukhatme 1995; Huang et al. 1998) and, as a transcription factor, could influence many genes required to cope with iron deficiency. Egr1 is implicated in cell proliferation and death, depending on the context (Shaulian and Karin 2001). Some studies reported that upregulation of Egr1 prevents cell cycle progression in tumor cells, and others reported that Egr1 activation increases tumor growth (de Belle et al. 1999; Scharnhorst et al. 2000; Akutagawa et al. 2008). Our data for mouse Hepa1c1c7 cells treated with 100 μM DFO for 24 h showed significant decreases in viability and increases in caspase 3/7 activities, suggesting that DFO induced apoptotic cell death. Thus, continuous upregulation of Egr1 might contribute to apoptosis. In a recent report, constitutive expression of Egr1 induced apoptosis in cardiac fibroblast cells (Zins et al. 2014). A number of human tumor cells lines including fibroblastoma and hepatocellular carcinoma lines have decreased Egr1 expression (Liu et al. 1996; Hao et al. 2002). Egr1 inhibits the growth and reverses the cancerous phenotype of v-sis transformed NIH3T3 cells (Huang et al. 1994). The mechanism of action of Egr1 for causing apoptosis appears to require the DNA-binding activity of Egr1 (Huang et al. 1995). Egr1 activation of the expression of transforming growth factor beta1 (TGF-β1) is suggested to be involved in its growth inhibition activity (Liu et al. 1996). In addition, transactivation of p53 and downregulation of bcl-2 involve apoptosis-inducing effects of Egr1(Liu et al. 1998; Huang et al. 1998). Whether Egr1 induces apoptotic cell death under conditions of iron-deficient signals or as part of defective iron-dependent cellular process is not clear.
DFO-mediated activation of Egr1 appears to be regulated in part at the transcriptional levels. Egr1 mRNA increased over 20-fold with DFO treatment, whereas luciferase activity indicating transcriptional activity of the Egr1 promoter showed only a 2.7-fold induction. Thus, the results raised the possibility of additional regulatory systems. For instance, a positive feedback mechanism by Egr1 might have further increased its transcription levels. Egr1 gene has a Egr1-binding site in its promoter region at −624 to –616, which was not included in our Egr1 luciferase construct. In addition, we cannot exclude the possibility of an enhancer region in the Egr1 gene contributing to DFO-mediated induction of Egr1.
Different types of intracellular signals induce Egr1 expression, depending on the stimuli (Pagel and Deindl 2011). Various signals can activate TCFs via phosphorylation by at least one of three mitogen-activated protein kinases (MAPKs), including ERK1/2 and JNK (Cavigelli et al. 1995). Phosphorylated ERK translocates into the nucleus and activates the transcriptional activity of Elk-1 (Pagel and Deindl 2011). Elk-1-binding sites (GGAA/T) are close to SRF-binding sites (CC (A/T)6 GG termed CArG box) (Mo et al. 2000). Cooperative activation of Elk-1 and SRF in forming a ternary complex with DNA increases expression of target genes including Egr1(Wasylyk et al. 1998). Our results were consistent with previous findings that the ERK pathway is essential for transcriptional activation of IEGs such as Egr1 (Gille et al. 1992). DFO induced ERK1/2 activation. When ERK signals were blocked by PD98059, attenuation in Egr1 expression levels was observed, suggesting that DFO signaled through ERK kinases to activate Egr1 expression. Significant elevation of ERK1/2 signals was accompanied by phosphorylation of Elk-1, a downstream target of ERK. When the Elk-1 mutant pElk-1S383A, which lacks a phosphorylation site for transcriptional activation (Yang et al. 1998), was overexpressed, the promoter activity of Egr1 was significantly downregulated in DFO-treated cells. Consistent with ERK and Elk-1 as major signaling pathways in inducing Egr1 expression, the ERK1/2 and Elk-1 signal cascades appeared to be important in DFO-induced Egr1 expression. MEK inhibitors including PD98059 were reported to have off-target effects by decreasing calcium influx into cells upon agonist stimulation (Wauson et al. 2013). Thus, there was also possibility that changes in intracellular calcium concentration by DFO might have played a role in the regulation of Egr1 expression.
DFO is used to mimic hypoxic conditions because iron has a key role in the action of prolyl hydroxylases, which counteract the activity of hypoxia-inducible factors (Chan et al. 2002). Thus, Egr1 activation by DFO could be partly due to hypoxia-inducible factor activation. Hypoxic effects on Egr1 expression appear to be context dependent. Low oxygen at 1 % increases Egr1 expression in U-2OS cells (Nishi et al. 2002), whereas glioblastoma cell lines do not upregulate expression of Egr1 in response to low oxygen levels (Said et al. 2009). Thus, the effect of the iron chelator DFO on prolyl hydroxylases might have been caused by hypoxic conditions, which in turn induced the expression of Egr1. However, how low oxygen increased the expression of Egr1 in hepatoma cells warrants further studies.
