

Abstract

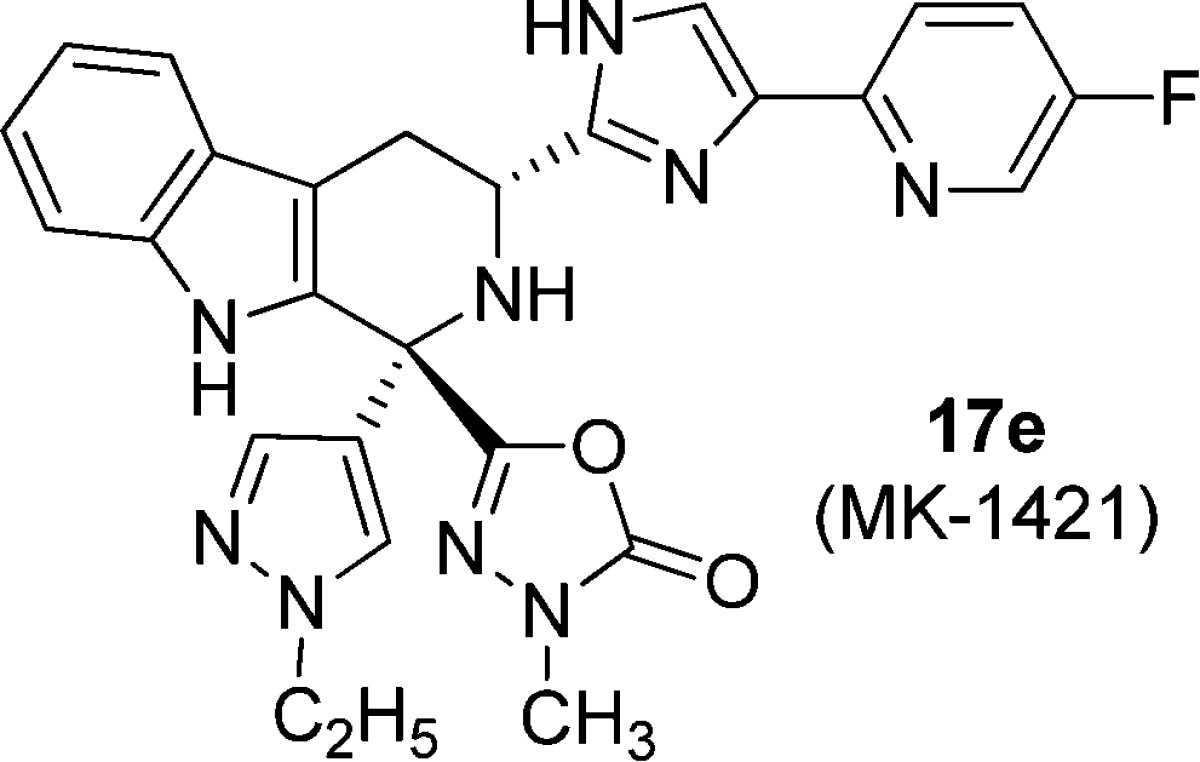
The imidazolyl-tetrahydro-β-carboline class of sstr3 antagonists have demonstrated efficacy in a murine model of glucose excursion and may have potential as a treatment for type 2 diabetes. The first candidate in this class caused unacceptable QTc interval prolongation in oral, telemetrized cardiovascular (CV) dogs. Herein, we describe our efforts to identify an acceptable candidate without CV effects. These efforts resulted in the identification of (1R,3R)-3-(4-(5-fluoropyridin-2-yl)-1H-imidazol-2-yl)-1-(1-ethyl-pyrazol-4-yl)-1-(3-methyl-1,3,4-oxadiazol-3H-2-one-5-yl)-2,3,4,9-tetrahydro-1H-β-carboline (17e, MK-1421).
