

Abstract

Inhibitors of the protein methyltransferase Enhancer of Zeste Homolog 2 (EZH2) may have significant therapeutic potential for the treatment of B cell lymphomas and other cancer indications. The ability of the scientific community to explore fully the spectrum of EZH2-associated pathobiology has been hampered by the lack of in vivo-active tool compounds for this enzyme. Here we report the discovery and characterization of EPZ011989, a potent, selective, orally bioavailable inhibitor of EZH2 with useful pharmacokinetic properties. EPZ011989 demonstrates significant tumor growth inhibition in a mouse xenograft model of human B cell lymphoma. Hence, this compound represents a powerful tool for the expanded exploration of EZH2 activity in biology.
Keywords: Methyltransferase, PRC2, EZH2, B cell lymphoma, KARPAS-422, xenograft, in vivo chemical probe
Inhibitors of Enhancer of Zeste Homolog 2 (EZH2) target the catalytic center of a multiprotein complex known as polycomb repressive complex 2 (PRC2). The PRC2 complex is responsible for methylating a specific histone lysine referred to as H3K27.1,2 In multiple human cancers, hyper-trimethylation of H3K27 results in the aberrant silencing of genes that otherwise control cell proliferation and induce differentiation.3 Moreover, genetic alterations of PRC2 components have been documented in both hematologic and solid tumors.4 For instance, EZH2 change-of-function mutations (affecting residues Y646, A682, and A692) are found in subsets of B cell non-Hodgkin lymphoma (NHL) where they confer an oncogenic dependency on EZH2. Hence, EZH2 mutant-bearing, diffuse large B cell lymphoma (DLBCL) cell lines can be effectively killed by EZH2 inhibitors in vitro and in vivo.5 Intriguingly, we have also shown that sensitivity to EZH2 inhibition in EZH2 mutant DLBCL cell lines of germinal center origin (GCB) can be enhanced by combination with prednisone, the glucocorticoid-agonist component of the standard chemotherapy regimen CHOP; this sensitivity can be extended to EZH2 wild-type GCB and to inhibitor-refractory, EZH2 mutant GCB cell lines.6 In stark contrast, myeloid malignancies and T cell leukemia7 bear mutations in EZH2 and other PRC2 components that lead to a loss of function of the complex, thus exemplifying the biological complexity of the role of H3K27 methylation in cancers.8 As a result of this complexity and the importance of EZH2 as a therapeutic target for specific human cancers, research in the field has continued to expand in recent years thereby driving the need for well-characterized chemical probes.
As we have described previously, the pyridone-benzamide core represents a highly optimized feature for binding to EZH2 in a SAM-competitive manner. This is a common feature in known indazole EZH2 inhibitors (e.g., EPZ005687,9UNC-199910), indole EZH2 inhibitors (e.g., GSK-126,11EI1,12CPI-16913) and EPZ-6438, our EZH2 inhibitor presently undergoing clinical trials.4 In pyridone-containing EZH2 inhibitors, pyridone oxidation is a common site of metabolism. Recently, there have been a series of published reports that propose a pyridone-replacement chemotype, 4-amino-2,2′,6,6′-tetramethyl-piperidine, as one means to avoid this issue and to expand the breadth of EZH2 inhibitors.15,15b To date, however, compounds with this novel substitution have not demonstrated potency equivalent to pyridone-containing inhibitors and have not reported in vivo activity. Because the pyridone plays such a role in binding and therefore potency, our continued research in this area has focused on modifications to other regions of our chemical scaffold to further investigate the impact on in vivo activity. What is needed today are potent, bioavailable tool compounds that can be made widely available to the greater research community to augment our collective understanding of the role of EZH2 in pathobiology. Herein we describe the discovery, characterization, and in vivo profile of such a chemical probe, EPZ011989.
