

Abstract

We report the discovery of a benzimidazole series of CYP11B2 inhibitors. Hit-to-lead and lead optimization studies identified compounds such as 32, which displays potent CYP11B2 inhibition, high selectivity versus related CYP targets, and good pharmacokinetic properties in rat and rhesus. In a rhesus pharmacodynamic model, 32 produces dose-dependent aldosterone lowering efficacy, with no apparent effect on cortisol levels.
Keywords: aldosterone synthase, CYP11B2, hypertension
Aldosterone is a steroid hormone that promotes increased blood pressure, inflammation, fibrosis, and organ damage.1−4 The final three rate-limiting steps of its biosynthesis are catalyzed by the mitochondrial cytochrome P450 enzyme aldosterone synthase (CYP11B2). Inhibition of CYP11B2 should lead to lower circulating plasma aldosterone levels, and thus may be an effective strategy for the treatment of a variety of ailments, including hypertension and heart failure.
Biological targets that are structurally related to CYP11B2 include CYP11B1 (>93% homology), and CYPs 17, 19, and 21. CYP11B1 catalyzes the biosynthesis of cortisol, an important regulator of glucose metabolism, while CYPs 17, 19, and 21 catalyze the formation of hormones such as estrone and testosterone. To be used safely in the clinic, CYP11B2 inhibitors need to display high selectivity versus these related targets, as well as the major hepatic CYP enzymes.
A variety of small molecule CYP11B2 inhibitors have recently been reported in the literature.5,6 One of these, LCI-699, has been shown to lower aldosterone levels and blood pressure in the clinic, thereby validating this mechanism as a treatment for hypertension.7−9 In cell-based in vitro assays, LCI-699 inhibits CYP11B2 with only modest, 4-fold selectivity vs CYP11B1. In the clinic, LCI-699 produces an undesired, dose-limiting impairment of cortisol response, presumably as a result of its CYP11B1 inhibition. Clinical candidates that inhibit CYP11B2 more selectively than LCI-699 are thus desired as improved antihypertensive therapies.
Our goal is to discover potent and selective CYP11B2 inhibitors as treatments for hypertension. Toward that end, we initiated screening of the Merck sample collection and identified benzimidazole 1 (Table 1) as a starting point for optimization studies. The modification of 1, described herein, has led to the discovery of potent benzimidazole CYP11B2 inhibitors that display high selectivity vs related CYPs, drug-like pharmacokinetic (PK) and physiochemical properties, and robust aldosterone lowering in a rhesus pharmacodynamic model.
Table 1. Effect of Benzimidazole N-Substitution on CYP11B2 and B1 Inhibition and LLE.
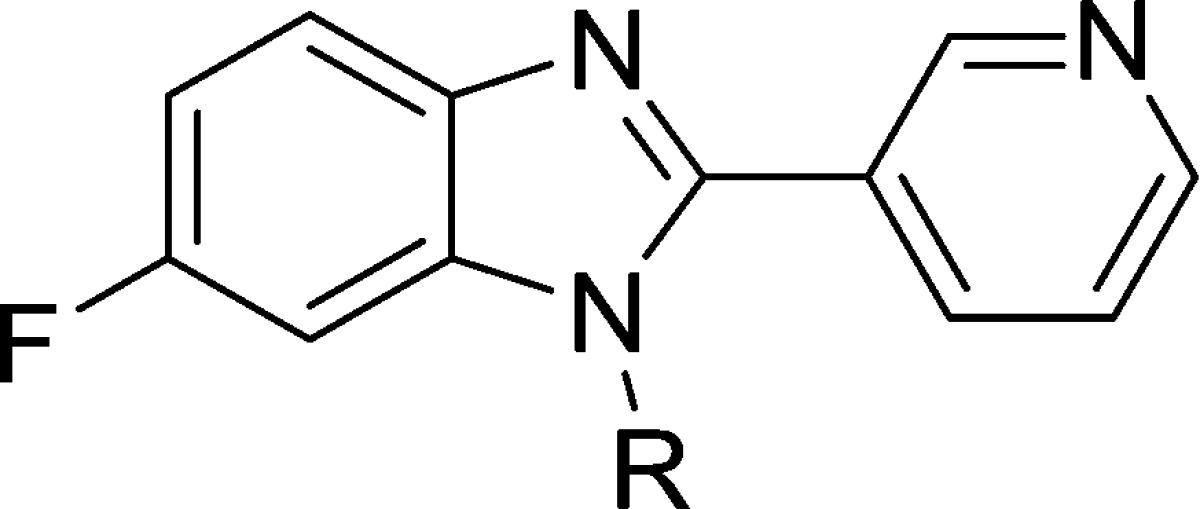
| compd | R | CYP11B2a (IC50, nM) | CYP11B1a (IC50, nM) | B1/B2b | LLEc |
|---|---|---|---|---|---|
| 1 | Me | 12 | 677 | 56 | 5.39 |
| 2 | H | >10000 | 1987 | <1 | n.d. |
| 3 | Et | 35 | 2101 | 60 | 4.57 |
| 4 | i-Pr | 138 | 3923 | 28 | 3.60 |
| 5 | c-Pr | 4 | 401 | 100 | 5.38 |
| 6 | (Me)c-Pr | 16 | 2857 | 179 | 4.57 |
| 7 | t-Bu | >5000 | >10000 | n.d. | n.d. |
| 8 | c-Bu | >5000 | >10000 | n.d. | n.d. |
| 9 | CH2CF3 | 561 | >8333 | >15 | 2.71 |
| 10 | Bn | 607 | >8333 | >14 | 2.10 |
Assay details reported in Supporting Information.