Keywords: sstr3, antagonist, type 2 diabetes, tetrahydro-β-carboline
We recently reported the discovery of a potent, selective sstr3 (somatostatin subtype-3 receptor) antagonist 1 (MK-4256) as a potential treatment for type 2 diabetes.1 This tetrahydro-β-carboline derivative is characterized by excellent sstr3 potency and subtype selectivity, a good pharmacokinetic (PK) profile in preclinical species, and exceptional potency in an oral glucose tolerance test (oGTT) in mice. On the basis of this attractive profile, compound 1 was advanced as a preclinical development candidate. However, in cardiovascular (CV) safety evaluation in dogs, 1 exhibited unexpected potentiation of the QTc interval. Initial studies indicated that the CV effects were not caused by binding to the sstr3 receptor.2 Nicotinic acid derivative 2 is a potent sstr3 antagonist devoid of ion channel binding that exhibited little to no effects on the QTc interval in dogs, thus providing evidence that the observed cardiovascular effects were likely associated with ion channel binding and not sstr3 (Table 1). Herein, we describe our efforts to identify an acceptable clinical candidate in this class of tetrahydro-β-carboline sstr3 antagonists with reduced QTc increase potential.
Table 1. Binding to h-sstr3 and hERG Channel by Tetrahydro-β-carbolines 1 and 2a.
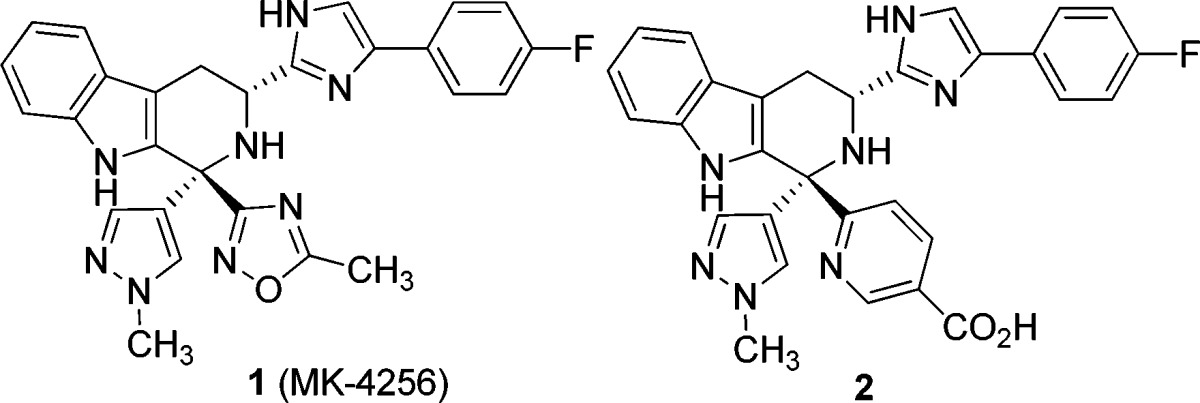
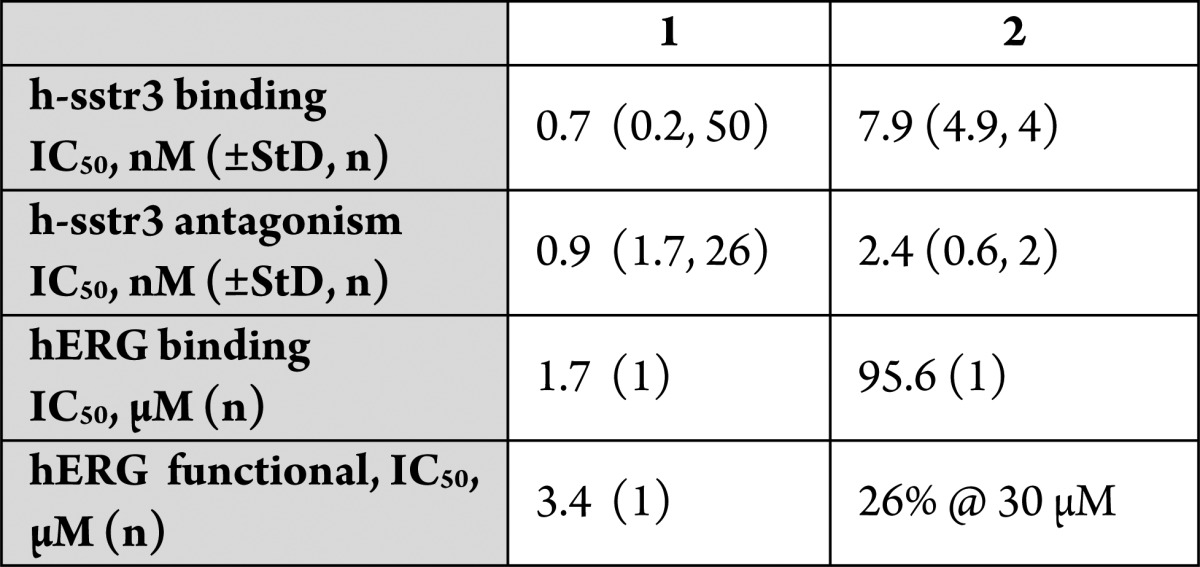
StD = standard deviation; n = number of determinations; nd = not determined.
