

Abstract
Two patients are described, a mother and son, who were initially clinically diagnosed with hereditary spastic paraparesis. This was rectified after very long chain fatty acid testing confirmed adrenomyeloneuropathy (AMN). The son's initial symptoms were characteristic of AMN (the commonest phenotype) but progressed to show symptoms of cerebral involvement. This evolution from non-cerebral to cerebral AMN is recognised in the medical literature and is increasingly important to consider in light of the availability of potential treatments such as haematopoietic stem cell transplantation.
Background
Adrenoleucodystrophy (ALD) is an X linked, neurodegenerative disease caused by a fault in the ABCD1 gene.1 Peroxisomal abnormalities cause very long chain fatty acids to accumulate in the central and peripheral nervous systems along with the adrenal glands. This results in demyelination, axonal neuropathy and adrenal dysfunction. Several ALD phenotypes exist encompassing childhood ALD, heterozygous carrier status, adrenomyeloneuropathy (AMN) and adult-onset cerebral ALD.2 ALD can be confused clinically with other neurological disorders due to its non-specific findings including spasticity, weakness and ataxia.
Case presentation
Case 1
In 1992, a 43-year-old woman with no relevant medical history, presented with a history of increasing stiffness in her legs. She had noticed a progressive decline in mobility, which was particularly noticeable when walking uphill, and in the dark. On examination, tone was significantly increased throughout, there was bilateral reduced power on hip flexion and positive Romberg's test. There was bilateral pin-prick impairment up to the mid-calf, bilateral absent toe vibration sensation, bilateral hyper-reflexia, ankle clonus and extensor plantars. Her gait was noticeably mildly spastic. An initial MRI of the brain and a spine scan showed a disc prolapse at C5/6 causing a slight indentation of the canal.
Over the next 12 years, mobility increasingly became an issue and the patient now needed a stick to walk. In 2004, as her son was also showing a gait disturbance, an initial clinical diagnosis of hereditary spastic paraparesis (HSP) was considered. A repeat MRI of the brain and spine scan showed mild spondylotic changes at C5/6 and C6/7.
However, a plasma very long chain fatty acid (VLCFA) test showed an increase in C26:0 levels and the C24:0/C22:0 ratio, which was consistent with ALD carrier status (figure 1). Repeat VLCFA testing was not undertaken as genetic analysis was performed to confirm.
Figure 1.
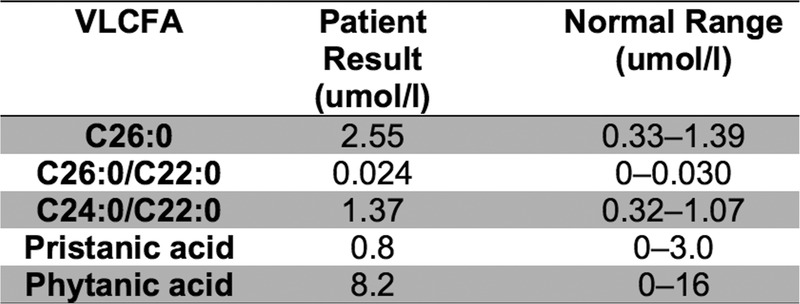
Very long chain fatty acid results for patient 1.
The genetic analysis confirmed a diagnosis of ALD by highlighting the presence of a missense mutation causing an amino acid change at P560L in the ABCD1 gene.
Case 2
A 28-year-old man presented with lower limb spasticity and weakness causing mobility disturbances. He was diagnosed with epilepsy at the age of eight after a ‘Jacksonian’ fit episode and tonic–clonic seizures. He was previously diagnosed with kyphoscoliosis and hallux valgus pronated feet, and had a tendency to fall. On examination, the patient had hyperextension of the joints and a slightly marfanoid body habitus. He had increased lower limb tone, bilateral hip flexion weakness and bilateral loss of vibration to the ankles. All reflexes were brisk with bilateral ankle clonus, extensor plantars and a stiff gait. No numbness was present.
In 2004, an initial MRI brain was normal and a MRI spine showed thoracic kyphosis. This was consistent with the suspected diagnosis of hereditary spastic paraparesis in line with the patient's mother's findings. However, on testing, the increase in plasma VLCFA levels was consistent with X linked LD. This was confirmed on repeat VLCFA testing (figure 2).
Figure 2.
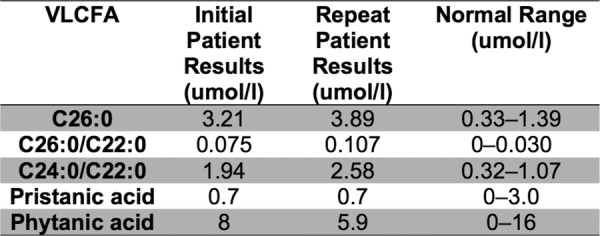
The initial and repeat very long chain fatty acid results for patient 2.
