

Abstract
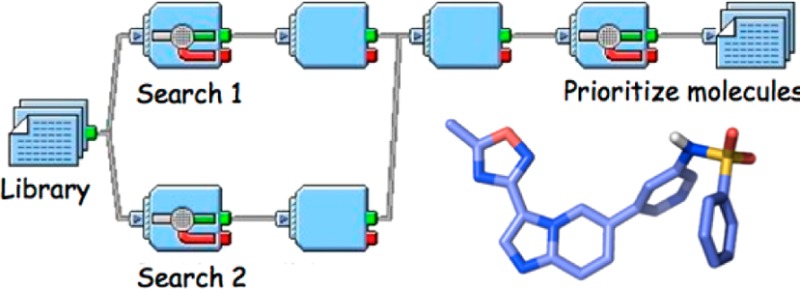
A method capable of identifying novel synthetic targets for small molecule lead optimization has been developed. The FRESH (FRagment-based Exploitation of modular Synthesis by vHTS) approach relies on a multistep synthetic route to a target series of compounds devised by a close collaboration between synthetic and computational chemists. It combines compound library generation, quantitative structure–acitvity relationship construction, fragment processing, virtual high throughput screening and display of results within the Pipeline Pilot framework. Outcomes enumerate tailored selection of novel synthetic targets with improved potency and optimized physical properties for an emerging compound series. To validate the application of FRESH, three retrospective case studies have been performed to pinpoint reported potent analogues. One prospective case study was performed to demonstrate that FRESH is able to capture additional potent analogues.
Keywords: FRESH, lead optimization, QSAR, ADME, virtual HTS, Pipeline Pilot
Successful lead optimization is a critical step in the preclinical stage of drug discovery. It requires novel and potent compounds to be synthesized, while multiple pharmacological properties are simultaneously refined to provide development candidates. From the estimated astronomical number of possible synthetic targets (∼1060),1 it is obvious that exploration of even a subsection of chemical space is not feasible. Thus, medicinal chemists usually investigate the chemical landscape around a lead molecule, which originates from intuition as well as low- and high-throughput screening. To increase the number of compounds investigated without significantly compromising synthesis throughput, modular synthesis is most often pursued. Accordingly, a core structure is preserved while one or more substitution patterns are explored by using different building blocks but the same or similar synthetic method. This strategy enables generation of a series of analogues in a relatively short time frame. Nevertheless, systematic synthetic exploration of chemical space around a lead is still limited.
Lead optimization is further complicated by the need to simultaneously balance potency improvement with optimization of drug-like properties. Thus, physical/ADMET factors like lipophilicity, solubility, metabolic stability and BBB permeability also require attention. Certain of these factors may not optimize in parallel, thereby forcing the drug to reside in a relatively narrow chemical space in which “conflicting” factors are tolerated in spite of their failure to achieve an ideal or near-ideal blend.
As implied above, medicinal chemists face numerous choices on the path to lead optimization. The chemical space around a drug lead, a small fraction of the total space, still represents a significant number of possible synthetic candidates. This is particularly important for academic institutions with limited funding, labor and supporting resources.2 A possible solution to this dilemma is to couple synthetic strategies and computational techniques in a campaign that attempts to simultaneously optimize potency, physical properties and ADME characteristics around a set of target molecules compatible with the principle of synthetic feasibility and the project’s goals. The fundamental design principle behind the FRESH program presented here is thereby captured. First, one constructs a diverse and tailored virtual compound library arising from a practical synthesis reformulated as an in silico modular synthesis. Second, a set of user-selected constraints are applied to library members to identify novel structures predicted to show improvements in potency and physical/ADMET properties. Specific examples evaluated in different contexts are detailed below.
The ability to process large databases of input is crucial for the success of FRESH. The main workflow protocol is constructed with the Pipeline Pilot platform,3 which offers excellent speed performance and efficient use of FRESH to probe great swaths of chemical space. Structural data from other programs are incorporated into the main workflow by a single shared file format (.sdf).
Application of the FRESH program consists of four major steps. Since potency is frequently a primary selection criterion, the program is complemented initially with one or more predictive quantitative structure–activity expressions (QSARs) established from existing data. The QSAR training set is generally derived from published data deposited in external libraries such as ChEMBL or BindingDB.4,5 Furthermore, previously tested in-house compounds can also be added as additional data points. Various receptor or ligand-based methods like Glide, MM-GBSA and ECFP (Extended Connectivity Finger Prints)-Bayesian, which estimate the interaction energies between small molecules and proteins, are methods available for evaluating binding affinity. The QSARs generated in this step are applied by FRESH for subsequent potency estimates of novel structures (see below).