An initial assessment of the phenyl-imidazolyl-tetrahydro-β-carboline structure suggested that it might contain the proposed hERG channel pharmacophore: a basic amine centered between two broad hydrophobic domains.3,4 The pKa of the tetrahydro-β-carboline nitrogen would be expected to play an important part in the binding to the ion channel through a putative π–cation interaction.5
Lowering of the nitrogen pKa or its replacement was explored through a series of simple analogues (Table 2). These compounds were prepared by methods previously described involving a Pictet–Spengler cyclization of the phenyl imidazolyl tryptamine with the appropriate aldehyde or ketone.6,7
Table 2. Binding to h-sstr3 and hERG Channel by Modified Tetrahydro-β-carboline Analoguesa.
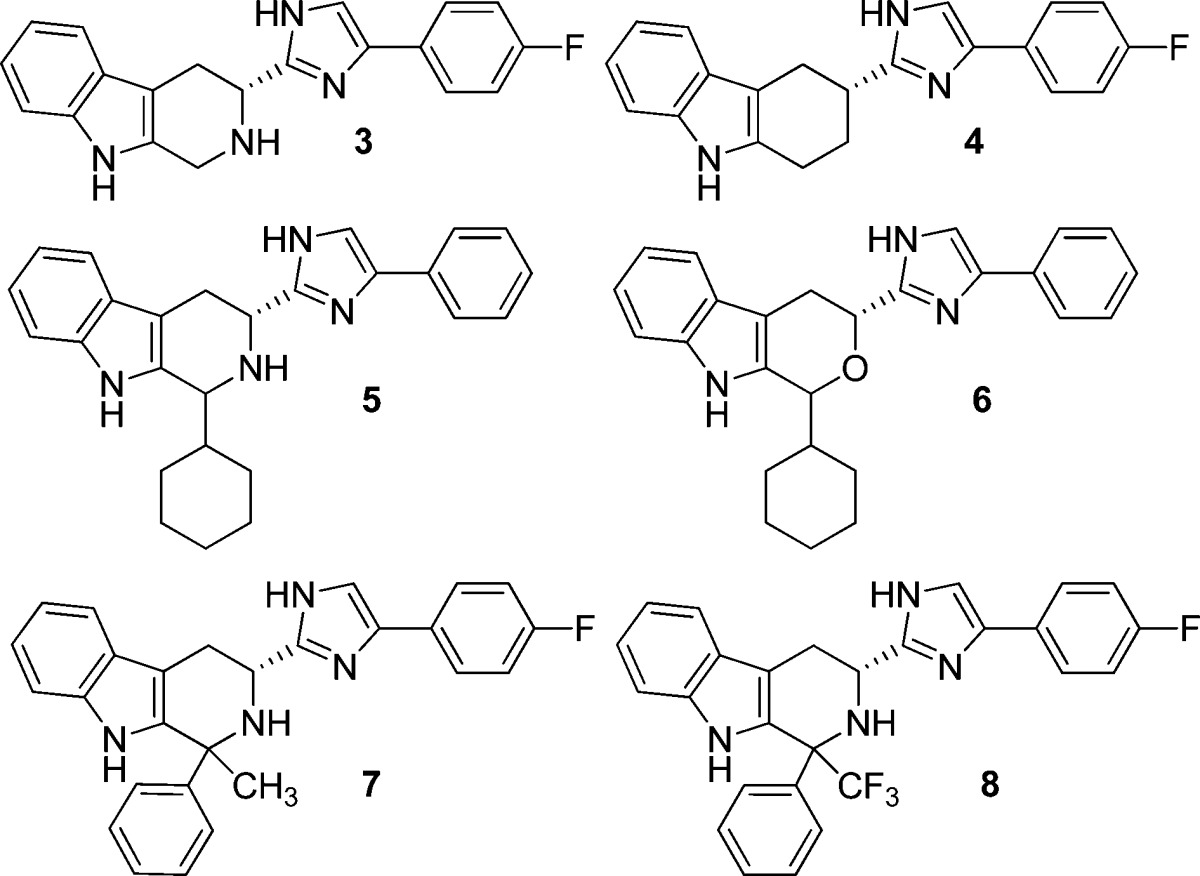
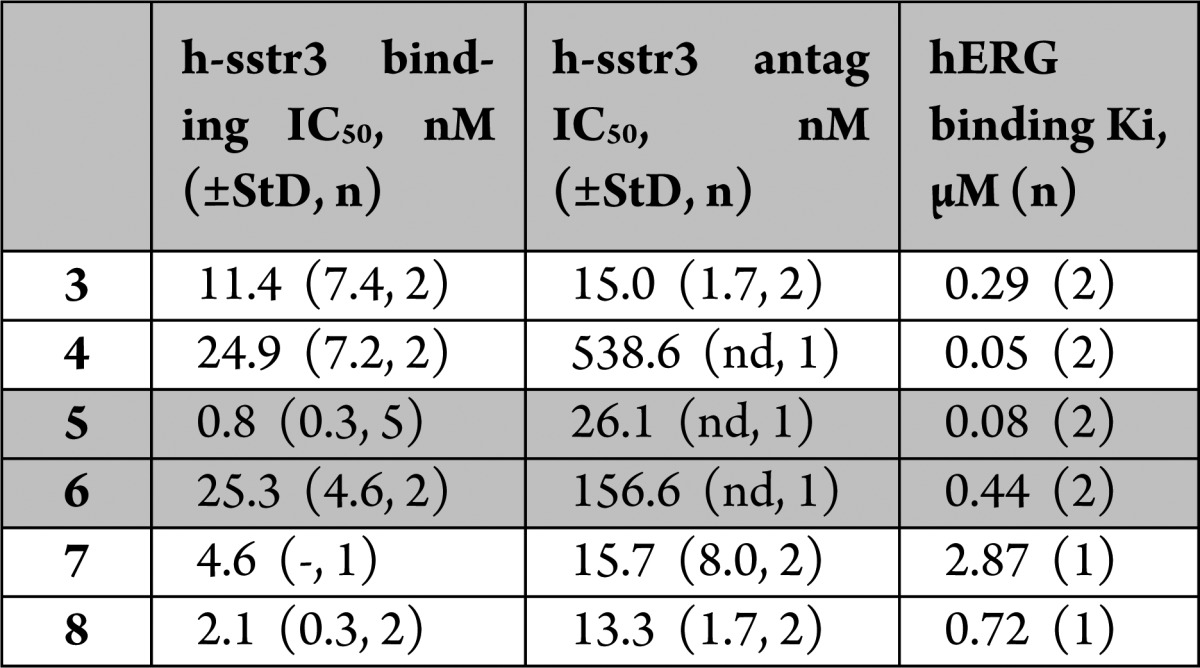
StD = standard deviation; n = number of determinations; nd = not determined.