Genetic analysis confirmed the diagnosis by identifying an amino acid change at P560L in the ABCD1 gene.
Over the next few years, the patient's mobility became progressively worse with nocturnal episodes of cramps and restless legs. Urinary incontinence became an issue with a spastic bladder resulting in weekly nocturnal enuresis.
The patient started to report forgetfulness and depression. In 2013, a MRI brain showed evidence of increased signal (figure 3). In the past year, the patient's wife had noticed significant cognitive decline presenting as memory loss, echolalia and severe mood swings involving suicidal thoughts.
Figure 3.
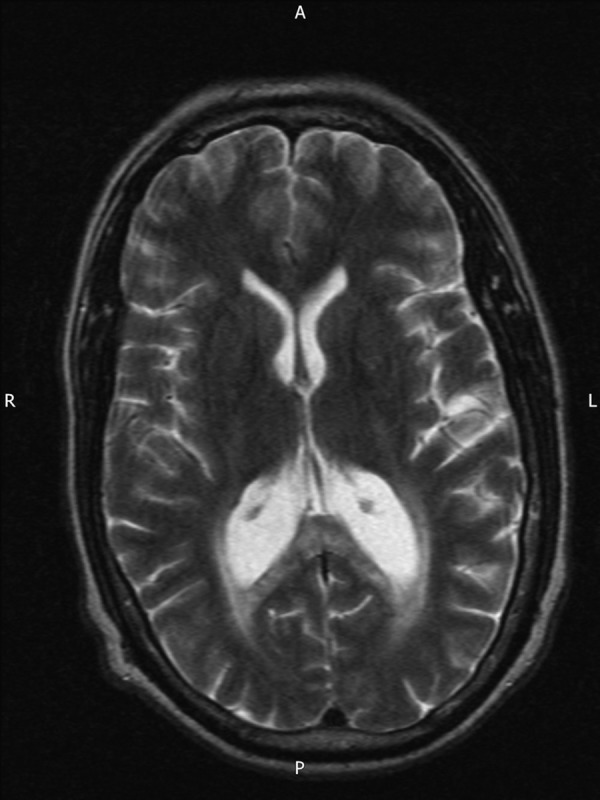
A T2-weighted axial MRI brain (patient 2), undertaken in 2013, showing areas of high signal surrounding the occipital horns of the lateral ventricle extending to the splenium of the corpus callosum.
A 2014 MRI showed evidence of a slight increase in the white matter changes, particularly posteriorly and in the peritrigonal areas (figure 4A, B). Additionally, bearing in mind the patient's age, the ventricles were prominent and consistent with posterior periventricular atrophy. A fluid-attenuated inversion recovery coronal scan showed prominent bilateral periventricular rim changes (figure 4C). Two months later, a follow-up MRI with gadolinium contrast showed a small area of increased enhancement (figure 4D).
Figure 4.

MRI brain (patient 2) undertaken in 2014: (A and B) T2-weighted axial views showing slight progression of white matter signal abnormalities. (C) Fluid-attenuated inversion recovery coronal scan with prominent periventricular rim hyper-intensity. (D) Gadolinium-enhanced T1-weighted axial scan with an area of increased enhancement (indicated by the arrow).
Outcome and follow-up
Patient 1
Until recently, this patient was able to work 3 days a week. Recent nerve conduction studies and electromyography showed no evidence of generalised peripheral neuropathy. However, her mobility has progressively declined and she is now reliant on a walking stick. Her most recent Synacthen test showed an adequate cortisol reserve level of 899 nmol/L. She has no postural symptoms and does not currently require steroid treatment.
Patient 2
Despite a decline in cognition, this patient still retains capacity. Regular MRIs are arranged to monitor his condition.
Discussion
Two cases are described involving a mother and son who were clinically thought to have HSP. One reason for the delay in diagnosis is the perceived rarity of X linked adrenoleucodystrophy (X-ALD). However, with a birth incidence of 1 in 17 000 newborns, it is the most common peroxisomal condition.3 The incidence of heterozygous female carriers is estimated to be 1 in 14 000.4 Despite the numbers of symptomatic carriers being as low as 14% when aged below 40, a sharp increase in numbers means the chance of symptoms occurring escalates to 88% in those aged 60 years plus.5 Manifestations of the disease tend to be milder in women, as it is X linked and can resultantly be harder to diagnosis.
