

Abstract

A main challenge in the development of new agents for the treatment of Pseudomonas aeruginosa infections is the identification of chemotypes that efficiently penetrate the cell envelope and are not susceptible to established resistance mechanisms. Siderophore-conjugated monocarbams are attractive because of their ability to hijack the bacteria’s iron uptake machinery for transport into the periplasm and their inherent stability to metallo-β-lactamases. Through development of the SAR we identified a number of modifications to the scaffold that afforded active anti-P. aeruginosa agents with good physicochemical properties. Through crystallographic efforts we gained a better understanding into how these compounds bind to the target penicillin binding protein PBP3 and factors to consider for future design.
Keywords: Pseudomonas aeruginosa, β-lactam, penicillin binding protein, siderophore, monocarbam, structure-guided design
One of the largest challenges in the discovery of new Gram-negative antibacterial agents is the identification of chemotypes that can penetrate the cell envelope.1 The pathogens, such as Pseudomonas aeruginosa (P. aeruginosa), have evolved complex cell architectures including an outer membrane composed of charged lipopolysaccharides, a thin peptidogylcan layer, and an inner membrane made up of phospholipids.2 Identifying molecules with the necessary features to enable permeation has been a challenge.
β-Lactams are one chemotype that have been successful in the treatment of P. aeruginosa.3 Since the discovery of penicillin, β-lactams have been used in the clinic and are still widely prescribed. One of the most effective classes of β-lactams for the treatment of P. aeruginosa infections are the carbapenems (e.g., meropenem, 1, Figure 1).4 Their effectiveness stems from their ability to acylate their penicillin-binding protein (PBP) targets, aided by high permeability into P. aeruginosa through the outer membrane porins.5 However, they are currently being challenged in the clinic by the increasing emergence of broad-spectrum serine β-lactamases and metallo-β-lactamases which hydrolyze most β-lactams and render them ineffective.6 There is one class of β-lactams that is stable to the metallo-β-lactamases: the monocyclic β-lactams represented by aztreonam (2). Aztreonam however has limited effectiveness against P. aeruginosa presumably due to its poor permeation of the outer membrane, β-lactamase susceptibility, and high propensity for efflux (P. aeruginosa MIC90 ≥ 1024 μg/mL, n = 20 panel).3,7
Figure 1.

Gram-negative active β-lactams.
To address the poor activity of 2, we focused on improving permeation by introducing siderophore mimics to promote uptake.8−10 Siderophores are small Fe-chelating molecules synthesized and secreted by bacteria to scavenge iron from the host.9,11 The iron-complexed siderophores are then brought into the cell via the iron uptake machinery, enabling the bacteria to access the much needed nutrient. It has been shown that introduction of iron-chelating groups to a monocyclic β-lactam scaffold can produce highly potent P. aeruginosa agents that are stable to metallo-β-lactamases.7,12−16
Known examples of monocyclic β-lactams incorporating iron-chelating groups are monocarbams U-78608 (3, Figure 1),16 MC-1 (4a), and MC-8 (4b).14 These compounds have been shown to be potent inhibitors of P. aeruginosa PBP3 and resistant to hydrolysis by β-lactamases.17 Some limitations to this class have been shown to be poor hydrolytic stability and high human plasma protein binding (PPB).14 Here we present our efforts to further develop the SAR of the monocarbams, exploring previously unexplored areas of the scaffold. Through crystallography efforts we were able to offer hypotheses to explain our data.
Initial studies focused on varying the side chain of the triazolone, attempting to introduce polar functionalities that could enable additional interactions with the target PBP and improve the activity. These analogs were synthesized utilizing a known three-step coupling procedure14 (Scheme 1) from β-lactam 5(14,18) and a substituted triazolone (6a–j). A subsequent two-step deprotection provided the target molecules.
Scheme 1. Synthesis of Side Chain Analogs.

(a) (i) 6a–j, MSTFA, THF; (ii) 5, CSI, DCM, 0 °C; (iii) THF, 0 °C to rt, 38–78%; (b) Pd black, H2, EtOH, 28–77%; (c) TFA, DCM, 0 °C, 29–90%.
As can be seen in Table 1, all the analogs (7a–j) demonstrated excellent P. aeruginosa MICs and PBP3 acylation rate constants equivalent to 2. These examples also demonstrated good hydrolytic stability; however, the free fraction did not increase, with observed values between 3% and 9% free.