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3-(ethyl((trans)-4-((2-methoxyethyl)(methyl)amino)cyclohexyl)amino)-2-methyl-5-(3-morpholinoprop-1-yn-1-yl)benzamide (EPZ011989, Figure 2) was discovered through modification of the pyran substituent in EPZ-6438 (Figure 1). In this position, a trans-N,N-dimethylcyclohexylamine substituent appears to maintain biochemical activity, but when combined with the morpholine side-chain, the cellular activity of the resulting dibasic compound is negatively impacted. A series of compounds were designed to attenuate the pKa of the amine components, and a balance of properties and potency was achieved through the addition of a methoxyethyl group to the cyclohexylamine (ca. pKa = 9.8)16 combined with the replacement of the second benzene ring in EPZ-6438 with an acetylene linker to modify the adjacent morpholine (ca. pKa = 5.7).
Figure 2.

SAR affords a potent, stable EZH2 inhibitor.
Figure 1.

Representative reported EZH2 inhibitors.
These adjustments led to a compound, EPZ011989, that equipotently inhibits mutant and wild-type EZH2 with an inhibition constant (Ki) of <3 nM. EPZ011989 is also a specific EZH2 inhibitor with a >15-fold selectivity over EZH1 and >3000-fold selectivity relative to the Ki of 20 other histone methyltransferases (HMTs) tested. As evidenced by the human and rat liver microsomal turnover (HLM and RLM respectively, Figure 2), EPZ011989 also exhibits metabolic stability (Table 1). Furthermore, EPZ011989 reduces cellular H3K27 methylation in the Y641F, mutant-bearing human lymphoma cell line, WSU-DLCL2, with an IC50 below 100 nM. This functional response translates to activity in a long-term proliferation assay where EPZ011989 demonstrates an average lowest cytotoxic concentration (LCC) in WSU-DLCL2 cells of 208 nM (Figure 3).
Table 1. EPZ011989 in Vitro Data Summary.
| property | analysis | units | EPZ011989 | |
|---|---|---|---|---|
| EZH2 activity | biochemical Ki | nM | <3 (WT) <3 (Y646) | |
| ELISA H3K27me3 IC50a | nM | 94 ± 48 (n = 20) | ||
| liver microsome clearance | scaled microsomal clearanceb | mL/min/kg (%Eh)c | human | 6 ± 0.5(29) |
| rat | <10(<14) | |||
| mouse | <10(<11) | |||
| plasma protein binding | equilibrium dialysis | percent unbound | human | 97 ± 3 |
| rat | 91 ± 6 | |||
| mouse | 80 ± 11 | |||
| LCC | 11d proliferation WSU-DLCL2 | nM | 208 ± 75 (n = 4) | |
| predicted efficacious plasma exposured | nM (ng/mL) | human | 214(130) | |
| rat | 223(135) | |||
| mouse | 260(158) | |||
Enzyme-linked immunoassay measure of cellular methyl mark reduction.
Scaled according to the well-stirred liver model.
Percent hepatic extraction.
Plasma protein binding corrected LCC.
Figure 3.
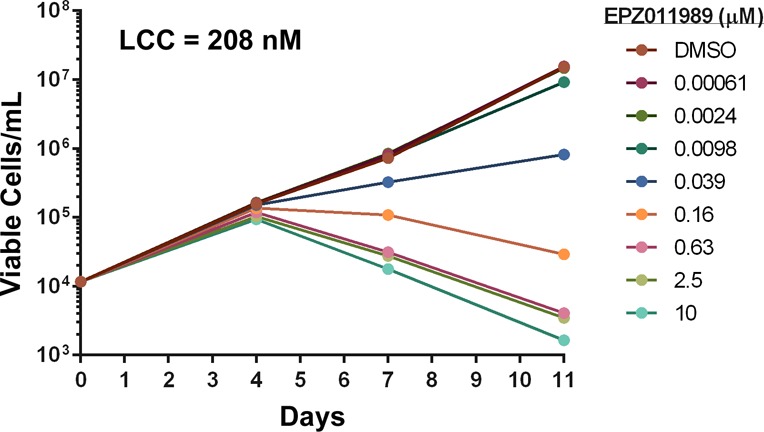
Effect of EPZ011989 concentration on the proliferation of WSU-DLCL2 cells in culture over an 11-day period.