Ratio of hCYP11B1 IC50/hCYP11B2 IC50.
Ligand lipophilic efficiency; LLE = pIC50 – a Log P98.
Compounds were synthesized as described in the Supporting Information section, and were tested in cell-based assays for inhibition of human CYP11B2 and CYP11B1. The preparation of these and related compounds has also been reported previously.10
Taking screening hit 1 as our starting point, we synthesized a series of compounds wherein the effects of benzimidazole N-substitution were examined. Compounds in this series were optimized with respect to CYP11B2 potency, selectivity vs CYP11B1 and other CYPs, lipophilic ligand efficiency (LLE), and PK properties.11 As shown in Table 1, screening hit 1 is a potent CYP11B2 inhibitor that displays moderate, 56-fold selectivity vs CYP11B1. In contrast, the unsubstituted parent compound in this series (compound 2, R = H) shows no inhibition of CYP11B2. When compared to 1, compounds bearing larger acyclic N-alkyl substituents such as ethyl (compound 3) or isopropyl (compound 4) are less active, with potency decreasing as the size of the N-substituent increases. Interestingly, N-cyclopropyl analog 5 is considerably more potent than the closely related isopropyl analog 4, suggesting that the cyclopropyl group may participate in a beneficial π-type interaction with the protein. Compounds 6 and 7 show an analogous relationship, with the (methyl)cyclopropyl analog 6 exhibiting much more potent CYP11B2 inhibition than the sterically similar tert-butyl analog 7. Finally, larger analogs that incorporate a methylene spacer such as 9 and 10 are also less potent when compared with truncated analogs such as 1 or 3. Taken together, these data suggest that binding space at this position is quite limited and that a small, cyclopropyl-containing group provides an optimal solution for potency, B2/B1 selectivity, and LLE.
Selected compounds from this set displayed promising profiles in a rat pharmacokinetic assay.12 As shown in Table 2, screening hit 1 exhibited moderate oral bioavailability and high plasma clearance. In comparison, cyclopropyl analog 5 displayed reduced plasma clearance and significantly higher oral bioavailability and exposure. As the intrinsic clearance rates of 1 and 5 are essentially identical, the reduced plasma clearance of 5 may derive from its increased binding to plasma protein. When compared to 5, methylcyclopropyl analog 6 showed significantly higher intrinsic clearance, suggesting that the additional methyl group may be a primary site of metabolic oxidation. From the initial set of compounds shown in Table 1, cyclopropyl analog 5 appeared to provide the best combination of PK profile, CYP11B2 inhibition, B2/B1 selectivity, and LLE, and the cyclopropyl substituent was thus selected for incorporation in subsequent analogs.
Table 2. Effect of Benzimidazole N-Substitution on Rat Pharmacokinetic Profilea.
| compd | PPBb | Mic Clintc | F (%)d | AUCN (po)e | Clpf |
|---|---|---|---|---|---|
| 1 | 25 | 30 | 37 | 0.56 | 59 |
| 5 | 15 | 28 | 77 | 1.82 | 28 |
| 6 | 19 | 82 | 32 | 0.20 | 131 |
Assay details reported in Supporting Information.
Protein binding in rat plasma, reported as % free compound.
Intrinsic clearance in rat liver microsomes (mL/min/kg).
Bioavailability.
Area under the curve, normalized for dose (μM·h·kg/mg).
Plasma clearance (mL/min/kg).