The second step constructs a virtual molecular fragment library for a practical modular synthetic scheme conceived by a collaborating chemist for a given target series, an in silico mimic of the wet-lab synthesis. In the benchtop procedure, building block compounds are purchased from commercial vendors, and the corresponding products are obtained by implementing the synthetic scheme. In the FRESH virtual library construction, building block structures are queried against a virtual library of synthetic intermediates. The source of such a library may be a commercial compound electronic database provided by various vendors like Chem-Navigator, Zinc, Maybridge, a pharmaceutical company’s electronic inventory or a research laboratory’s list of all previously acquired compounds. Thus, the building block structures assembled at this step are considered to be either immediately available or easily obtained and then covalently attached to the core structure.
As stated previously, favorable physical/ADMET properties are crucial components of a successful drug discovery campaign. In step 3, FRESH makes use of these features as additional filters by utilizing established criteria such as the Lipinski “Rule of Five”, Jorgensen’s “Rule of Three” and Morelli, Bourgeas, and Roche’s “Rule of Four”, among others.6−8 For certain scaffolds that obviously violate these rules, the property selection criteria can be modified or simply dropped from the FRESH filtering scheme.
The fourth and final step of the FRESH protocol is the processing and merging of the calculated results, selection of structures that satisfy the desired properties and elimination of known compounds. Members of the final list of prioritized structures are regarded as highly attractive candidates for synthesis.
To validate the FRESH program, three case studies have been performed. The purpose of these exercises is to demonstrate how FRESH can independently capture highly potent compounds from diverse projects reported in the literature. The three cases were chosen based on five criteria: (1) The protein target involved in each case has confirmed or potential therapeutic benefit. (2) The data analyzed is recent, reported within the past 5 years. (3) The drug-lead targets are derived from modular synthesis around a core structure. (4) The seed compound or core structure should not already be a potent ligand; that is, the IC50 or Ki value must be above 100 nM. (5) The literature contains at least one potent compound with an IC50 or Ki value less than 10 nM; none of which were used in a QSAR employed as a guide by FRESH.
It is important to acknowledge that all QSAR methods, whether they are ligand-based or receptor-based, have limitations. A universal QSAR method that can accurately predict the potency of all the unknown compounds in a given project currently does not exist. Considering the shortcomings and limitations of QSAR methods, at least two independent QSAR evaluations are generally used in FRESH to select target compounds when data depth permits it.
All three cases described below require the evaluation of potency. Two receptor-based approaches, Glide and MM-GBSA, were chosen as scoring functions for cases involving an explicit protein structure. A third approach takes ECFP as the molecular portrait and Bayesian statistics (an estimate of probability based on the presence/absence of specific features from the binary data input) as the modeling method. One significant advantage of this 2D-based approach is the speed of calculation. The ECFP descriptor is particularly useful within FRESH when probing a large chemical space. To maximize computational efficiency in the prioritization of candidate structures, QSAR selections based on ECFP are placed in the task-stream ahead of the more computationally expensive score generators like Glide or MM-GBSA.
Phosphatidylinositol 3-kinase (PI3K) phosphorylates the 3-hydroxyl group of the inositol ring of phosphatidylinositol and converts phosphatidylinositol (3,4)-biphosphate (PIP2) to phosphatidylinositol (3,4,5)-triphosphate (PIP3). PI3K is a crucial component in a number of pathways, which regulates cell proliferation, survival, chemotaxis and differentiation.9−11 Among all the isoforms, PI3Kα is an interesting cancer therapeutic target. It has been recognized that up-regulation of the PI3K signaling pathway promotes angiogenesis and is associated with development of human cancers. In addition, the pathway has been implicated in conferring resistance to conventional therapies.12 Currently, there is no FDA approved anti-PI3K drug available.
The present PI3K-based case study originates from the 2011 report by Kim et al.13 in which the R2 extension of scaffold 1 in Figure 1 has been examined. The initial hit compound (R2 = H) is a 360 nM (IC50) agent that emerged from scaffold screening at a concentration of 10 μM. Various aromatic substituents within the R2 group modified by Suzuki coupling were tested.