The LCC parameter, when corrected for plasma protein-binding, predicts an efficacious plasma level in mouse for EPZ011989 of 158 ng/mL. The LCC-predicted exposure was used as a benchmark to enable the selection of doses bracketing this value for in vivo studies. The pharmacokinetics in SCID mice following oral administration of 125, 250, 500, and 1000 mg/kg indicated that the 1000 mg/kg dose provided coverage over the LCC for 24 h, while the 250 and 500 mg/kg doses provided coverage over this value for approximately 8 h (Figure 4). This confirmed that, with appropriate formulation, sustained exposure above the LCC could be achieved in vivo.
Figure 4.
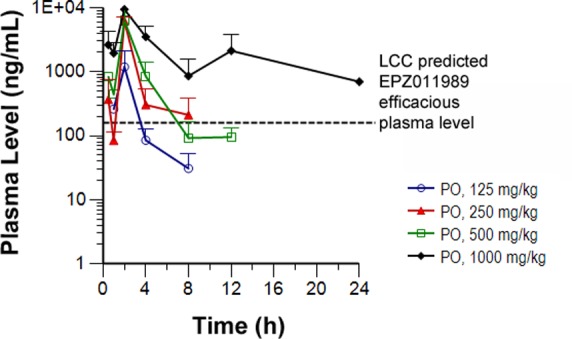
Single dose PK in SCID mice following oral administration of 125, 250, 500, and 1000 mg/kg dosed as suspensions in 0.5% w/v methyl cellulose and 0.1% Tween-80 acidified with 1 mol equiv of HCl. LCC predicted efficacious plasma level for compound EPZ011989 (158 ng/mL) is shown by a horizontal, dashed line.
On the basis of the preliminary PK results, we conducted a 7-day PK study with pharmacodynamic (PD) measurement of H3K27 methylation in bone marrow at 125, 250, 500, 750, and 1000 mg/kg dosed twice-daily (BID). All of the doses were well tolerated for the length of the study. The results from this experiment are shown in Figure 5, where the trough exposure represents the Cmin at 12 h after the first dose on day 7 of test article administration. The dose of EPZ011989 that achieves complete coverage over the predicted efficacious plasma level was determined to be 500 mg/kg BID (Figure 5a). The exposures at 500 mg/kg BID (mean Cmin = 150 ng/mL) correspond well with the above PK experiment and with the PD results in bone marrow, where we observed complete ablation of the methyl mark by the end of day 7 (Figure 5b).
Figure 5.

(a) Pharmacokinetic analysis of day 7 plasma samples for EPZ011989. (b) Pharmacodynamic analysis of histone methyl mark in bone marrow tissue at day 7 of dosing EPZ011989.
The above results demonstrate that formulation of the free base of EPZ011989 in HCl-acidified vehicle was suitable to complete target engagement proof-of-concept studies. After completion of this initial PK/PD study, we performed a salt screen, which identified the d-tartrate salt (DTAL), an amorphous solid with low hygroscopicity, as an alternative for future work. We conducted a rat PK experiment at 30, 100, and 300 mg/kg (Table 2) and found that this optimized salt form provided sustained oral exposure over the rat predicted efficacious plasma exposure (135 ng/mL) for approximately 10 h after a single dose (Figure 6).
Table 2. Summary of Rat PK for EPZ011989.
| EPZ011989
Rat PK | ||||||
|---|---|---|---|---|---|---|
| dose (mg/kg) |
route |
t1/2 (h) |
tmax (h) |
Cmax (ng/mL) |
AUCinf (h·ng/mL) |
time above LCC (h) |
| 30 | p.o. | 4.7 | 2 | 240 | 970 | 4 |
| 100 | p.o. | 3.9 | 2.7 | 1600 | 5600 | 8 |
| 300 | p.o. | 3.7 | 2.7 | 2900 | 10000 | 10 |
Figure 6.