With an improved N-substituent in place, we next optimized substitution at each position on the benzimidazole phenyl ring. Because the initial screening hit 1 was fluorinated, we began by substituting fluorine at each ring position (Table 3). Of the monofluorinated derivatives 12, 13, 5, and 14, the 6-fluoro analog 5 displayed CYP11B2 inhibition and LLE that were significantly improved relative to both the other monofluorinated compounds and to the parent, unsubstituted compound 11. A broader examination of substitution at the 5-position revealed that larger lipophilic groups such as trifluoromethyl (compound 15) as well as a variety of polar groups (compounds 16–18) were all poorly tolerated. At the 6-position, incorporation of larger lipophilic groups like trifluoromethyl (compound 20) again afforded compounds with reduced potency and LLE. However, small polar groups such as cyano (compound 21) and methoxy (compound 22) were better tolerated here than at the 5-position, with cyano in particular providing a potent and selective compound. Of the multiple difluorinated derivatives that were prepared, the 5,6-difluoro analog 23 offered the best combination of CYP11B2 inhibition, B2/B1 selectivity, and LLE (other difluoro compounds not shown). Finally, several other disubstituted analogs were synthesized, and of those, the highly potent 5-fluoro-6-chloro derivative 24 displayed optimal B2/B1 selectivity, while the 5-fluoro-6-cyano compound 25 exhibited optimal LLE.
Table 3. Effect of Benzimidazole Substitution on CYP11B2 and B1 Inhibition and LLE.
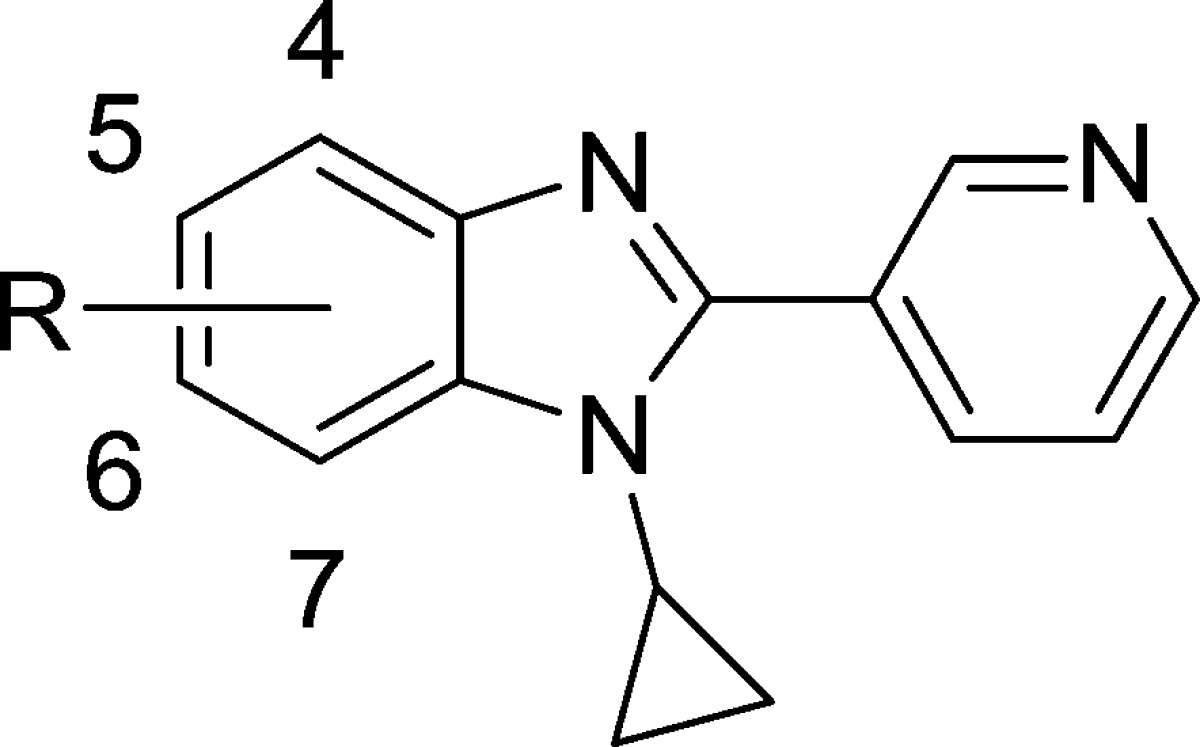
| compd | R | CYP11B2a (IC50, nM) | CYP11B1a (IC50, nM) | B1/B2b | LLEc |
|---|---|---|---|---|---|
| 11 | H | 49 | >8333 | >170 | 4.49 |
| 12 | 4-F | 219 | >8333 | >38 | 3.64 |
| 13 | 5-F | 28 | 3984 | 142 | 4.53 |
| 5 | 6-F | 3.8 | 400 | 105 | 5.38 |
| 14 | 7-F | 107 | >10000 | >93 | 3.95 |
| 15 | 5-CF3 | >10000 | >10000 | n.d. | n.d. |
| 16 | 5-OCH3 | >1000 | >10000 | n.d. | n.d. |
| 17 | 5-CN | 1768 | >10000 | >6 | 3.06 |
| 18 | 5-SO2CH3 | >1000 | >10000 | n.d. | n.d. |
| 19 | 6-Cl | 2.2 | 477 | 215 | 5.18 |
| 20 | 6-CF3 | 254 | 1831 | 7 | 2.84 |
| 21 | 6-CN | 1.2 | 374 | 309 | 6.22 |
| 22 | 6-OCH3 | 24 | 1980 | 83 | 4.82 |
| 23 | 5,6-di-F | 2.1 | 370 | 176 | 5.45 |
| 24 | 5-F-6-Cl | 0.4 | 334 | 835 | 5.71 |
| 25 | 5-F-6-CN | 0.6 | 117 | 195 | 6.32 |
Assay details reported in Supporting Information.