Another reason for the difficultly in diagnosing X-ALD is the non-specific nature of manifesting features. Common features in men and in women include the gradual onset of gait abnormalities, incontinence and chronic myelopathy. Brain and spine imaging is frequently normal (unless there is cerebral involvement). Adrenal insufficiency, although rare in women, is present in up to 80% of boys (overtly in 43% of cases).6 Various differential diagnoses are mistaken for X-ALD.7 These include multiple sclerosis, HSP, spinal cord pathologies and Addison's disease (when adrenal dysfunction is involved). Additionally, cerebral involvement can manifest in various ways, mimicking psychiatric disorders such as schizophrenia and dementia. A relevant family history can strongly hint at a more accurate diagnosis. However, as described in both patients, clinically, HSP was seen as more likely until the VLCFA-positive results returned indicating the importance of VLCFA analysis in HSP differential diagnosis. It is important to note that 10–20% of heterozygous carriers have normal levels of VLCFA.8 Thus, a normal level does not exclude X-ALD and a mutation analysis should be undertaken to confirm a suspected diagnosis.
In cases 1 and 2, patients with AMN, the most common phenotype of X-ALD, are described. Symptoms usually manifest in the third and fourth decade but AMN is compatible with a life expectancy into the ninth decade. However, this X-ALD phenotype has been shown to evolve and 45% of male patients with AMN will show brain involvement on an MRI at some stage.9 In addition, 19% of male patients with AMN will develop cerebral demyelination that leads to severe cognitive decline.3 Subsequently, symptoms and prognosis is similar to that of adult onset cerebral X-ALD; a rarer phenotype with increased mortality. Symptoms are often described as akin to those of schizophrenia with dementia.10 At this stage, MRI brain findings are often helpful in monitoring the progression of cerebral demyelination. It is important to confirm cerebral involvement and monitor the extent of progression.
There is no specific treatment for the majority of patients with X-ALD although many centres have adapted guidelines suggested by a 2012 paper by Engelen et al3 for the diagnosis, follow-up and management. The treatments for a majority of patients currently focus on dietary constraints and symptomatic relief. Both our patients were referred to a dietician who, along with an endocrinologist, devised a dietary plan to reduce VLCFA levels. This plan consisted of Lorenzo's oil, essential fatty acids, a very low fat diet and vitamin supplementation. Both patients were reluctant to start this onerous diet and have not, to date. Studies have reported that Lorenzo's oil coupled with a low fat diet decreases levels of VLCFAs (specifically hexacosanoic acid).11 However, the use of Lorenzo's oil has not been shown to reduce or delay neurological symptoms in patients exhibiting cerebral involvement.12
Owing to the failings of dietary treatment alone, experimental therapies are currently being tried with varying success rates. Postmortem brains of patients with X-ALD showed signs of oxidative stress.13 Studies have shown that hexacosanoic acid (26:0) excess increases free radical production and inflammatory cytokines. The mechanism as to why this occurs is unknown but the endpoint of this chain of events is axonal degeneration and demyelination. In order to remove the oxidative stress, antioxidant treatments have been trialled in ABCD1 null mice.14 A triple therapy consisting of N-Acetyl-Cysteine, α-tocopherol and α-lipoic acid was administered. This resulted in a halt in axonal degeneration and onset of motor symptoms. The study concluded that antioxidant therapy had reversed oxidative damage and, hence, halted clinical progression.15 A trial in humans is currently underway whereby the same cocktail of antioxidants is being administered.
Allogeneic haematopoietic stem cell (HSC) transplantation has been used as a treatment for children with X-ALD for decades with the first reports dating back to 1984. These initial trials were not effective and at the time some concluded that the treatment might have in fact increased cerebral deterioration. A 2000 study involving children with cerebral X-ALD disagreed by showing that HSC transplantation can reverse the demyelinating process when undertaken at an early enough stage.15 However, a recently published long-term follow-up found that three of five patients with cerebral ALD who underwent HSC transplantation as children developed signs of myelopathy during adulthood.16 Trials on adult patients with cerebral involvement have begun recently and studies are only just starting to be published. The first report of an adult-onset patient with cerebral X-ALD treated with bone marrow transplantation described an improvement in sensory disturbance, gait and cognitive function.17 A 2-year follow-up showed a maintained level of improvement, and motor function was found to be near normal. Another published trial looked at HSC transplantation in nine patients with adult cerebral X-ALD.18 Despite two patients dying soon after treatment due to infections, the transplantation halted central nervous system inflammation in six of the remaining seven patients. Subsequently, HSC transplantation is currently the only successfully tested option to prevent rapid cognitive decline and death in patients with cerebral X-ALD.
ALD is underdiagnosed due to a lack of awareness and the range of clinical presentations. It is paramount to diagnose the patient early in order to identify serious complications such as adrenal dysfunction and cerebral involvement. Adrenocortical deficiency can be lethal if left untreated. In addition, cerebral involvement is a bad prognostic indicator. However, potential treatments are currently being tested in clinical trials. Early diagnosis is becoming increasingly important when considering potential treatment options.
Patient's perspective.
Patient 1
It is 33 years since I first noticed something not quite right in the way my legs moved, and at first it was a slight issue. Over the next 20 years I saw a very slow deterioration and was able to work and walk with reasonable ease. Now at the age of 65, mobility and balance have become quite a problem for me, despite the fact that I try to keep active in many ways.