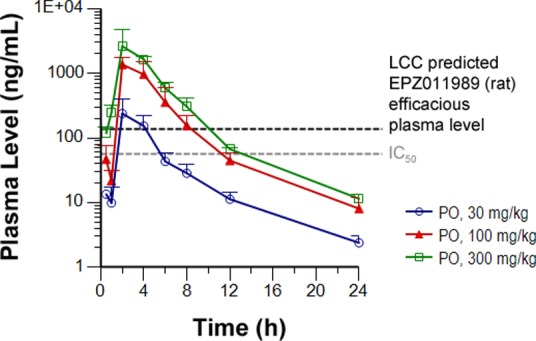
PK after oral dosing of EPZ011989 DTAL at doses of 30, 100, and 300 mg/kg.
To demonstrate further the utility of EPZ011989 as an in vivo tool compound, we evaluated the antitumor activity of the optimized d-tartrate salt form in the treatment of subcutaneous EZH2 mutant KARPAS-422 human DLBCL xenografts. Homogenous suspensions of 250 and 500 mg/kg in 0.5% methyl cellulose and 0.1% Tween-80 were dosed orally to implanted SCID mice for 21 days, BID. On the basis of the PD study, we expected to see tumor regression with the 500 mg/kg dose; however, EPZ011989 administration induced significant tumor regression at both doses, with nominal effect on mean body weights over the course of the study period (Figure 7). Evaluation of PD in tumor samples on day 7 demonstrated robust H3K27 methyl mark reduction for EPZ011989 at the 250 and 500 mg/kg dose over the 12 h time-course (Figure 8). Notably, the exposure for EPZ011989 at 3 h postdose is an order of magnitude higher in this experiment, using the DTAL salt, than for the corresponding dose in the PK/PD study (Figure 9). As a result, at a dose of 250 mg/kg, EPZ011989 remains over the predicted efficacious plasma levels for a minimum of 6 h, though not for the full 12 h time interval. Examination of the data in Figure 8 reveals that, even though plasma exposure does not remain over the LCC, tumor methyl mark levels do not rebound during the dosing interval at 500 nor 250 mg/kg. This suggests that the observed efficacious exposure of EPZ011989 required for tumor growth inhibition is even lower than the level predicted by the LCC and that persistent methyl mark inhibition likely accounts for the resultant antitumor activity at lower exposure. Additional PK and xenograft studies are underway to see if this observation with EPZ011989 holds in the exploration of an expanded set of cancer types associated with EZH2 mutation and dysfunction.
Figure 7.

Robust tumor growth inhibition seen at 250 and 500 mg/kg BID EPZ011989.
Figure 8.

Methyl mark reduction observed in tumor tissue over time on day 7 of EPZ011989 administration.
Figure 9.

Total and free plasma exposure time courses for EPZ011989 in the KARPAS-422 xenograft study. Values measured postdose on day 7 of 21.
Through SAR studies on the pyridone-benzamide scaffold we have discovered an in vivo tool compound with which to further the study of the role of the PRC2 complex in biology and in preclinical models of disease. We have characterized EPZ011989, a compound with oral exposure and metabolic stability that is able to elicit robust methyl mark inhibition and antitumor activity. The PK/PD and in vivo activity data for this compound were highlighted to enable collaborative research in the field. Samples of EPZ011989 can be made available upon request.
Acknowledgments
We thank Rahul Nagawade for his contribution to the synthesis of EPZ011989.