Table 1. P. aeruginosa Antibacterial Activity and Physicochemical Properties of Siderophore-Conjugated Monocarbam Analogs.
| PAO1 MICa | PBP3ckon | human PPBd,g | hydrolytic stabilitye,g | |
|---|---|---|---|---|
| compd | (μg/mL) | (M–1 s–1) | (% free) | t1/2 (h) |
| 1 | 1 | 4.9 × 104 | NT | 95 ± 2.6 |
| 2 | 4 | 2.6 × 105 | 62 ± 8.8 | >150f |
| 3 | 0.78b | 1.7 × 105 | 5 ± 0.1 | NT |
| 4a | 0.78b | 8.1 × 104 | 6 ± 0.5 | NT |
| 4b | 0.25 | 7.2 × 104 | 4 ± 0.9 | 117 ± 17 |
| 7a | 0.13 | 3.2 × 105 | 6 ± 0.2 | 86 ± 13 |
| 7b | 0.25 | 1.7 × 105 | 9 ± 2.2 | 93 ± 12 |
| 7c | 0.25 | 6.2 × 105 | 7 ± 0.5 | 100 ± 8.5 |
| 7d | 1 | >4.0 × 105 | 3 ± 0.04 | >150f |
| 7e | 1 | 5.4 × 105 | 4 ± 0.1 | 87 ± 5.7 |
| 7f | 0.5 | 4.5 × 105 | 4 ± 0.3 | 94 ± 8.4 |
| 7g | 0.5 | 2.5 × 105 | 9 ± 0.2 | 71 ± 23 |
| 7h | 0.5 | 4.1 × 105 | 8 ± 0.1 | 61 ± 9.1 |
| 7i | 0.5 | 3.0 × 105 | 8 ± 0.2 | NT |
| 7j | <0.6 | >4.0 × 105 | NT | 102 ± 7.1 |
| 9a | 1 | 2.5 × 105 | 20 ± 9.3 | 102 ± 7.1 |
| 9b | 4 | 1.5 × 105 | 12 ± 1.0 | 82 ± 7.1 |
| 10a | 0.25 | 5.0 × 105 | 27 ± 8.7 | 85 ± 4.7 |
| 10b | 1 | 3.2 × 105 | 6 ± 1.5 | 118 ± 23 |
PAO1 = P. aeruginosa ARC545.
MICs reported in μM.
P. aeruginosa PBP3.
n = 3, standard deviation.
pH = 7.4, 37 °C.
Not determined.
NT = not tested.
Next, modifications to the iron-chelating group (siderophore) were explored to determine if there was any effect on activity or physicochemical properties. The syntheses of 1,3-dihydroxypyridin-4-one monocarbam derivatives have never been reported in the literature. However, the catechol-isostere has been shown to be optimal for other classes of monocyclic β-lactams.15,19 Synthesis of these analogs was achieved via peroxide oxidation of the protected 3-hydroxypyrid-4-one to provide the N-oxides 8a and 8b. The addition of acid was required for the oxidation because of the poor solubility of the triazolone intermediate. Subsequent coupling of the triazolones to the core 5 and deprotection (Scheme 2) provided the target molecules.
Scheme 2. Synthesis of 1,3-Dihydroxypyridin-4-one Derivatives.

(a) 6a: m-CPBA, TFA, DCM, 69%. Or 6b: 50% H2O2, AcOH/THF 1:1, 60 °C, 34%. (b) (i) MSTFA, THF; (ii) CSI, DCM, 0 °C; (iii) THF, 0 °C to rt, 24% 8a, 48% 8b; (c) Pd black, H2, EtOH, 58% 8a, 46% 8b; (d) TFA, DCM 0 °C, 54% 9a, 38% 9b.
As can be seen in Table 1 for both 9a and 9b, no change was observed in target affinity as compared to the des-hydroxy analogs 7a and 7b. Whereas with the 3-hydroxypyridin-4-one analogs, the MICs were equivalent for the different triazolone substitutions, there was differing activity for the analogs with the 1,3-dihydroxypyridin-4-one. Both 9a and 9b afforded elevated MICs, with 9b showing a 4-fold increase. One highlight of these compounds was a 3-fold improvement in free fraction for 9a, unprecedented for the triazolone class of compounds.
It has been established that for the monocyclic β-lactam and cephalosporin classes, the aminothiazole provides optimal interactions with the P. aeruginosa PBP3 enzyme; however there are known changes that are tolerated.20−22 Using known methods, we synthesized propylsulfone thiadiazole 10a and chlorothiazole 10b (Figure 2) to investigate if there would be any changes in the physicochemical properties.23 The most dramatic change observed was for thiadiazole 10a (Table 1). The P. aeruginosa MIC was similar to the parent 7b; however, the acylation rate was increased 2-fold presumably due to improved interactions of the thiadiazole with the PBP3 enzyme as compared to the thiazole. The free fraction increased 3-fold to 27% free, a 5-fold improvement in PPB as compared to the reference monocarbams 3, 4a, and 4b, highlighting that small changes can significantly affect physicochemical properties.
Figure 2.

Modified thiazole siderophore-conjugated monocarbams.