The examples in Table 2 should be considered in pairs: compounds 3 and 4 compare the tetrahydro-β-carboline nitrogen replaced by carbon. There is a small loss of sstr3 binding, a large loss in receptor antagonism, and a significant gain in hERG binding. Compounds 5 and 6 highlight the effect of oxygen replacement for nitrogen. The large changes observed in all three assays reflect the large structural change of replacing a basic amine with a neutral, polar oxygen. Finally, C-1 substituted analogues 7 and 8 both retain the nitrogen, but the presence of the trifluoromethyl group in 8 would be expected to dramatically lower the pKa of the nitrogen. sstr3 activities are retained, but there is actually an increase in hERG channel binding. Taken together, the results in Table 2 suggest that a strategy of replacing or attenuating basicity of the tetrahydro-β-carboline nitrogen would not be fruitful.
The structure–activity relationship (SAR) of the phenyl-imidazole moiety in 5 was examined for effects on sstr3 and hERG activities (Table 3). Phenyl-imidazoles are reported to have hERG activity.8 An exploration of imidazole substitution and replacement, including N-methylation, iso-imidazolyl, isoxazolyl, and oxadiazolyl, was not fruitful and will be reported separately. The three pyridyl isomers 9, 10, and 11 afforded a loss of sstr3 binding relative to the phenyl analogue 5 but generally retained similar antagonist activities. Changes to hERG channel binding were modest. The pyrazine analogue 12 offered no advantages in terms of potency or selectivity. The addition of a 4-fluoro substituent had previously been shown to increase sstr3 potency and was added to the 2-pyridyl group in 9 to afford 13, which seemed to improve selectivity of receptor antagonism vs hERG activity.1 The 4-fluoro-2-pyridyl moiety was incorporated into 1 to afford 14, which again only showed a modest improvement in hERG selectivity. Compound 14 had a similar profile to 1 in the mouse oGTT model (∼82% inhibition at 1 mg/kg po). In a patch clamp assessment of hERG activity, 14 afforded an IC50 = 12 μM (IC20 ≈ 3 μM),9 representing a 3–4-fold improvement over 1. Unfortunately, 14 also exhibited a greater increase in the QTc interval in dogs than 1 at similar plasma concentrations. Clearly a 3–4-fold improvement in hERG activity was insufficient to minimize or eliminate the QTc prolongation.
Table 3. Binding to h-sstr3 and hERG Channel by Heteroaryl-imidazolyl-tetrahydro-β-carbolinesa.