Figure 1.

PI3K inhibitor scaffold 1 and analogue 2.
The QSAR scoring functions for evaluating potency were established as described above in the description of FRESH. The performance of QSAR scores was evaluated by the area under the curve (AUC) of the receiver operating characteristic (ROC) curve. Among them, the Bayesian score has the highest AUC (0.93, Figure S1), followed by the MM-GBSA score (0.72, Figure S2) and the Glide score (0.64, Figure S3). All three QSAR scores were implemented in FRESH for this case.
According to the synthesis route, the aromatic R2 moiety is attached to the core structure by Suzuki coupling in which arylboronic acids (or bromides) are the building blocks. The corresponding fragments were obtained from the latter by querying the “Zinc bb now” database. The fragments were covalently linked to the core structure 1 in Figure 1 to generate structures of all possible PI3Kα inhibitors in this subclass.
Synthetic targets were then chosen as described in the introduction and experimental procedures. Examination of the prioritized structure list revealed the third best structure with an R2 oxadiazole ring to be literature analogue 2 (Figure 1). With an IC50 = 2 nM, the compound is a potent inhibitor for PI3Kα and featured in the Table of Contents graphic of the original paper.12 Thus, the method successfully captures a known potent molecule among the top five in the final FRESH list.
Carbonic anhydrases (CAs) are ubiquitously expressed in all organisms. This family of enzymes catalyzes the reversible hydration of CO2 to bicarbonate and a proton. CAs are categorized as metalloenzymes since the catalytic center contains a functional zinc ion. Such proteins (including HDAC-1 below) are generally regarded as difficult targets for virtual screening exercises. The enzymes are involved in many physiological processes, and their inhibitors have been explored clinically for various therapeutic purposes such as antiglaucoma, anticonvulsant, antiobesity, pain-relief and antitumor activities. The physiologically dominant isoform, CA II, is one of the most extensively studied proteins among all known protein targets.14
Pacchiano et al. have investigated CA inhibitors with the ureido-benzenesulfonamide scaffold 3 shown in Figure 2, an application of the popular sulfonamide moiety.15,16 The R1 group contains at least one phenyl ring. The corresponding synthetic building blocks for R1 are isocyanates or acid chlorides.
Figure 2.
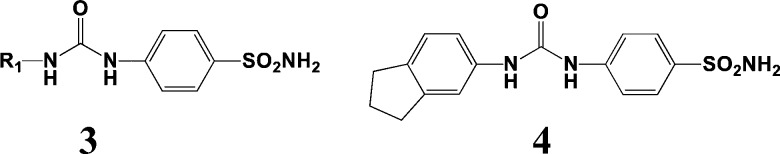
CA II inhibitor scaffold 3 and analogue 4.
Construction of a FRESH protocol is similar to the case study for PI3Kα inhibitors. Since the CA II work involved two previous reports, the publication cutoff year for mining prospective inhibitor candidates in this case study was selected to be the earlier one (2010). The corresponding Bayesian model furnished an AUC of 0.88 for the ROC curve (Figure S4), indicating an excellent separation of actives and inactives. The AUC for Glide scores is 0.63 (Figure S5), which is acceptable. The MM-GBSA score failed to provide a separation of activity classes (AUC < 0.6, figure not shown) and was excluded from the FRESH potency analysis.
The two CA II reported studies did not explicitly specify a starting compound suitable for use as a reference for FRESH QSAR scores. However, since the authors investigated mainly phenyl substitution effects on activity, the unsubstituted phenyl compound (R1 = Ph, Ki = 3.7 μM) was selected as the reference standard. Application of the FRESH target selection scheme provided candidate targets with indane 4 (Ki = 8.9 nM,16 Figure 2) ranked fourth in the final list. FRESH successfully identified a potent CA II inhibitor along with several other previously unconsidered potential CA II blockers (see below) once the zinc dication was incorporated into the docking scheme (see experimental procedures).
HDAC enzymes catalyze the removal of acetyl groups from acetylated lysine amino acids on histone proteins in the nucleosome. This allows histones to be more tightly wrapped around DNA and consequently regulates DNA expression. HDAC enzymes fall into four different classes. Among these, Class I and II isozymes have been associated with uncontrolled tumor growth.17 An example knockdown study on HDACs performed by Glaser et al. suggested that HDAC 1 is essential to the proliferation and survival of mammalian carcinoma cells.18 In 2006, the FDA approved suberoylanilide hydroxamic acid (SAHA) for the treatment of cutaneous T-cell lymphoma.19 This work validated HDAC inhibitors as a strategy for cancer therapy.