Introduction of a methyl substituent to the 4-position of the β-lactam core has been shown to increase the hydrolytic stability of the monocarbams.14,24 However, besides the trans-methyl 5, no other substitutions have been investigated in the monocarbam series. Wanting to develop this SAR further, a number of substituted β-lactam cores were targeted. The synthesis of the β-lactam cores is depicted in Scheme 3.
Scheme 3. Synthesis of 4-Substituted β-Lactam Cores.

(a) NaBH4, MeOH, 0 °C, 90%; (b) MsCl, pyridine, DCM, 0 °C to rt, 71%; (c) NaI, acetone, reflux, >95%; (d) (i) H2, 10% Pd/C, MeOH; NaOAc; (ii) 23, MeOH, rt, 50%; (e) TBSCl, imid, DMF, quant; (f) DIBAL-H, toluene, −78 °C, 88%; (g) CH2CHMgBr, DCM, −78 °C, 34%; (h) 2 N HCl, MeOH, 43%; (i) (i) 20 mol % TEMPO, NaHCO3, NaClO; (ii) MeI, K2CO3, DMF, 0 °C, 57% 17, 59% 21; (j) (i) NH2OH·HCl, KOH, MeOH, 0 °C; (ii) Ac2O, 0 °C, 56% 18, 28% 22; (k) (i) DIAD, PPh3, THF; (ii) NaHCO3, MeOH/H2O, 0 °C, 60% 18, 30% 22; (l) (i) TFA, DCM, 0 °C to rt ; (ii) 23, 4-methylmorpholine, EtOH, toluene, 13% 14b, 52% 14c, 36% 14e; (m) 10% Pd/C, H2 (1 atm); EtOH, quant 14d, quant 14f; (n) CH3NHOMe·HCl, EDC·HCl, 4-methylmorpholine, DCM, −15 °C, 76%; (o) TBSCl, imid, DMF, 0 °C, 76%; (p) CH2CHCH2MgBr, THF, −78 °C, quant; (q) NaBH4, EtOH, −78 °C, 49%.; (r) TBAF, THF, 78%.
Synthesis of the cis-analogue (3S,4R)-3-amino-4-methylazetidin-2-one 14a began with known chiral ester 11(25) which was converted to iodide 12 in three steps (Scheme 3). Pd-mediated reduction with simultaneous removal of the Cbz group afforded intermediate 13, which was treated in situ with activated ester 23 to provide the substituted β-lactam core 14a.
Synthesis of vinyl and ethyl substituted cores began with Boc-d-serine methyl ester (15). Silylation of the primary alcohol with TBSCl followed by DIBAL-H reduction of the ester and vinyl Grignard addition to the resultant aldehyde produced alcohol 16 as a ∼1:1 mixture of diastereomers. Alcohol 16 was then deprotected and oxidized to provide 17. Following procedures developed by Miller and co-workers,1817 was converted to the hydroxamate and cyclized under Mitsunobu conditions to provide β-lactam 18. The diastereomeric mixture was subsequently deprotected and condensed with activated ester 23 to afford the coupled β-lactams 14b and 14c which were separable by silica gel chromatography. The vinyl β-lactams could be reduced under hydrogenation conditions to provide the ethyl substituted core as demonstrated with β-lactam 14d.
The synthesis of allyl substituted β-lactam 14e began with d-serine-OH (19). Introduction of the allyl was achieved through allyl Grignard addition to a Weinreb amide to provide ketone 20. Alcohol 21 was synthesized as a 9:1 mixture of diastereomers (favoring the depicted isomer) in three steps. Cyclization of 21, employing the previously described conditions, and subsequent coupling with activated ester 23 afforded β-lactam 14e, which could be reduced to produce propyllactam 14f.
Assembly of the monocarbams could be achieved using one of three methods depending on the β-lactam substitution (Scheme 4). cis-Methyl 14a and cis-vinyl 14b were coupled utilizing the already described methods to give 24a and 25b after a two-step deprotection sequence (Scheme 4). For vinyl analogue 24b, after monocarbam formation, a global BCl3 deprotection was employed to prevent reduction of the olefin. This method was also utilized to couple known β-lactam core 14g.26,27 Alternatively, to avoid exposure of the fully coupled monocarbams to hydrogenation conditions, the benzyl groups on triazolone 6b could be removed prior followed by in situ treatment with MSTFA to afford the trisilylated intermediate 26. This material was not isolated but directly treated with the chlorosulfonamide intermediate of 14c–f to afford moderate yields of debenzylated coupled material (27c–f). Subsequent TFA deprotection produced the final targets (24c–f).
Scheme 4. Synthesis of Monocarbams with 4-Substituted β-Lactams.