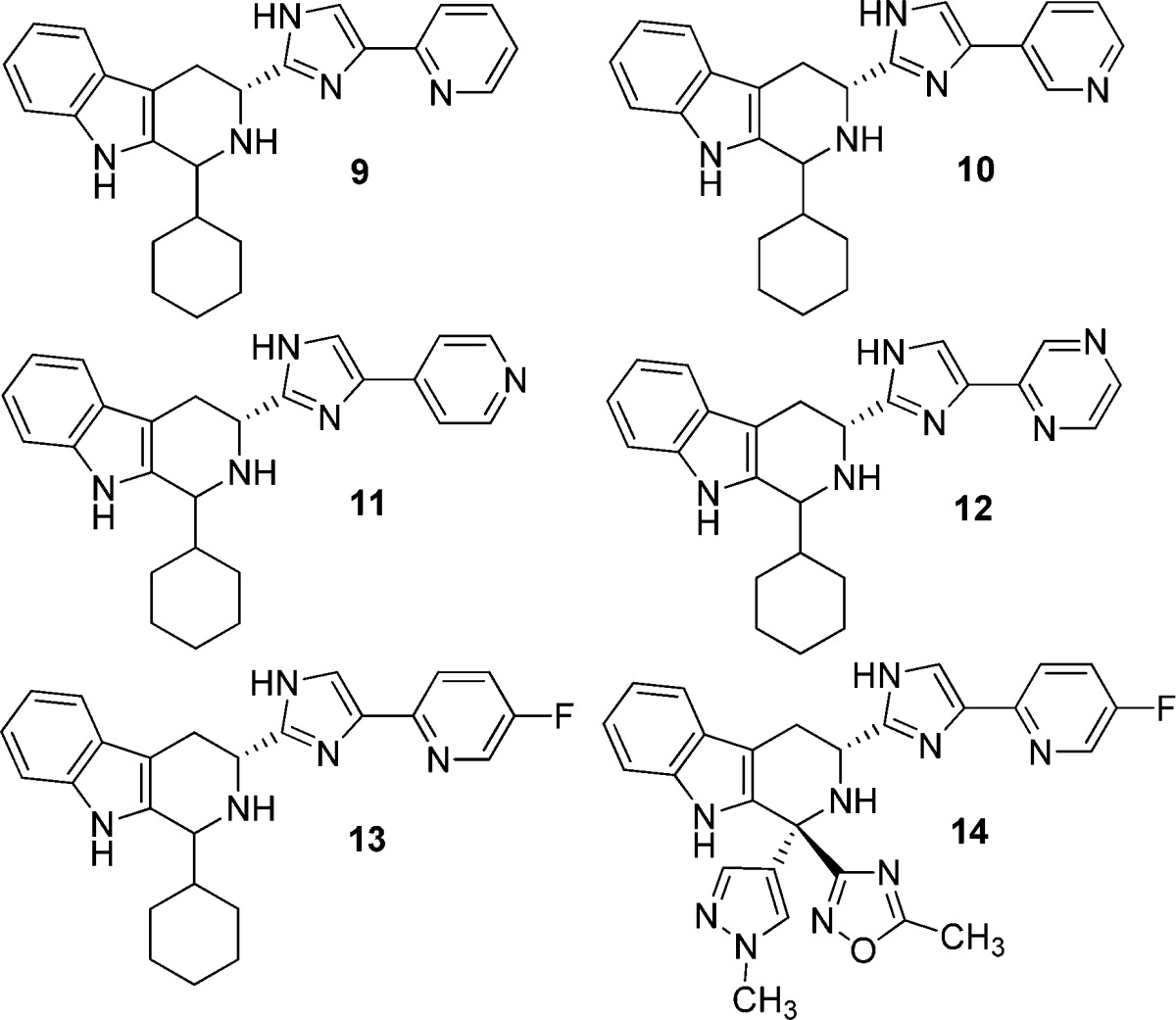
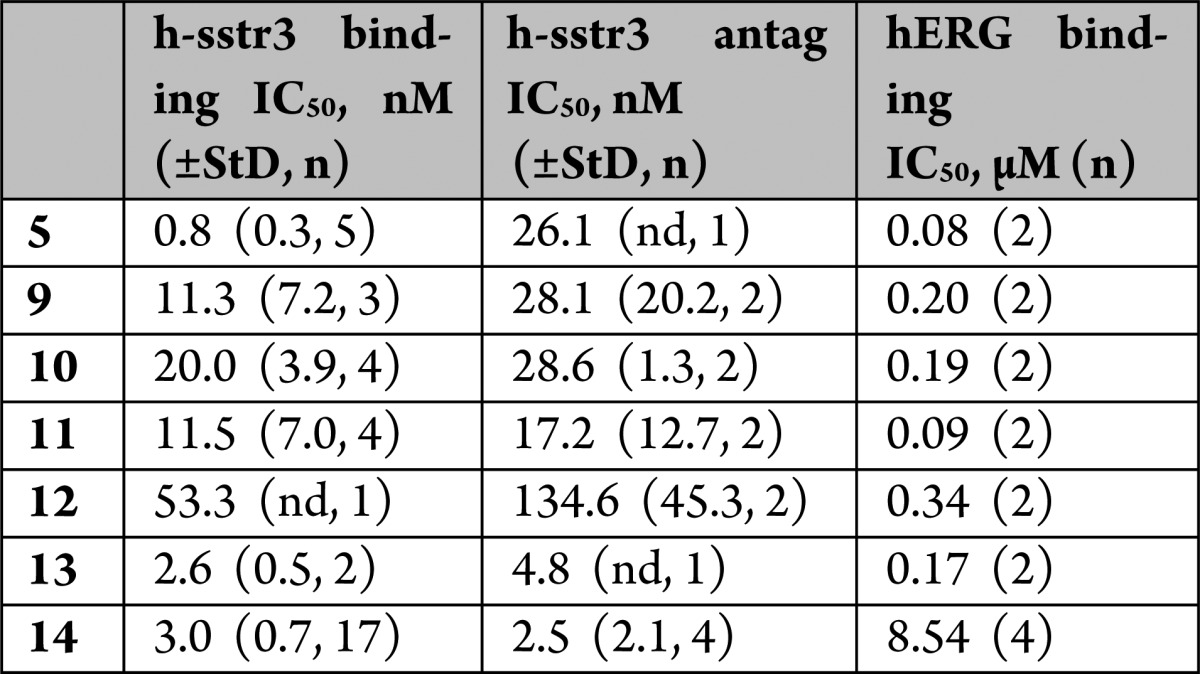
StD = standard deviation; n = number of determinations; nd = not determined.
In the discovery phase of 1 (MK-4256), it was found that hERG channel binding could be greatly reduced by placement of a variety of heteroaryls at the C-1 position of the tetrahydro-β-carboline scaffold.1 In particular, substituted pyrazoles and oxadiazoles afforded an attractive balance between sstr3 potencies, pharmacokinetic profiles, and off-target activities. The effect of simple substitution on the pyrazole and 1,2,4-oxadiazole rings is shown in Table S1 (see Supporting Information). Compound 15a loses some sstr3 potency, while compounds 15b–d retain a sstr3 profile similar to that of 14, but no improvement in hERG channel binding overall. The more polar substituents in 15e and 15f afford a loss of potency on the receptor.
Effects on substitution of the isomeric 1,3,4-oxadiazole analogues 16 are shown in Table S1. Simple alkyl substitution in 16a–c afforded a similar profile to 14. The more polar heteroatom replacements in 16d, 16e, and 16g with hydroxyl, sulfhydryl, and amino, respectively, retain sstr3 potency and trend toward reduced hERG channel binding. Alkylation of the heteroatoms in 16f, 16h, and 16i return hERG activity toward that of 14. Alkylation of the hydroxyl analogue 16d did not give O-alkylation but rather N-alkylation to afford the oxadiazolones 17. This was a fortuitous result as these compounds generally retained sstr3 activities with much reduced hERG binding. QSAR modeling of hERG binding from Table S1 suggested a correlation with hydrophobicity (more hydrophobicity led to worse hERG), which might be addressed prospectively via PSA and cLogP (see Supporting Information). While the trends present in the data set provide a path away from low micromolar hERG binders, the statistical significance of the model indicated the need to include a more diverse compound set which would likely represent a more dynamic range in measured hERG.