The present HDAC case study is based on a report by Wang et al. in 2010.20 The authors explored the urea scaffold 5 shown in Figure 3. The right-hand part of the scaffold is a hydroxamic acid functionality designed to bind to the zinc dication within the catalytic pocket. The length of the aliphatic chain linker was varied, and the left-hand sector of the scaffold consisted of at least one aromatic ring. The corresponding building blocks for R1 were aryl acid chlorides.
Figure 3.
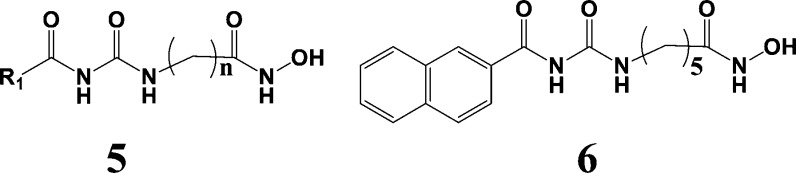
HDAC1 inhibitor scaffold 5 and the naphthalenyl analogue 6 (n = 5).
We elected to treat this case as a ligand-based example, instead of an X-ray structure-based analysis,21 to demonstrate the usefulness of FRESH under circumstances where only limited structural information is available. In addition to manipulating R1 group information, the FRESH application was formulated to vary the linker length from 1 to 7 simultaneously with variations in R1 to intentionally increase the complexity of potent inhibitor identification.
Construction of the FRESH protocol was similar to the previous two case studies on PI3Kα and CA II. However, since only ligand-based methodology was allowed in this study, the Glide and MM-GBSA scores were not utilized. The Bayesian score with the ECFP descriptor was the only SAR-type filter applied in this case. It provided an AUC of 0.87 (Figure 6S). FRESH-searching delivered the naphthalene analogue 6 (Figure 3) among the top five hits with an IC50 = 6 nM, the most potent HDAC blocker yet reported. Consistent with its performance in the previous case studies, FRESH likewise pinpoints a highly potent inhibitor along with a novel set of potentially potent alternative structures.
To validate FRESH methodology in the current time frame, we further explored the final list of predictions from the CA II case study. The purpose of the work was to examine if FRESH can provide additional potent molecules that were not previously discovered.
After verifying that chemical intermediates were indeed commercially available, five compounds composed of inexpensive building blocks were selected from the FRESH final list (∼40 structures) and synthesized. The compounds (>95% pure) were subjected to the CA II potency assay described previously and performed in Florence, Italy.14,15 The Ki values for all five novel inhibitors are less than 10 nM, with two entries less than 1 nM (Table 1). The Ki for the most potent 9 (0.3 nM) is the lowest among agents for this scaffold and seven times more potent than that previously reported (2.1 nM).15 One additional point of interest is that sulfonamides are able to establish a strong interaction by complexation with the metal cation. Nonetheless, as demonstrated herein, synthetic manipulation at eight to ten heavy atoms from the SO2N can lead to significant improvements in Ki.
Table 1. Novel CA II Inhibitors from Family 3 of Figure 2.
| compd | Ri moiety | Ki (nM) |
|---|---|---|
| 7 | 2-napththalenyl | 2.6 |
| 8 | 3-isopropoxyphenyl | 3.9 |
| 9 | 3-isopropylphenyl | 0.3 |
| 10 | 3-chloro-4-fluorophenyl | 2.7 |
| 11 | 3-chloro-4-methylphenyl | 0.77 |
Several QSAR studies have been performed for PI3Kα, HDAC 1, and CA II. However, most were retrospective in that potency correlations were generated subsequent to biodata collection and not used to guide chemical synthesis.22−24 Hage-Melim et al. proposed PI3K synthesis candidates based on physicochemical and pharmacokinetic properties,25 while Tang et al.26 and Zhao et al.27 performed prospective screening studies for HDAC. However, the screening pools in these studies were restricted to known compound databases such as Binding DB, PDB, ZINC, WDI, and PubMed, while novel chemical space attainable by a broadened synthesis campaign was not probed. By contrast, FRESH has been fashioned to include not only a well-conceived QSAR and handpicked physical properties to select synthesis candidates from both available compound collections and synthetic reachable space but also any number of user-defined ADMET factors in the final prediction of synthetic targets. Furthermore, the CA II application of FRESH herein has been complemented by synthesis and bioassay to demonstrate the practical utility of the methodology (Table 1).