(a) (i) 14a or 14b, MSTFA, THF; (ii) 6b, CSI, DCM, 0 °C; (iii) THF, 0 °C − rt; (b) Pd black, H2, EtOH; (c) TFA, DCM 0 °C; (d) BCl3, DCM; (e) (i) Pd black, H2 (1 atm), THF; (ii) MSTFA, THF; (f) (i) 5, CSI, DCM, 0 °C; (ii) THF, 0 °C − rt.
The data for these analogs can be seen in Table 2. For the alkyl-substituted β-lactams the observed SAR indicated the trans-β-lactams were favored. We observed higher acylation rate constants and better MICs for all the trans-alkyl substituted β-lactams (kon ≥ 1.7 × 105 M–1 s–1) as compared to the cis (compare 7b to 24a, 24c to 24b, 24d to 25b). In fact, as the cis-substituent became larger, the acylation rate decreased. For example, the acylation rate constants for the cis-allyl (24e) and cis-propyl (24f) derivatives were 200-fold lower. The lower affinity for the enzyme is reflected in higher P. aeruginosa MICs for these analogs.
Table 2. Antibacterial Activity and Hydrolytic Stability of C4-Substituted β-Lactam Analogs.
| PAO1aMIC | PBP3bkon | hydrolytic stabilityc | ||
|---|---|---|---|---|
| compd | (μg/mL) | (M–1 s–1) | t1/2 (h) | |
| 7b | trans-methyl | 0.5 | 1.7 × 105 | 93 ± 12 |
| 24a | cis-methyl | 1 | 1.6 × 104 | 76 ± 6.8 |
| 24b | cis-vinyl | 2 | 1.9 × 104 | 38 ± 4.1 |
| 25b | cis-ethyl | 1 | 8.8 × 103 | 94 ± 13 |
| 24c | trans-vinyl | 0.5 | 2.1 × 105 | 53 ± 2.8 |
| 24d | trans-ethyl | 0.5 | 3.6 × 105 | 98 ± 12 |
| 24e | cis-allyl | 8 | 5.9 × 103 | 126 ± 25 |
| 24f | cis-propyl | 4 | 7.4 × 103 | >150d |
| 24g | dimethyl | 8 | 1.1 × 104 | >150d |
PAO1 = P. aeruginosa ARC545.
P. aeruginosa PBP3.
pH = 7.4, 37 °C.
Not determined.
The stereochemistry had no effect on the hydrolytic stability of these compounds. The data indicated the stability was driven by the size and activating ability of the substituent. The stereochemical matched pairs (7b to 24a, 24c to 24b, 24d to 25b) had about equivalent stability. The least stable pair was the vinyl substituted cores possibly because the vinyl group provides additional activation of the β-lactam. As the steric congestion on the core increased, the hydrolytic half-life increased, as can be seen with cis-propyl 24f and disubstituted core 24g which did not exhibit any hydrolysis under the assay conditions. These stability trends were the same as what has been observed with monocyclic-β-lactams in relation to β-lactamase stability.28
To gain further insight into these SAR trends, we obtained the crystal structures of P. aeruginosa PBP3 with two trans-substituted cores, 7b and 24c, and the cis-substituted 24e (Figure 3). All three compounds were covalently bound to residue Ser294 of PBP3. Expectedly, the binding modes of these compounds are similar (Figure 3). The aminothiazole containing left-hand side of each analog is oriented equivalently with the thiazole, oxyiminopropylcarboxylic acid, and amide substituents assuming the same orientation. As observed previously with other crystallographic structures of P. aeruginosa PBP3 in complex with aminothiazole containing β-lactams, the aminothiazole group forms multiple interactions with the protein.13,29,30 In addition, the amide carbonyl of these compounds formed a hydrogen bond with residue Asn351, and an ordered water molecule. The right-hand sides of these compounds, consisting of the triazolone and siderophore moieties, also show a consistent binding mode. The triazolone carbonyl interacts with the backbone amide of residue Gly535. The propylmethylsulfone side chain is directed toward the solvent and forms limited interactions with the protein. Although the sulfone oxygens form a hydrogen bond with a conserved water molecule, no additional interactions are made with the protein. These limited interactions may explain the similar PBP3 acylation rate constants observed with analogs 7a–j. The siderophore group is also oriented toward solvent and forms limited direct interactions with the PBP3 enzyme, which is further supported by the observation that that there is no difference in acylation rate constants between the 3-hydroxypyridin-4-ones (7a and 7b) and 1,3-dihydroxypyridin-4-ones (9a and 9b).
Figure 3.