Ratio of hCYP11B1 IC50/hCYP11B2 IC50.
Ligand lipophilic efficiency; LLE = pIC50 – a Log P98.
With refinements to the benzimidazole core complete, we conducted a final round of optimization on the pyridine ring, focusing our efforts on reducing clearance and further improving PK in this series. Metabolite identification studies of partly optimized compounds such as 5 had implicated the pyridine ring as the primary site of oxidative metabolism. In an attempt to block any pyridyl N-oxidation, we substituted each position adjacent to the pyridine nitrogen, but found that incorporation of even small groups such as methyl at these positions resulted in a complete loss of potency, presumably due to interference with binding of the pyridine to the CYP11B2 heme (results not shown). Given that, we focused subsequent optimization efforts at positions nonadjacent to the pyridine nitrogen (R3 and R4 in Table 4). In an effort to reduce metabolic oxidation, we introduced electron-withdrawing groups that would not only block those positions specifically but also deactivate the ring generally, as well as sterically bulky groups that could block access to the ring. As shown in Table 4, incorporation of an electron-withdrawing fluorine at R4 was tolerated (compound 26) and yielded a compound that was nearly as potent as the unsubstituted parent 23 despite its presumably diminished capacity to bind the CYP heme. Other electron-withdrawing groups such as nitrile (compound 27) were somewhat less well tolerated, while electron donating groups such as methoxy (compound 28) imparted improved potency and selectivity. Comparing compounds 26, 29, and 30, one sees that conversion of the R2 substituent from fluorine (26) to chlorine (29) or nitrile (30) again improved potency and LLE in the series where R4 = fluorine. Introduction of a bulky tertiary alcohol group at R4 as in 31 generated improvements in potency, LLE, and clearance (see Table 6 for effects on PK). In the tertiary alcohol series, fluorination at R1 provided a nearly 10-fold boost in potency as well as a gain in LLE (compare compounds 31 and 32). Moving around the ring, substitution of the R3 position with methyl (compound 33) or trifluoromethyl (compound 34) provided compounds that were only slightly less potent than parent 23. Additional substitution with fluorine at R4 afforded compound 35, which was 20-fold more potent and nearly 10-fold more selective than 33. Interestingly, incorporation of fluorine at R4 yielded far greater benefit when R3 was also substituted with methyl than when it was unsubstituted (compare 33 and 35 versus 23 and 26), suggesting that the R3 methyl might alter the dihedral angle about the benzimidazole–pyridine bond, thus altering the binding pocket environment into which the fluorine is projected. Substitution of both R3 and R4 with methyl provided a very potent CYP11B2 inhibitor (compound 36); subsequent conversion of the R3 methyl to methoxy (compound 37) then produced a compound nearly as potent and also 10-fold more B2/B1 selective.
Table 4. Effect of Pyridine Substitution on CYP11B2 and B1 Inhibition and LLE.