A point raised by a Reviewer is that the SAR for the compounds reported in Table 1 is rather flat. The nanomolar Ki values are necessarily flat since, in the FRESH validation, we have reported the synthesis and activities of only those compounds predicted to be most active. As verified by Table 1, the predictions were borne out. The CA II blockers selected correspond to those with the best Bayesian QSAR scores and ADMET properties. The “flat SAR” actually indicates good performance of the QSAR selection scheme in FRESH for this case study. Notably, these compounds were not present in the training sets used for QSAR establishment. Furthermore, the activity spread for the latter training set, hardly flat, runs from 0.2 nM to greater than 20 μM, a range of more than 100,000-fold.
As demonstrated by all three retro-analyzed case studies, the FRESH workflow is able to independently provide a set of synthetic candidates containing at least one potent and previously reported compound. The ranks of each of these substances are among the top five candidates derived by FRESH. Further exploration of the CA II case reveals five additional potent CA II inhibitors from the treatment. Since both CA II and HDAC 1 are metalloenzymes with a cationic catalytic center, one additional apparent asset of the method is the ability to handle such proteins so long as molecular manipulation is distal to metal binding. We conclude that the FRESH methodology is a useful scheme for assisting drug hit-to-lead optimization. Further examples of its application, particularly as it pertains to subsequent iteration steps for individual cases such as those described here, are in progress. An example protocol is available upon request.
Experimental Procedures
Details for QSAR generation and validation, fragment preparation, molecule selection, drug-like property calculations, and syntheses are described in the Supporting Information.
CA II Assay Testing. All new CA II analogues were evaluated as previously reported.15
Acknowledgments
We are grateful to Professor Dennis Liotta (Department of Chemistry, Emory University) for support and encouragement.
Glossary
ABBREVIATIONS
- FRESH
fragment-based exploitation of modular synthesis by virtual HTS
- HTS
high-throughput screening
- ADMET
absorption distribution metabolism excretion toxicity
- BBB
blood–brain barrier
- MM-GBSA
macromodel generalized Born surface area
- ECFP
extended connectivity finger prints
- QSAR
quantitative structure–activity relationship
- PI3K
phosphatidylinositol 3-kinase
- PIP2
phosphatidylinositol (3,4)-biphosphate
- PIP3
phosphatidylinositol (3,4,5)-triphosphate
- AUC
area under curve
- ROC
receiver operating characteristic
- CA
carbonic anhydrase
- HDAC
histone deacetylase
- SAHA
suberoylanilide hydroxamic acid
Supporting Information Available
ROC plots from Bayesian analysis, the detailed filtering scheme, syntheses of CA II analogues with characterization data and purity determination and computational details. This material is available free of charge via the Internet at http://pubs.acs.org.
FRESH development was generously supported by NIH/NCI R01 Ca180805-01.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bohacek R. S.; McMartin C.; Guida W. C. The art and practice of structure-based drug design. Med. Res. Rev. 1996, 16, 3–50. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L. Challenges for Academic Drug Discovery. Angew. Chem., Int. Ed. 2012, 51, 11680–11684. [DOI] [PubMed] [Google Scholar]
- BIOVIA, Dassault Systèmes.