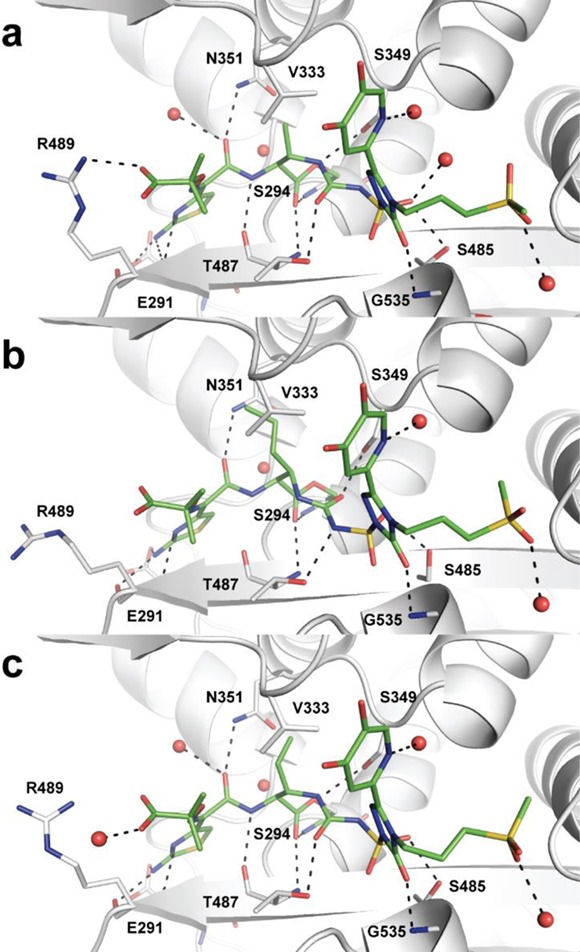
Binding site for liganded P. aeruginosa PBP3 complexes: (a) 7b; (b) 24e; (c) 24c. Compounds are shown as stick models with green carbon atoms, blue nitrogen atoms, red oxygen atoms, and yellow sulfur atoms. All residues within ∼3.2 Å of the covalently bound compound are shown. Water molecules that coordinate additional interactions are shown as red spheres. Hydrogen bonds are depicted as black dashes.
Though a number of similarities are observed among 7b, 24c, and 24e, some distinct differences between the crystal structures were observed. SAR for the monocyclic β-lactams indicates the oxyiminopropylcarboxylic acid is important for P. aeruginosa activity, and most structures indicate an interaction with residue Arg489.29 It is of note that for cis-allyl analog 24e, the Arg is rotated away and no such interaction exists. This could be a reason for the lower affinity of the allyl analog. However, the electron density in this region is poorly defined.
Another key difference observed was in the orientation of the urea of the acylsulfonamide. Trans-analogs 7b and 24c have similar orientations with the urea carbonyl forming a hydrogen bond with the amide residue of Thr487, and the nitrogen from the β-lactam ring also makes a hydrogen bond with the hydroxyl of residue Ser349. The C4 substitutents (methyl and vinyl) are oriented in such a way as to allow van der Waals interactions with residue Val333. The allyl analog 24e is bound with the urea portion of the molecule in a flipped orientation as compared to the trans-compounds. In this case the β-lactam nitrogen forms a hydrogen bond with residue Thr487 and the urea carbonyl is hydrogen bonded to the hydroxyl of residue Ser349. Presumably the flipped orientation is a result of preventing a steric clash between the allyl substituent and Val333.
It is noted that 4a which has no C4 substituent binds in the same orientation as the cis-allyl analog 24e and also forms hydrogen bonds with residues Thr487 and Ser349 (Figure 4).29 However, 4a has a 10-fold better acylation rate constant than 24e (8.1 × 104 h versus 5.9 × 103 h). This indicates the urea orientation is not a reflection of good affinity for PBP3 but just the preferred conformation driven by the stereochemistry of the substituents on the core. Instead a contribution to the lower acylation rate constant of 24e could be a steric clash with the protein in the initial encounter complex, prior to acylation.
Figure 4.
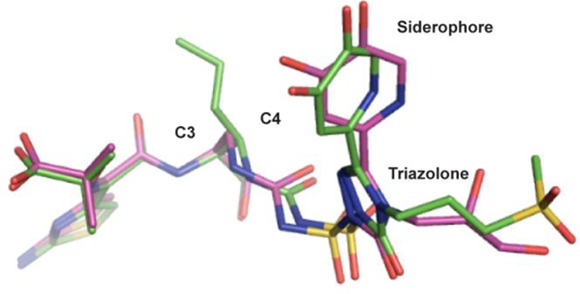
Overlay of P. aeruginosa PBP3 crystal structures of 4a and 24e.
Further profiling of the best compounds indicates the monocarbams demonstrate excellent pharmacokinetic (PK) properties, similar to other classes of β-lactam antibiotics (Table 3).31
Table 3. Intravenous Rat PK of Selected Monocarbamsa.
| compd |
|||||
|---|---|---|---|---|---|
| 7a | 7b | 10a | 24c | 24d | |
| rat CL (mL min–1 kg–1) | 8.9 | 7.3 | 9.5 | 11 | 12 |
| t1/2 (h) | 2.5 | 3.0 | 2.0 | 2.0 | 2.0 |
| Vss (L/kg) | 0.23 | 0.21 | 0.26 | 0.18 | 0.35 |
Han Wistar rat (male), 1 mg/kg, n = 2.