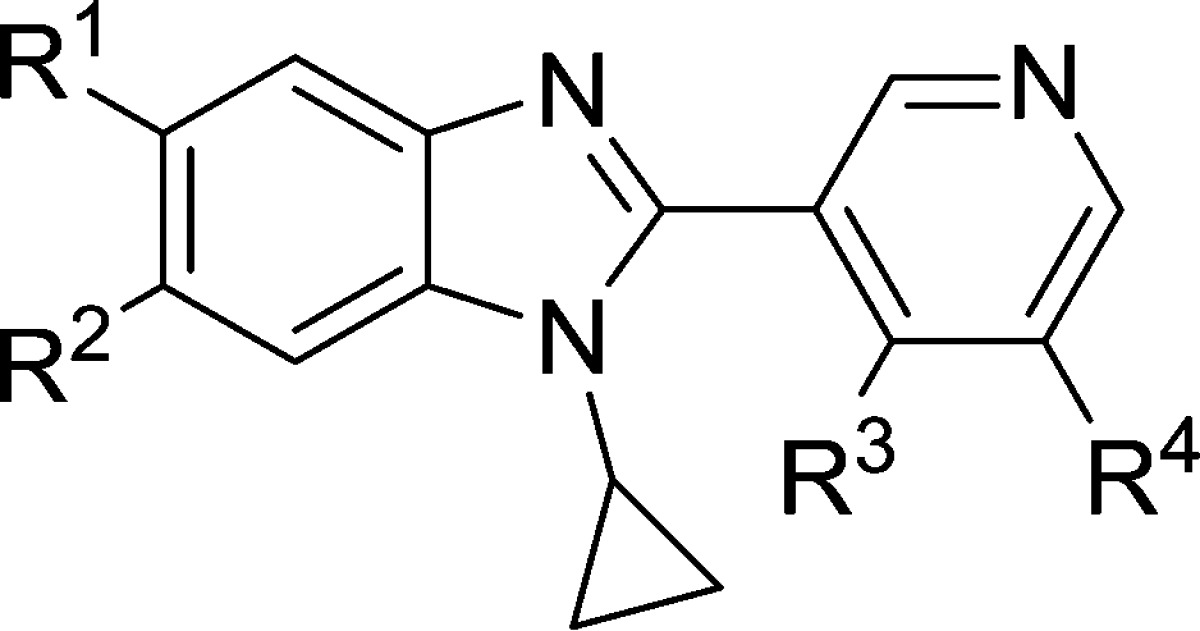
| compd | R1 | R2 | R3 | R4 | CYP11B2a (IC50, nM) | CYP11B1a (IC50, nM) | B1/B2b | LLEc |
|---|---|---|---|---|---|---|---|---|
| 23 | F | F | H | H | 2.1 | 370 | 176 | 5.45 |
| 26 | F | F | H | F | 4.7 | 435 | 93 | 5.01 |
| 27 | F | F | H | CN | 22.3 | 1552 | 70 | 4.54 |
| 28 | F | F | H | OCH3 | 0.4 | 250 | 606 | 6.12 |
| 29 | F | Cl | H | F | 0.8 | 251 | 296 | 5.20 |
| 30 | F | CN | H | F | 1.0 | 225 | 225 | 5.89 |
| 31 | F | F | H | C(CH3)2OH | 0.3 | 42 | 140 | 6.32 |
| 32 | H | F | H | C(CH3)2OH | 2.3 | 142 | 71 | 5.70 |
| 33 | F | F | CH3 | H | 10.2 | 723 | 71 | 4.28 |
| 34 | F | F | CF3 | H | 8.1 | 795 | 98 | 3.92 |
| 35 | F | F | CH3 | F | 0.5 | 258 | 553 | 5.38 |
| 36 | F | F | CH3 | CH3 | 0.2 | 17.7 | 74 | 5.50 |
| 37 | F | F | OCH3 | CH3 | 0.6 | 435 | 709 | 5.52 |
Assay details reported in Supporting Information.
Ratio of hCYP11B1 IC50/hCYP11B2 IC50.
Ligand lipophilic efficiency; LLE = pIC50 – a Log P98.
Table 6. Rat and Rhesus Pharmacokinetic Profiles of Selected Compoundsa.
| compd | species | PPBb | Mic Clintc | F (%)d | AUCN (po)e | Clpf |
|---|---|---|---|---|---|---|
| 23 | rat | 15 | <20 | 92 | 1.32 | 43 |
| 26 | rat | 14 | <20 | 86 | 2.10 | 37 |
| 26 | rhesus | 23 | <20 | 53 | 3.07 | 10 |
| 35 | rhesus | n.d. | <20 | 88 | 2.49 | 19 |
| 31 | rat | 14 | <20 | 94 | 6.55 | 7 |
| 32 | rat | 13 | <20 | 76 | 4.32 | 10 |
| 32 | rhesus | 15 | 22 | 15 | 1.53 | 5 |
Assay details reported in Supporting Information.
Plasma protein binding, reported as % free compound.
Intrinsic clearance in rat or rhesus liver microsomes (mL/min/kg).
Bioavailability.
Area under the curve, normalized for dose (μM·h·kg/mg).
Plasma clearance (mL/min/kg).