Iron can produce ROS, which act as signaling molecules for gene expression (Templeton and Liu 2003). Egr1 is upregulated by the presence of hemin, a source of heme. Hemin activates ROS, which further induces ERK-dependent activation of Elk-1 and Egr1 expression (Hasan and Schafer 2008). The redox-sensitive activation of Ras is suggested to be involved in ROS-mediated activation of Egr1 (Hasan and Schafer 2008; Kuster et al. 2005). In our study, the iron chelator DFO significantly lowered the production of ROS, suggesting that ROS was not a factor in induction of Egr1. Thus, our data provide evidence for the possibility of iron deprivation in increasing Egr1 expression.
Taken together, our findings suggested that iron deficiency increased Egr1 expression in a time-dependent and dose-dependent manner that involved the activation of the ERK1/2 pathway and transcriptional activity of the SRF coregulator Elk-1. DFO-mediated activation of ERK lasted for 24 h, which could continuously activate Egr1 expression to trigger cellular apoptosis. However, the precise mechanism of action of the iron chelator in inducing Egr1 for apoptosis needs to be further investigated.
Electronic supplementary material
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013-11-2083).
Conflict of interest
None.
Abbreviations
- DFO
Desferrioxamine
- FAC
Ferric ammonium citrate
- Egr1
Early growth response 1
- ERK
Extracellular signal-regulated kinase
References
- Akutagawa O, et al. Early growth response-1 mediates downregulation of telomerase in cervical cancer. Cancer Sci. 2008;99(7):1401–1406. doi: 10.1111/j.1349-7006.2008.00835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JL, et al. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science. 1988;240(4854):924–928. doi: 10.1126/science.2452485. [DOI] [PubMed] [Google Scholar]
- Cavigelli M, et al. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 1995;14(23):5957–5964. doi: 10.1002/j.1460-2075.1995.tb00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DA, et al. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1alpha. J Biol Chem. 2002;277(42):40112–40117. doi: 10.1074/jbc.M206922200. [DOI] [PubMed] [Google Scholar]
- Collins JF, et al. Identification of differentially expressed genes in response to dietary iron deprivation in rat duodenum. Am J Physiol Gastrointest Liver Physiol. 2005;288(5):G964–G971. doi: 10.1152/ajpgi.00489.2004. [DOI] [PubMed] [Google Scholar]
- de Belle I, et al. p53 and Egr-1 additively suppress transformed growth in HT1080 cells but Egr-1 counteracts p53-dependent apoptosis. Oncogene. 1999;18(24):3633–3642. doi: 10.1038/sj.onc.1202696. [DOI] [PubMed] [Google Scholar]
- Ganz T, Nemeth E. Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta. 2006;1763(7):690–699. doi: 10.1016/j.bbamcr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823(9):1434–1443. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/S0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- Gille H, Sharrocks AD, Shaw PE. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature. 1992;358(6385):414–417. doi: 10.1038/358414a0. [DOI] [PubMed] [Google Scholar]
- Hao MW, et al. Transcription factor EGR-1 inhibits growth of hepatocellular carcinoma and esophageal carcinoma cell lines. World J Gastroenterol. 2002;8(2):203–207. doi: 10.3748/wjg.v8.i2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan RN, Schafer AI. Hemin upregulates Egr-1 expression in vascular smooth muscle cells via reactive oxygen species ERK-1/2-Elk-1 and NF-kappaB. Circ Res. 2008;102(1):42–50. doi: 10.1161/CIRCRESAHA.107.155143. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117(3):285–297. doi: 10.1016/S0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Huang RP, et al. Suppression of v-sis-dependent transformation by the transcription factor, Egr-1. Oncogene. 1994;9(5):1367–1377. [PubMed] [Google Scholar]
- Huang RP, et al. Egr-1 negatively regulates human tumor cell growth via the DNA-binding domain. Cancer Res. 1995;55(21):5054–5062. [PubMed] [Google Scholar]
- Huang RP, et al. Suppression of human fibrosarcoma cell growth by transcription factor, Egr-1, involves down-regulation of Bcl-2. Int J Cancer. J Int Cancer. 1998;77(6):880–886. doi: 10.1002/(SICI)1097-0215(19980911)77:6<880::AID-IJC14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Huang RP, et al. Suppression of human fibrosarcoma cell growth by transcription factor, Egr-1, involves down-regulation of Bcl-2. Int J Cancer. 1998;77(6):880–886. doi: 10.1002/(SICI)1097-0215(19980911)77:6<880::AID-IJC14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Johansen FE, Prywes R. Serum response factor: transcriptional regulation of genes induced by growth factors and differentiation. Biochim Biophys Acta. 1995;1242(1):1–10. doi: 10.1016/0304-419x(94)00014-s. [DOI] [PubMed] [Google Scholar]