Compound 7a has been shown to have a P. aeruginosa MIC90 equal to 1 μg/mL (n = 20) and to be efficacious against multiple P. aeruginosa strains in vivo.32 Profiling of the resistance mechanisms indicates the monocarbam utilizes the siderophore receptors for uptake. Details of these studies will be reported elsewhere.
In conclusion, through our synthetic efforts we have explored and developed the SAR of the triazolone-based siderophore-conjugated monocarbams. Introduction of sulfone and sulfonamide side chains afforded highly potent and stable inhibitors of P. aeruginosa; however, there was no increase in free fraction. Through modifications of the siderophore and thiazole moieties, compounds with free fractions of >20% were produced. β-Lactams with substituted cores indicated trans-substituted β-lactam cores are favored for efficient inhibition of P. aeruginosa PBP3, and the hydrolytic stability of the compounds is controlled by the size and electronics of the substitution. Cocrystal structures of three analogs covalently bound to P. aeruginosa PBP3 were obtained. We observed some key differences between the structure of allyl analog 24e and the structures of the methyl and vinyl analogs 7b and 24c, including lack of a salt bridge between Arg489 and the oxyiminopropylcarboxylic acid and a flipped conformation of the urea of the acylsulfonamide. By comparison with the structure of 4a, we concluded the final orientation of the urea has no relation to the acylating efficiency of the molecule and instead is driven by the stereochemistry of the β-lactam substituent. These results highlight the challenges of structure-based drug design with covalent inhibitors, since the final complex does not reflect the initial encounter complex which is most likely the major factor in determining enzyme affinity.
Acknowledgments
We thank Camil Joubran and Sharon Tentarelli for generating characterization data; Sara Patey, Robert Giacobbe, Linda Otterson, and Marie Potter for generating MIC data; Adam Shapiro for support with the PBP3 acylation assay; D. Krishnaswamy and his colleagues at Anthem Biosciences Pvt, Ltd. for the synthesis of intermediates; Baolei Zheng and his colleagues at Pharmaron for synthesis of intermediates and final compounds.
Glossary
Abbreviations
- PBP3
penicillin binding protein 3
- MSTFA
N-methyl-N-(trimethylsilyl)trifluoroacetamide
- THF
tetrahydrofuran
- CSI
chlorosulfonyl isocyanate
- DCM
dichloromethane
- TFA
trifluoroacetic acid
- m-CPBA
3-chloroperbenzoic acid
- MsCl
methanesulfonyl chloride
- TBSCl
tert-butyldimethylsilyl chloride
- DMF
N,N-dimethylformamide
- imid
imidazole
- DIBAL-H
diisobutylaluminum hydride
- TEMPO
2,2,6,6-tetramethylpiperidine 1-oxyl
- DIAD
diisopropyl azodicarboxylate
- EDC·HCl
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- TBAF
tetrabutylammonium fluoride
- CL
clearance
- Vss
volume of distribution at steady state
Supporting Information Available
Crystallography details, P. aeruginosa PBP3 acylation assay, hydrolytic stability assay, microbiology assays, pharmacokinetic studies, synthesis of compounds 7a–j, 9a, 9b, 10a, 10b, 24a–g, 25b. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ K.E.M.-B.: Moderna Therapeutics, 200 Technology Square, Cambridge, MA 02139, U.S.
Author Present Address
⊥ B.D.: Life Sciences Solutions, Thermo Scientific, Inc., 5791 Van Allen Way, Carlsbad, CA 92008, U.S.
Author Present Address
# E.L.M.: Agios Pharmaceuticals, 38 Sidney St., Cambridge, MA 02139, U.S.
Author Present Address
∇ M.Z.: Novartis Institutes for BioMedical Research, Inc., 250 Massachusetts Avenue, Cambridge, MA 02139, U.S.
The authors declare no competing financial interest.