Compounds from this series displayed high selectivity versus CYPs 17, 19 and 21, and hepatic CYPs 3A4, 2C9, and 2D6. As shown in Table 5, compounds in this series consistently displayed no inhibition of CYP17 or CYP21. Inhibition of CYP19 was observed, though compounds generally exhibited at least 500-fold selectivity or better for CYP11B2 vs CYP19. Compounds with monosubstituted pyridine heme-binding domains such as 26, 31, and 32 typically showed no inhibition of CYPs 3A4, 2C9, and 2D6, and this held true whether the pyridine was substituted with a lipophilic group as in 26, or a more polar group as in 31 or 32. Representative data for inhibition of CYP3A4 are shown in Table 5. Compounds with disubstituted heme binding domains such 35 did display hepatic CYP inhibition, although even in these cases selectivity vs CYP11B2 was >1000-fold. The results shown in Table 5 are representative of the series as a whole, and indicate high selectivity versus related CYP targets.
Table 5. Activity of Selected Compounds at Related CYP Enzyme Targets.
| compd | CYP17a (IC50, nM) | CYP19a (IC50, nM) | CYP21a (IC50, nM) | CYP3A4a (IC50, nM) |
|---|---|---|---|---|
| 26 | >10000 | 4513 | >10000 | >50000 |
| 31 | >10000 | 1268 | >10000 | >50000 |
| 32 | >10000 | 1021 | >10000 | >50000 |
| 35 | >10000 | n.d. | >10000 | 8235 |
Assay details reported in Supporting Information.
A benchmark benzimidazole also displayed high selectivity versus a broad panel of biological targets. Compound 26 was assayed for activity against 114 receptors, enzymes, and ion channels, and showed mid-micromolar binding activity against just one target (adrenergic α2B receptor, IC50 = 2.49 μM). At a concentration of 10 μM, 26 displayed <40% activity against all other targets in the screen. Compound 26 was also screened against a collection of >100 kinase enzymes, and at a concentration of 10 μM, exhibited <40% activity against the entire panel. Finally, compound 26 exhibited an IC50 > 30 μM in functional assays that measure block of Nav1.5 and Cav1.2 ion channels, and in a binding assay that measures displacement of the known hERG blocker MK-0499.13 The lack of ion channel block observed with compound 26 is representative of the series as a whole, and suggests reduced risk for cardiovascular adverse effects.
Key compounds from this series exhibited good pharmacokinetic profiles in both rat and rhesus (Table 6).12 When compared to compound 5 (Table 2), compound 23, which contains an additional fluorine at the benzimidazole 5-position, displays a comparable rat PK profile, albeit with a slightly higher rate of plasma clearance. Compound 26, which adds a meta-fluorine to the pyridine, shows improvements in plasma clearance, half-life, and oral exposure as a result. Compound 26 also displays an acceptable PK profile in rhesus, with low plasma clearance, good oral exposure, and a half-life of 7.4 h. Compound 35 displays a broadly similar rhesus PK profile, though it suffers from a higher rate of plasma clearance, perhaps as a result of oxidative metabolism at the pyridyl 4-methyl group. The best overall rat PK profiles from this set belong to compounds 31 and 32, which both contain a tertiary alcohol substituent at the pyridine 3-position. These compounds display excellent oral bioavailability and oral exposure in rat. They also exhibit plasma clearance rates that are notably lower than their unsubstituted or meta-fluoro substituted counterparts (compare 31 to 23 or 26, and 32 to 5). Their improved clearance rates may result from steric blockade, as the bulky alcohol group may inhibit or prevent metabolizing enzymes from accessing the pyridine. Compound 32 also displays a good rhesus PK profile, with low plasma clearance and acceptable oral exposure.
Compounds from this series exhibited potent inhibition of human and rhesus CYP11B2, but only weak inhibition of rat CYP11B2. Given the low identity/homology between human and rat CYP11B2 (∼80%) and the higher homology between human and rhesus CYP11B2 (∼95%), this is perhaps not surprising. Consequently, these compounds could not be tested in known rodent models of hypertension, and were instead evaluated in a recently described rhesus pharmacodynamic (PD) model.14
Several compounds from the current study exhibited profiles promising enough to warrant testing in the PD model. However, as the model was resource-intensive, throughput was restricted. From the set of promising compounds, compound 32 was identified earliest and was thus the compound chosen for proof-of-concept PD testing.