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, 1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorrisen R. N.; Gilson M. K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009, 42, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli X.; Bourgeas R.; Roche P. Chemical and structural lessons from recent successes in protein–protein interaction inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 1–7. [DOI] [PubMed] [Google Scholar]
- Cain R. J.; Ridley A. J. Phosphoinositide 3-kinases in cell migration. Biol. Cell. 2009, 101, 13–29. [DOI] [PubMed] [Google Scholar]
- Sawyer C.; Sturge J.; Bennett D. C.; O’Hare M. J.; Allen W. E.; Bain J.; Jones G. E.; Vanhaesebroeck B. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110delta. Cancer Res. 2003, 63, 1667–1675. [PubMed] [Google Scholar]
- Price J. T.; Tiganis T.; Agarwal A.; Djakiew D.; Thompson E. W. Epidermal growth factor promotes MDA-MB-231 breast cancer cell migration through a phosphatidylinositol 3′-kinase and phospholipase C-dependent mechanism. Cancer Res. 1999, 59, 5475–5478. [PubMed] [Google Scholar]
- Akinleye A.; Avvaru P.; Furqan M.; Song Y.; Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J. Hematol. Oncol. 2013, 6, 88–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim O.; Jeong Y.; Lee H.; Hong S. S.; Hong S. Design and synthesis of imidazopyridine analogues as inhibitors of phosphoinositide 3-kinase signaling and angiogenesis. J. Med. Chem. 2011, 54, 2455–2466. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. [DOI] [PubMed] [Google Scholar]
- Pacchiano F.; Aggarwal M.; Avvaru B. S.; Robbins A. H.; Scozzafava A.; McKenna R.; Supuran C. T. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureidobenzenesulfonamides and correlate to inhibitor potency. Chem. Commun. 2010, 46, 8371–8373. [DOI] [PubMed] [Google Scholar]
- Pacchiano F.; Carta F.; McDonald P. C.; Lou Y.; Vullo D.; Scozzafava A.; Dedhar S.; Supuran C. T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [DOI] [PubMed] [Google Scholar]
- De Ruijter A. J.; Van Gennip A. H.; Caron H. N.; Kemp S.; Van Kuilenburg A. B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser K. B.; Li J.; Staver M. J.; Wei R.-Q.; Albert D. H.; Davidsen S. K. Role of Class I and Class II histone deacetylases in carcinoma cells using siRNA. Biochem. Biophys. Res. Commun. 2003, 310, 529–536. [DOI] [PubMed] [Google Scholar]
- Richon V. M. Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95 (Suppl 1), S2–S6. [Google Scholar]
- Wang H.; Lim Z. Y.; Zhou Y.; Ng M.; Lu T.; Lee K.; Sangthongpitag K.; Goh K. C.; Wang X.; Wu X.; Khng H. H.; Goh S. K.; Ong W. C.; Bonday Z.; Sun E. T. Acylurea connected straight chain hydroxamates as novel histone deacetylase inhibitors: Synthesis, SAR, and in vivo antitumor activity. Bioorg. Med. Chem. Lett. 2010, 20, 3314–3321. [DOI] [PubMed] [Google Scholar]
- Wang D. Computational Studies on the Histone Deacetylases and the Design of Selective Histone Deacetylase Inhibitors. Curr. Top. Med. Chem. 2009, 9, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran T.; Lu T.; Yuan H.; Liu H.; Wang J.; Zhang W.; Leng Y.; Lin G.; Zhuang S.; Chen Y. A selectivity study on mTOR/PI3Kα inhibitors by homology modeling and 3D-QSAR. J. Mol. Model. 2012, 18, 171–186. [DOI] [PubMed] [Google Scholar]
- Melagraki G.; Afantitis A.; Sarimveis H.; Igglessi-Markopoulou O.; Supuran C. T. QSAR study on para-substituted aromatic sulfonamides as carbonic anhydrase II inhibitors using topological information indices. Bioorg. Med. Chem. 2006, 14, 1108–1114. [DOI] [PubMed] [Google Scholar]
- Yang J. S.; Chun T.; Nam K.; Kim H. M.; Han G. Structure-Activity Relationship of Novel Lactam Based Histone Deacetylase Inhibitors as Potential Anticancer Drugs. Bull. Korean Chem. Soc. 2012, 33, 2063–2066. [Google Scholar]
- Hage-Melim L. I.; Santos C. B. R.; Poiani J. G.; de Menezes Vaidergom M.; Eduardo S. M.; Manzolli E. S.; de Paula da Silva C. H. T. Computational Medicinal Chemistry to Design Novel Phosphoinositide 3-Kinase (PI3K) Alpha Inhibitors in View of Cancer. Curr. Bioact. Compd. 2014, 10, 147–151. [Google Scholar]
- Tang H.; Wang X. S.; Huang X. P.; Roth B. L.; Butler K. V.; Kozikowski A. P.; Jung M.; Tropsha A. Novel inhibitors of human histone deacetylase (HDAC) identified by QSAR modeling of known inhibitors, virtual screening, and experimental validation. J. Chem. Inf. Model. 2009, 49, 461–476. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Xiang Y.; Song J.; Zhang Z. A novel two-step QSAR modeling work flow to predict selectivity and activity of HDAC inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 929–933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.