Supplementary Material
References
- Silver L. L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy T. J.; Kahne D.; Walker S. The bacterial cell envelope. Cold Spring Harbor Perspect. Biol. 2010, 2, a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K.; Macielag M. J. New beta-lactam antibiotics and beta-lactamase inhibitors. Expert Opin. Ther. Pat. 2010, 20, 1277–1293. [DOI] [PubMed] [Google Scholar]
- Rossolini G. M.; Mantengoli E. Treatment and control of severe infections caused by multiresistant Pseudomonas aeruginosa. Clin. Microbiol. Infect. 2005, 11 (Suppl. 4), 17–32. [DOI] [PubMed] [Google Scholar]
- Nikaido H.; Vaara M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985, 49, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. F.; Meroueh S. O.; Mobashery S. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [DOI] [PubMed] [Google Scholar]
- Sykes R. B.; Koster W. H.; Bonner D. P. The new monobactams: chemistry and biology. J. Clin. Pharmacol. 1988, 28, 113–119. [DOI] [PubMed] [Google Scholar]
- Mislin G. L.; Schalk I. J. Siderophore-dependent iron uptake systems as gates for antibiotic Trojan horse strategies against Pseudomonas aeruginosa. Metallomics 2014, 6, 408–420. [DOI] [PubMed] [Google Scholar]
- Braun V.; Hantke K. Recent insights into iron import by bacteria. Curr. Opin. Chem. Biol. 2011, 15, 328–334. [DOI] [PubMed] [Google Scholar]
- Foley T. L.; Simeonov A. Targeting iron assimilation to develop new antibacterials. Expert Opin. Drug Discovery 2012, 7, 831–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N.; Guillier M.; Barnard T. J.; Buchanan S. K. TonB-dependent transporters: Regulation, structure, and function. Annu. Rev. Microbiol. 2010, 64, 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr J.; Brown M. F.; Aschenbrenner L.; Caspers N.; Che Y.; Gerstenberger B. S.; Huband M.; Knafels J. D.; Lemmon M. M.; Li C.; McCurdy S. P.; McElroy E.; Rauckhorst M. R.; Tomaras A. P.; Young J. A.; Zaniewski R. P.; Shanmugasundaram V.; Han S. Siderophore receptor-mediated uptake of lactivicin analogues in gram-negative bacteria. J. Med. Chem. 2014, 57, 3845–3855. [DOI] [PubMed] [Google Scholar]
- Brown M. F.; Mitton-Fry M. J.; Arcari J. T.; Barham R.; Casavant J.; Gerstenberger B. S.; Han S.; Hardink J. R.; Harris T. M.; Hoang T.; Huband M. D.; Lall M. S.; Lemmon M. M.; Li C.; Lin J.; McCurdy S. P.; McElroy E.; McPherson C.; Marr E. S.; Mueller J. P.; Mullins L.; Nikitenko A. A.; Noe M. C.; Penzien J.; Plummer M. S.; Schuff B. P.; Shanmugasundaram V.; Starr J. T.; Sun J.; Tomaras A.; Young J. A.; Zaniewski R. P. Pyridone-conjugated monobactam antibiotics with gram-negative activity. J. Med. Chem. 2013, 56, 5541–5552. [DOI] [PubMed] [Google Scholar]
- Flanagan M. E.; Brickner S. J.; Lall M.; Casavant J.; Deschenes L.; Finegan S. M.; George D. M.; Granskog K.; Hardink J. R.; Huband M. D.; Hoang T.; Lamb L.; Marra A.; Mitton-Fry M.; Mueller J. P.; Mullins L. M.; Noe M. C.; O’Donnell J. P.; Pattavina D.; Penzien J. B.; Schuff B. P.; Sun J.; Whipple D. A.; Young J.; Gootz T. D. Preparation, Gram-negative antibacterial activity, and hydrolytic stability of novel siderophore-conjugated monocarbam diols. ACS Med. Chem. Lett. 2011, 2, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page M. G.; Dantier C.; Desarbre E. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant gram-negative bacilli. Antimicrob. Agents Chemother. 2010, 54, 2291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbachyn M. R.; Tuominen T. C. Synthesis and structure-activity relationships of monocarbams leading to U-78608. J. Antibiot. (Tokyo) 1990, 43, 1199–1203. [DOI] [PubMed] [Google Scholar]
- McPherson C. J.; Aschenbrenner L. M.; Lacey B. M.; Fahnoe K. C.; Lemmon M. M.; Finegan S. M.; Tadakamalla B.; O’Donnell J. P.; Mueller J. P.; Tomaras A. P. Clinically relevant Gram-negative resistance mechanisms have no effect on the efficacy of MC-1, a novel siderophore-conjugated monocarbam. Antimicrob. Agents Chemother. 2012, 56, 6334–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woulfe S. R.; Miller M. J. The synthesis of substituted 3(S)-(acylamino)-2-oxo-1-azetidinyl]thio]acetic acids. J. Org. Chem. 1986, 51, 3133–3139. [Google Scholar]