Glossary
ABBREVIATIONS
- EZH2
enhancer of zeste Homolog 2
- PRC2
polycomb repressive complex 2
- H3K27
histone 3 lysine 27
- NHL
non-Hodgkin lymphoma
- DLBCL
diffuse large B cell lymphoma
- SAR
structure–activity relationship
- %Eh
percent hepatic extraction
- LCC
lowest cytotoxic concentration
- HMTs
histone methyltransferases
- HLM
human liver microsomes
- RLM
rat liver microsomes
- SCID
severe combined immune deficiency
- BID
twice-daily dosing
- PK
pharmacokinetics
- PD
pharmacodynamics
- DTAL
d-tartrate salt
- p.o.
oral dosing
- SEM
standard error of the mean
- H3K27me3
histone 3 lysine 27 trimethyl
Biography
John E. Campbell received his Ph.D. in Organic Chemistry from the University of Wisconsin−Madison in 2003 for the total synthesis of unnatural macrocycle ionophores under the supervision of Steve D. Burke. After returning to the East Coast for postdoctoral training in transition metal catalysis at Boston College with Amir H. Hoveyda, he entered the pharmaceutical industry as a bench chemist at Sepracor, Inc. (now Sunovion), developing small molecule candidates for the treatment of depression and schizophrenia. After 6 years and participating in three Investigational New Drugs (INDs), John moved to Epizyme where he now holds the title of Principal Scientist, and his research focus centers on the development of small molecule inhibitors of chromatin-modifying enzymes, searching for therapeutics for genetically defined cancers.
Supporting Information Available
Detailed biological assay information, and procedures and characterization data for the synthesis of EPZ011989. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States.
Author Present Address
⊥ Raze Therapeutics, Suite 303, 25 First Street, Cambridge, Massachusetts 02141, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): J.E.C., K.W.K., S.K.K., N.M.W., H.K., T.J.W., A.R., C.R.K., N.R., M.P.S., N.J.W., J.J.S., R.C., M.P.M., and R.A.C. have ownership interest (including patents) in Epizyme.
Supplementary Material
References
- Kuzmichev A.; Nishioka K.; Erdjument-Bromage H.; Tempst P.; Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R.; Wang L.; Xia L.; Erdjument-Bromage H.; Tempst P.; Jones R. S.; Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Simon J. A.; Lange C. A. Roles of the EZH2 Histone Methyltransferase in Cancer Epigenetics. Mutat. Res. 2008, 647, 21–29. [DOI] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Porter-Scott M.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M.; Kuntz K. W.; Keilhack H. Durable Tumor Regression in Genetically Altered Malignant Rhabdoid Tumors by Inhibition of Methyltransferase EZH2. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeringer C. J.; Porter-Scott M.; Kuntz K. W.; Knutson S. K.; Pollock R. M.; Richon V. R.; Copeland R. A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (HEK27) in human B cell Lymphomas. Proc. Natl. Acad. Sci. U.S.A. 2011, 107, 20980–20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Johnston L. D.; Klaus C. R.; Wigle T. J.; Iwanowicz D.; Littlefield B. A.; Porter-Scott M.; Smith J.; Moyer M. P.; Copeland R. A.; Pollock R. M.; Kuntz K. W.; Raimondi A.; Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocoericoid Receptor Agonists in Models of Germinal Center Non-Hodgekin Lymphomas. PLoS One 2014, 9, e111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P.; Tsirigos A.; Van Vlierberghe P.; Nedjic J.; Trimarchi T.; Sol Flaherty M.; Ferres-Marco D.; da Ros V.; Tang Z.; Siegle J.; Asp P.; Hadler M.; Rigo I.; De Keersmaecker K.; Patel J.; Huynh T.; Utro F.; Poglio S.; Samon J. B.; Paietta E.; Recevskis J.; Rowe J. M.; Rabadan R.; Levine R. L.; Brown S.; Pflumio F.; Dominguez M.; Ferrando A.; Aifantis I. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Robertson G. P.; Zhu J. A novel human homologue of Drosophila polycomblike gene is up-regulated in multiple cancers. Gene 2004, 343, 69–78. [DOI] [PubMed] [Google Scholar]