Compound 32 exhibited dose-dependent aldosterone lowering efficacy in a rhesus PD model. In this model, monkeys are anesthetized to maintain stable baseline levels of aldosterone. Thus, although compound 32 displayed a PK profile suitable for oral dosing, the use of anesthetized animals necessitated i.v. dosing. Compound 32 was given at i.v. doses ranging from 0.001 to 0.3 mg/kg. Plasma levels of aldosterone and 11-deoxycortisol (11-DOC), a biosynthetic precursor of cortisol, were quantified by LC–MS at time points up to 180 min postdose, allowing the determination of AUCs for aldosterone and 11-DOC.15 Plasma concentrations of compound 32 were measured at 90 min postdose, and free (unbound) concentrations were calculated based on 32’s 7% free fraction in rhesus plasma.
When dosed i.v. at 0.01 and 0.3 mg/kg, compound 32 produced reductions in aldosterone AUC of 62% and 95%, respectively, compared to baseline (Figure 1). Compound 32 achieved these effects at free plasma concentrations 90 min postdose of 12 and 27 nM, respectively. The reduction in aldosterone AUC produced by a 0.3 mg/kg dose of compound 32 is comparable to that of a 1.0 mg/kg i.v. dose of the positive control Fadrozole, a pan-CYP19/CYP11B2/CYP11B1 inhibitor that has been shown to reduce blood pressure in rodent models of hypertension.16−18 At doses up to 0.3 mg/kg, compound 32 does not produce an increase in 11-DOC AUC, suggesting that CYP11B1, the enzyme that converts 11-DOC to cortisol, has not been significantly inhibited. In contrast, a 1.0 mg/kg dose of Fadrozole, which displays potent inhibition of both CYP11B2 and B1, elicits a significant increase in 11-DOC AUC. These data demonstrate that, in an acute setting, compound 32 is capable of producing significant reductions in rhesus plasma aldosterone levels with no apparent effect on cortisol levels.
Figure 1.
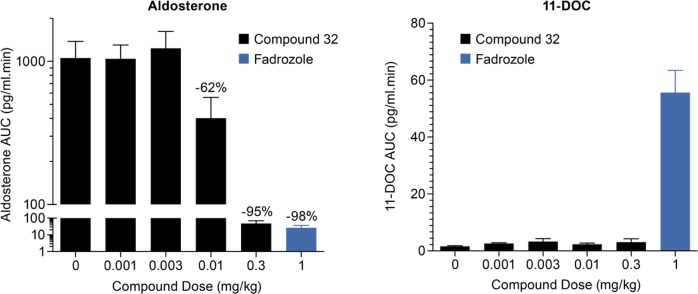
Effects of compound 32 on plasma AUCs of aldosterone and 11-DOC in rhesus monkeys.
At a free plasma concentration of 12 nM, compound 32 produces a 62% reduction in aldosterone AUC, which is consistent with its potent inhibition of rhesus CYP11B2 (IC50 = 0.3 nM). Additionally, at free plasma concentrations up to 27 nM, no reduction in 11-DOC AUC and no apparent inhibition of CYP11B1 are observed. Although that plasma concentration approaches compound 32’s IC50 for inhibition of rhesus CYP11B1 (IC50 = 41 nM), higher concentrations are apparently required to affect reductions in 11-DOC AUC in this acute setting.
Reductions in plasma aldosterone levels have been shown to produce reductions in blood pressure in the clinic. In a Phase II clinical trial, the aldosterone synthase inhibitor LCI-699 produced a mean 34% reduction in plasma aldosterone at 8 weeks, with concomitant reductions in systolic (−9.7 mmHg) and diastolic (−4.7 mmHg) blood pressure.5 In light of those results, the fact that compound 32 can reduce plasma aldosterone levels by >90% in rhesus suggests that a compound from this series could produce significant blood pressure reduction in the clinic.
In summary, we have reported the discovery of a structurally novel series of benzimidazole CYP11B2 inhibitors. Benchmark compound 32 from this series displays potent and selective inhibition of CYP11B2, and a good pharmacokinetic profile in rat and rhesus. In a rhesus pharmacodynamic model, compound 32 exhibits dose-dependent aldosterone lowering efficacy, with maximal reductions in plasma aldosterone AUC of >90%. Additional optimization in this series has been completed and will be reported in due course.