- Kautz L, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503–1509. doi: 10.1182/blood-2008-03-143354. [DOI] [PubMed] [Google Scholar]
- Kuster GM, et al. Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation. 2005;111(9):1192–1198. doi: 10.1161/01.CIR.0000157148.59308.F5. [DOI] [PubMed] [Google Scholar]
- Le NT, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta. 2002;1603(1):31–46. doi: 10.1016/s0304-419x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- Lee SM, Vasishtha M, Prywes R. Activation and repression of cellular immediate early genes by serum response factor cofactors. J Biol Chem. 2010;285(29):22036–22049. doi: 10.1074/jbc.M110.108878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Adamson E, Mercola D. Transcription factor EGR-1 suppresses the growth and transformation of human HT-1080 fibrosarcoma cells by induction of transforming growth factor beta 1. Proc Natl Acad Sci USA. 1996;93(21):11831–11836. doi: 10.1073/pnas.93.21.11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, et al. Suppression of growth and transformation and induction of apoptosis by EGR-1. Cancer Gene Ther. 1998;5(1):3–28. [PubMed] [Google Scholar]
- Loguinov AV, Mian IS, Vulpe CD. Exploratory differential gene expression analysis in microarray experiments with no or limited replication. Genome Biol. 2004;5(3):R18. doi: 10.1186/gb-2004-5-3-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Monroe JG. A ternary complex factor-dependent mechanism mediates induction of egr-1 through selective serum response elements following antigen receptor cross-linking in B lymphocytes. Mol Cell Biol. 1995;15(2):1086–1093. doi: 10.1128/mcb.15.2.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, et al. Structure of the elk-1-DNA complex reveals how DNA-distal residues affect ETS domain recognition of DNA. Nat Struct Biol. 2000;7(4):292–297. doi: 10.1038/74055. [DOI] [PubMed] [Google Scholar]
- Muthukkumar S, et al. Role of EGR-1 in thapsigargin-inducible apoptosis in the melanoma cell line A375-C6. Mol Cell Biol. 1995;15(11):6262–6272. doi: 10.1128/mcb.15.11.6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H, Nishi KH, Johnson AC. Early growth response-1 gene mediates up-regulation of epidermal growth factor receptor expression during hypoxia. Cancer Res. 2002;62(3):827–834. [PubMed] [Google Scholar]
- Organization WH. Worldwide prevalence of anaemia 1993–2005: WHO global database on anaemia. Geneva: World Health Organization; 2008. [Google Scholar]
- Pagel JI, Deindl E. Early growth response 1–a transcription factor in the crossfire of signal transduction cascades. Indian J Biochem Biophys. 2011;48(4):226–235. [PubMed] [Google Scholar]
- Said HM, et al. Egr-1 is not upregulated in response to hypoxic and oxygenation conditions in human glioblastoma in vitro. Mol Med Reports. 2009;2(5):757–763. doi: 10.3892/mmr_00000169. [DOI] [PubMed] [Google Scholar]
- Scharnhorst V, et al. EGR-1 enhances tumor growth and modulates the effect of the Wilms’ tumor 1 gene products on tumorigenicity. Oncogene. 2000;19(6):791–800. doi: 10.1038/sj.onc.1203390. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- Templeton DM, Liu Y. Genetic regulation of cell function in response to iron overload or chelation. Biochim Biophys Acta. 2003;1619(2):113–124. doi: 10.1016/S0304-4165(02)00497-X. [DOI] [PubMed] [Google Scholar]
- Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434(3):365–381. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasylyk B, Hagman J, Gutierrez-Hartmann A. Ets transcription factors: nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem Sci. 1998;23(6):213–216. doi: 10.1016/S0968-0004(98)01211-0. [DOI] [PubMed] [Google Scholar]
- Wauson EM, et al. Off-target effects of MEK inhibitors. Biochemistry. 2013;52(31):5164–5166. doi: 10.1021/bi4007644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, Munro HN. Induction of ferritin subunit synthesis by iron is regulated at both the transcriptional and translational levels. J Biol Chem. 1988;263(18):8938–8942. [PubMed] [Google Scholar]
- Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science. 1999;283(5402):676–679. doi: 10.1126/science.283.5402.676. [DOI] [PubMed] [Google Scholar]
- Yang SH, et al. The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol Cell Biol. 1998;18(2):710–720. doi: 10.1128/mcb.18.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordy JS, Muise-Helmericks RC. Signal transduction and the Ets family of transcription factors. Oncogene. 2000;19(55):6503–6513. doi: 10.1038/sj.onc.1204036. [DOI] [PubMed] [Google Scholar]
- Zhang DL, et al. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009;9(5):461–473. doi: 10.1016/j.cmet.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zins K, et al. Egr-1 upregulates Siva-1 expression and induces cardiac fibroblast apoptosis. Int J Mol Sci. 2014;15(1):1538–1553. doi: 10.3390/ijms15011538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.