- Mitton-Fry M. J.; Arcari J. T.; Brown M. F.; Casavant J. M.; Finegan S. M.; Flanagan M. E.; Gao H.; George D. M.; Gerstenberger B. S.; Han S.; Hardink J. R.; Harris T. M.; Hoang T.; Huband M. D.; Irvine R.; Lall M. S.; Megan Lemmon M.; Li C.; Lin J.; McCurdy S. P.; Mueller J. P.; Mullins L.; Niosi M.; Noe M. C.; Pattavina D.; Penzien J.; Plummer M. S.; Risley H.; Schuff B. P.; Shanmugasundaram V.; Starr J. T.; Sun J.; Winton J.; Young J. A. Novel monobactams utilizing a siderophore uptake mechanism for the treatment of gram-negative infections. Bioorg. Med. Chem. Lett. 2012, 22, 5989–5994. [DOI] [PubMed] [Google Scholar]
- Breuer H.; Cimarusti C. M.; Denzel T.; Koster W. H.; Slusarchyk W. A.; Treuner U. D. Monobactams—structure-activity relationships leading to SQ 26,776. J. Antimicrob. Chemother. 1981, 8 (Suppl. E), 21–28. [DOI] [PubMed] [Google Scholar]
- Kawabata K.; Yamanaka H.; Takasugi H.; Takaya T. Studies on Î2-lactam antibiotics. XIII. Synthesis and structure-activity relationships of 7Î2-(Z)-2-aryl-2-carboxymethoxyiminoacetamido]-3-vinylcephalosporins. J. Antibiot. 1986, 39, 404–14. [DOI] [PubMed] [Google Scholar]
- Yamanaka H.; Takasugi H.; Masugi T.; Kochi H.; Miyai K.; Takaya T. Studies on Î2-lactam antibiotics. VIII. Structure-activity relationships of 7Î2-(Z)-2-carboxymethoxyimino-2-arylacetamido]-3-cephem-4-carboxylic acids. J. Antibiot. 1985, 38, 1068–76. [DOI] [PubMed] [Google Scholar]
- Brown M. F.; Mitton-Fry M. J.; Han S.; Lall M.; Plimmer M. S.; Hud L.; Shanmugasundaram V.; Starr J.. Monobactams. WO 2012/073138, November 29, 2010.
- Mulchande J.; Martins L.; Moreira R.; Archer M.; Oliveira T. F.; Iley J. The efficiency of C-4 substituents in activating the beta-lactam scaffold towards serine proteases and hydroxide ion. Org. Biomol. Chem. 2007, 5, 2617–2626. [DOI] [PubMed] [Google Scholar]
- Sendai M.; Hashiguchi S.; Tomimoto M.; Kishimoto S.; Matsuo T.; Kondo M.; Ochiai M. Chemical modification of sulfazecin. Synthesis of 4-(substituted methyl)-2-azetidinone-1-sulfonic acid derivatives. J. Antibiot. (Tokyo) 1985, 38, 346–371. [DOI] [PubMed] [Google Scholar]
- Slusarchyk W. A.; Dejneka T.; Gougoutas J.; Koster W. H.; Kronenthal D. R.; Malley M.; Perri M. G.; Routh F. L.; Sundeen J. E.; et al. Î2-Lactam synthesis: chemospecific sulfonation and cyclization of the Î2-hydroxyvaline nucleus. Tetrahedron Lett. 1986, 27, 2789–92. [Google Scholar]
- Mattingly P. G.; Miller M. J. Titanium trichloride reduction of substituted N-hydroxy-2-azetidinones and other hydroxamic acids. J. Org. Chem. 1980, 45, 410–15. [Google Scholar]
- Cimarusti C. M.; Sykes R. B. Monocyclic Î2-lactam antibiotics. Med. Res. Rev. 1984, 4, 1–24. [DOI] [PubMed] [Google Scholar]
- Han S.; Zaniewski R. P.; Marr E. S.; Lacey B. M.; Tomaras A. P.; Evdokimov A.; Miller J. R.; Shanmugasundaram V. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 22002–22007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr J.; Brown M. F.; Aschenbrenner L.; Caspers N.; Che Y.; Gerstenberger B. S.; Huband M.; Knafels J. D.; Lemmon M. M.; Li C.; McCurdy S. P.; McElroy E.; Rauckhorst M. R.; Tomaras A. P.; Young J. A.; Zaniewski R. P.; Shanmugasundaram V.; Han S. Siderophore receptor-mediated uptake of lactivicin analogues in gram-negative bacteria. J. Med. Chem. 2014, 57, 3845–3855. [DOI] [PubMed] [Google Scholar]
- Turnidge J. D. The pharmacodynamics of beta-lactams. Clin. Infect. Dis. 1998, 27, 10–22. [DOI] [PubMed] [Google Scholar]
- Kim A.; Crandon J.; Gorseth E.; Blinn C.; Patey S.; Kutschke A.; Chen A.; Rooney M.; Benenato K.; Ehmann D.; Eakin A.; Nicolau D.. In vivo pharmacodynamics of a siderophore-conjugated monocarbam in Pseudomonas aeruginosa (PA): Assessing the risk of attenuated efficacy. Abstracts of Papers, Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), Washington DC, Sep 5–9, 2014; American Society of Microbiology: Washington, DC, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.