Glossary
ABBREVIATIONS
- 11-DOC
11-deoxycortisol
- AUC
area under curve
- BP
blood pressure
- CNS
central nervous system
- LLE
lipophilic ligand efficiency
- PK
pharmacokinetics
Supporting Information Available
Assay protocols, synthesis schemes, representative procedures for the synthesis of compounds 31, 32, 35, and related intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Carey R. M. Aldosterone and cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 194–198. [DOI] [PubMed] [Google Scholar]
- Shavit L.; Lifschitz M. D.; Epstein M. Aldosterone blockade and the mineralocorticoid receptor in the management of chronic kidney disease: current concepts and emerging treatment paradigms. Kidney Int. 2012, 81, 955–968. [DOI] [PubMed] [Google Scholar]
- Tomaschitz A.; Pilz S.; Ritz E.; Obermayer-Pietsch B.; Pieber T. R. Aldosterone and arterial hypertension. Nat. Rev. Endocrinol. 2010, 6, 83–93. [DOI] [PubMed] [Google Scholar]
- Gilbert K. C.; Brown N. J. Aldosterone and inflammation. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.; Yin L.; Hartmann R. W. Aldosterone synthase inhibitors as promising treatments for mineralocorticoid dependent cardiovascular and renal diseases. J. Med. Chem. 2014, 57, 5011–5022. [DOI] [PubMed] [Google Scholar]
- Cerny M. A. Progress toward clinically useful aldosterone synthase inhbitors. Curr. Top. Med. Chem. 2013, 13, 1385–1401. [DOI] [PubMed] [Google Scholar]
- Calhoun D. C.; White W. B.; Krum H.; Guo W.; Bermann G.; Trapani A.; Lefkowitz M. P.; Menard J. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension. Circulation 2011, 124, 1945–1955. [DOI] [PubMed] [Google Scholar]
- Azizi M.; Amar L.; Menard J. Aldosterone synthase inhibition in humans. Neprhol. Dial. Transplant. 2013, 28, 36–43. [DOI] [PubMed] [Google Scholar]
- Meredith E. L.; Ksander G.; Monovich L. G.; Papillon J. P. N.; Liu Q.; Miranda K.; Morris P.; Rao C.; Burgis R.; Capparelli M.; Hu Q.-Y.; Singh A.; Rigel D. F.; Jeng A. Y.; Beil M.; Fu F.; Hu C.-W.; LaSala D. Discovery and in vivo evaluation of potent dual CYP11B2 (Aldosterone Synthase) and CYP11B1 inhibitors. ACS Med. Chem. Lett. 2013, 4, 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt S. B.; Park M. K.; London C.; Xiong Y.; Bennett D. J.; Cai J.; Ratcliffe P.; Cooke A.; Carswell E.; MacLean J.; Saxena R.; Kulkarni B. A.; Gupta A.. Aldosterone Synthase Inhibitors. WO 2012012478, Jan 26, 2012.
- Lipophilic ligand efficiency (LLE) was calculated using the equation LLE = pIC50 – a Log P98, as first proposed by:Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- For detailed experimental protocols, see Supporting Information section.
- Wang J.; Della Penna K.; Wang H.; Karczewski J.; Connolly T. M.; Koblan K. S.; Bennett P. B.; Salata J. J. Am. J. Physiol. Heart Circ. Physiol. 2002, 284, H256. [DOI] [PubMed] [Google Scholar]
- Cai T.-Q.; Stribling S.; Tong X.; Xu L.; Wisniewski T.; Fontenot J. A.; Struthers M.; Akinsanya K. O. Rhesus monkey model for concurrent analyses of pharmacodynamics, pharmacokinetics and in vivo selectivity of aldosterone synthase inhibitors. J. Pharmacol. Toxicol. Methods 2015, 71, 137–146. [DOI] [PubMed] [Google Scholar]
- We had initially planned to monitor plasma levels of cortisol, but development of an LC–MS analytical method that cleanly resolved the cortisol signal proved challenging. Thus, rather than monitoring decreases in cortisol levels, we monitor increases in levels of 11-DOC, the direct biosynthetic precursor that is converted to cortisol by CYP11B1.
- Rigel D. F.; Fu F.; Beil M.; Hu C.-W.; Liang G.; Jeng A. Y. Pharmacodynamic and pharmacokinetic characterization of the aldosterone synthase inhibitor FAD286 in two rodent models of hyperaldosteronism: comparison with the 11beta-hydroxylase inhibitor metapyrone. J. Pharmacol. Exp. Ther. 2010, 334, 232–243. [DOI] [PubMed] [Google Scholar]
- LaSala D.; Shibanaka Y.; Jeng A. Y. Coexpression of CYP11B2 or CYP11B1 with adrenodoxin and adrenodoxin reductase for assessing the potency and selectivity of aldosterone synthase inhibitors. Anal. Biochem. 2009, 394, 56–61. [DOI] [PubMed] [Google Scholar]
- Huang B. S.; White R. A.; Jeng A. Y.; Leenen F. H. H. Role of central nervous system aldosterone synthase and mineralocorticoid receptors in salt-induced hypertension in Dahl salt-sensitive rats. Am. J. Physiol. Reg. Integr. Comp. Physiol. 2009, 296, R994–R1000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.