My exercise bike is used every day, I also do some Pilates exercises and for the past year I have been a member of a Tai Chi group especially adapted for the disabled. I still manage to do all my own housework and most of my gardening, drive a car and am socially very active.
The greatest sadness in my life is the fact that I have passed this terrible condition on to my younger son. Mentally this takes its toll on my life, but I try to remain positive at all times, enabling me to support him and his young family.
Patient 2
I fall over.
It is depressing that I cannot do things I used to or play sport like I used to. I get urgency to use the toilet. Wet the bed at times. Sex isn’t as easy as it used to be. I can’t remember as much.
Learning points.
Adrenoleucodystrophy presents non-specifically and subsequently can be difficult to diagnosis.
Cerebral involvement in patients with adrenoleucodystrophy is important to identify when considering potential treatments.
The effectiveness of haematopoietic stem cell transplantation has been shown in seven adult patients with cerebral involvement.
Despite dietary therapies not halting cerebral progression, novel treatments such as antioxidant therapy are showing promising initial results.
Footnotes
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain 1997;120:1485–508. 10.1093/brain/120.8.1485 [DOI] [PubMed] [Google Scholar]
- 2.Mosser J, Lutz Y, Stoeckel ME et al. The gene responsible for adrenoleukodystrophy encodes a peroxisomal membrane protein. Hum Mol Genet 1994;3:265–71. 10.1093/hmg/3.2.265 [DOI] [PubMed] [Google Scholar]
- 3.Engelen M, Kemp S, de Visser M et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis 2012;7:51 10.1186/1750-1172-7-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bezman L, Moser AB, Raymond GV et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol 2001;49:512–17. 10.1002/ana.101 [DOI] [PubMed] [Google Scholar]
- 5.Engelen M, Barbier M, Dijkstra I et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain 2014;137(Pt-3):693–706. 10.1093/brain/awt361 [DOI] [PubMed] [Google Scholar]
- 6.Dubey P, Raymond GV, Moser AB et al. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J Pediatr 2005;146:528–32. 10.1016/j.jpeds.2004.10.067 [DOI] [PubMed] [Google Scholar]
- 7.Bargiela D, Eglon G, Horvath R et al. An under-recognised cause of spastic paraparesis in middle-aged women. Pract Neurol 2013;14:182–4. 10.1136/practneurol-2013-000662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stradomska T, Tylki-Szymańska A. Adrenoleukodystrophy (X-ALD)—changes in serum VLCFA profile and clinical course of patients following bone marrow transplantation and administration of Lorenzo oil. Eur J Paediatr Neurol 2008;12:S73–4. 10.1016/S1090-3798(08)70253-8 [DOI] [Google Scholar]
- 9.van Geel BM, Bezman L, Loes DJ et al. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol 2001;49:186–94. [DOI] [PubMed] [Google Scholar]
- 10.Szpak GM, Lewandowska E, Schmidt-Sidor B et al. Adult schizophrenic-like variant of adrenoleukodystrophy. Folia Neuropathol 1996;34:184–92. [PubMed] [Google Scholar]
- 11.Berger J, Pujol A, Aubourg P et al. Current and future pharmacological treatment strategies in X-Linked adrenoleukodystrophy. Brain Pathol 2010;20:845–56. 10.1111/j.1750-3639.2010.00393.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moser H. Clinical and therapeutic aspects of adrenoleukodystrophy and adrenomyeloneuropathy. J Neuropathol Exp Neurol 1995;54:740–5. 10.1097/00005072-199509000-00017 [DOI] [PubMed] [Google Scholar]
- 13.Galea E, Launay N, Portero-Otin M et al. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim Biophys Acta 2012;1822:1475–88. 10.1016/j.bbadis.2012.02.005 [DOI] [PubMed] [Google Scholar]
- 14.López-Erauskin J, Fourcade S, Galino J et al. Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann Neurol 2011;70:84–92. 10.1002/ana.22363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro E, Krivit W, Lockman L et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet 2000;356:713–18. 10.1016/S0140-6736(00)02629-5 [DOI] [PubMed] [Google Scholar]
- 16.van Geel BM, Poll-The BT, Verrips A et al. Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis 2014;38:359–61. 10.1007/s10545-014-9797-1 [DOI] [PubMed] [Google Scholar]
- 17.Hitomi T, Mezaki T, Tomimoto H et al. Long-term effect of bone marrow transplantation in adult-onset adrenoleukodystrophy. Eur J Neurol 2005;12:807–10. 10.1111/j.1468-1331.2005.01055.x [DOI] [PubMed] [Google Scholar]
- 18.Köhler W, Kühl J. Hematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Neurology 2014;82:5.174. [Google Scholar]