- Knutson S. K.; Wigle T. J.; Warholic N. M.; Sneeringer C. J.; Allain C. J.; Klaus C. R.; Sacks J. D.; Raimondi A.; Majer C. R.; Song J.; Porter-Scott M.; Jin L.; Smith J. J.; Olhava E. J.; Chesworth R.; Moyer M.; Richon V. M.; Copeland R. A.; Keilhack H.; Pollock R. M.; Kuntz K. W. A Selective Inhibitor of EZH2 blocks H3K27 methylation and Kills Mutant Lymphoma Cells. Nat. Chem. Biol. 2012, 8, 890–896. [DOI] [PubMed] [Google Scholar]
- Konze K. D.; Ma A.; Li F.; Barsyte-Lovejoy D.; Parton T.; MacNevin C. J.; Liu F.; Gao C.; Huang X.-P.; Kuznetsova E.; Rougie M.; Jiang A.; Patterden S. G.; Norris J. L.; James L. I.; Roth B. L.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Hahn K. M.; Wang G. G.; Vedadi M.; Jin J. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S. K.; Tian X.; LaFrance L. V.; Duquenne C.; Suarez D. P.; Newlander K. A.; Romeril S. P.; Burgess J. L.; Grant S. W.; Brackley J. A.; Graves A. P.; Scherzer D. A.; Shu A.; Thompson C.; Ott H. M.; Van Aller G. S.; Machutta C. A.; Diaz E.; Jiang Y.; Johnson N. W.; Knight S. D.; Kruger R. G.; McCabe M. T.; Dhanak D.; Tummino P. J.; Creasy C. L.; Miller W. H. Identification of Potent, Selective Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett. 2012, 3, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W.; Chan H.; Teng L.; Li L.; Chuai S.; Zhang R.; Zeng J.; Li M.; Fan H.; Lin Y.; Gu J.; Ardayfio O.; Zhang J.-H.; Yan X.; Fang J.; Mi Y.; Zhang M.; Zhao T.; Feng G.; Chen Z.; Li G.; Ynag T.; Zhao K.; Liu X.; Yu Z.; Lu C. X.; Atadja P.; Li E. Selective inhibition of EZH2 by a small molecule inhibitor blocks tumor cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley W. D.; Arora S.; Busby J.; Balasubramanian S.; Gehling V.; Nasveschuk C. G.; Vaswani R. G.; Yuan C.-C.; Hatton C.; Zhao F.; Williamson K. E.; Iyer P.; Méndez J.; Campbell R.; Cantone N.; Garapaty-Rao S.; Audia J.; Cook A. S.; Dakin L. A.; Albrecht B. K.; Harmange J.-C.; Daniels D. L.; Cummings R. T.; Bryant B. M.; Normant E.; Trojer P. EZH2 Inhibitor Efficacy in Non-Hodgkin’s Lymphoma Does Not Require Suppression of H3K27 Monomethylation. Chem. Biol. 2014, 21, 1463–1475. [DOI] [PubMed] [Google Scholar]
- Garapaty-Rao S.; Nasvechuk C.; Gagnon A.; Chan E. Y.; Sandy P.; Busby J.; Balasubramanian S.; Campbell R.; Zhao F.; Bergeron L.; Audia J. E.; Albrecht B. K.; Harmange J.-C.; Cummings R.; Trojer P. Identification of EZH2 and EZH1 Small Molecule Inhibitors with Selective Impact on Diffuse Large B Cell Lymphoma Cell Growth. Chem. Biol. 2013, 20, 1329–1339. [DOI] [PubMed] [Google Scholar]
- Nasveschuk C. G.; Gagnon A.; Garapaty-Rao S.; Balasubramanian S.; Campbell R.; Lee C.; Zhao F.; Bergeron L.; Cummings R.; Trojer P.; Audia J. E.; Albrecht B. K.; Harmange J.-C. P. Discovery and Optimization of Tetramethylpiperidinyl Benzamides as Inhibitors of EZH2. ACS Med. Chem. Lett. 2014, 5, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- As calculated using ChemAxon JChem for Excel software. http://www.chemaxon.com/conf/Calculating_pKa_values_of_small_and_large_molecules.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.