The smaller alkyl substituents in 17b–e afforded sstr3 profiles similar to 1 but with >10-fold less hERG channel activities. More importantly, these analogues reduced glucose excursion in a mouse oGTT model after oral dosing at 1 mg/kg po. The only slightly larger alkyl groups in 17i–j once again brought back too much hERG binding. The dimethylaminoethyl derivative 17k also had significant hERG activity, whereas the acid 17l and amide 17m both had reduced ion channel binding but lost efficacy in the mouse oGTT evaluation. Both 17c and 17e had comparable profiles, but in a functional patch clamp hERG assay, the former compound afforded an IC50 = 12 μM vs 27 μM for 17e. Given its superior profile, compound 17e was chosen as a potential development candidate.
The configuration of 17e was determined to be (1R,3R) by 1H NMR (nOE) analysis of starting material 16d; the 3R position was assigned from the starting material d-tryptophan.10,10b The (1S,3R) diastereomer 17f and the (1SR,3S) isomer 17g were 10–100-fold less potent as an sstr3 antagonist compared to 17e. The binding profile of 17e vs sstr3 from other species and the other human sstr subtypes is shown in Table 4. These data clearly show that 17e is very potent vs sstr3 from other preclinical species and is highly subtype selective. No agonist activity was noted for any sstr subtype (data not shown).
Table 4. In Vitro Binding Profile of 17ea.
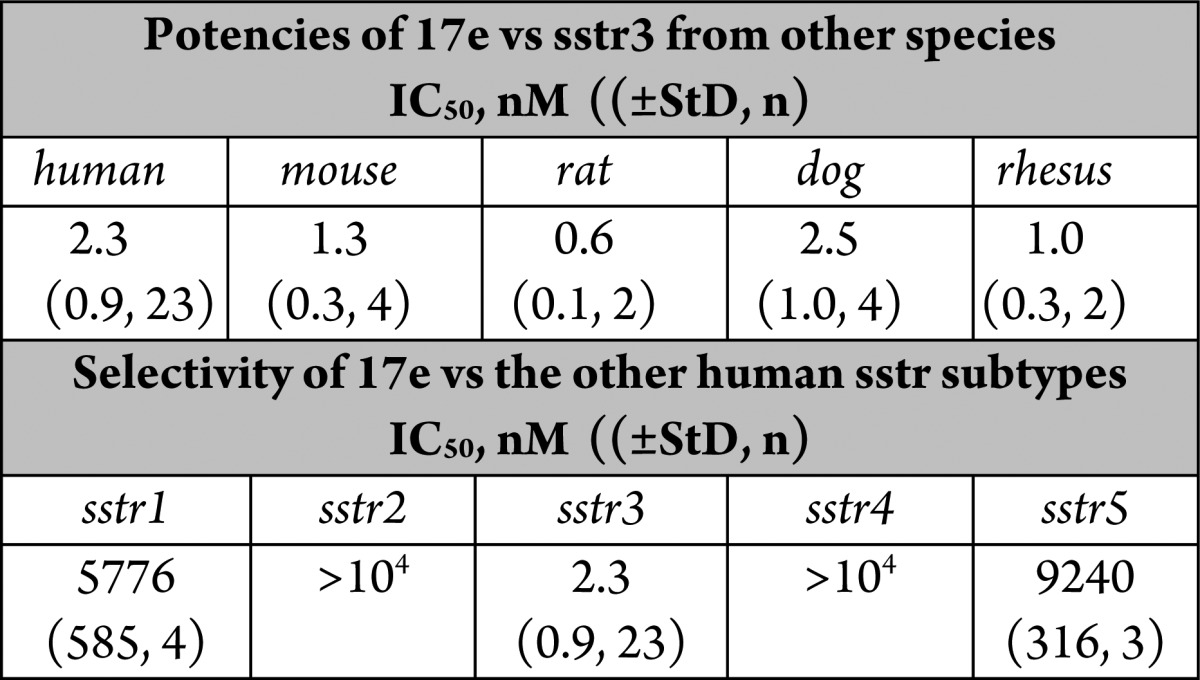
StD = standard deviation; n = number of determinations; nd = not determined.
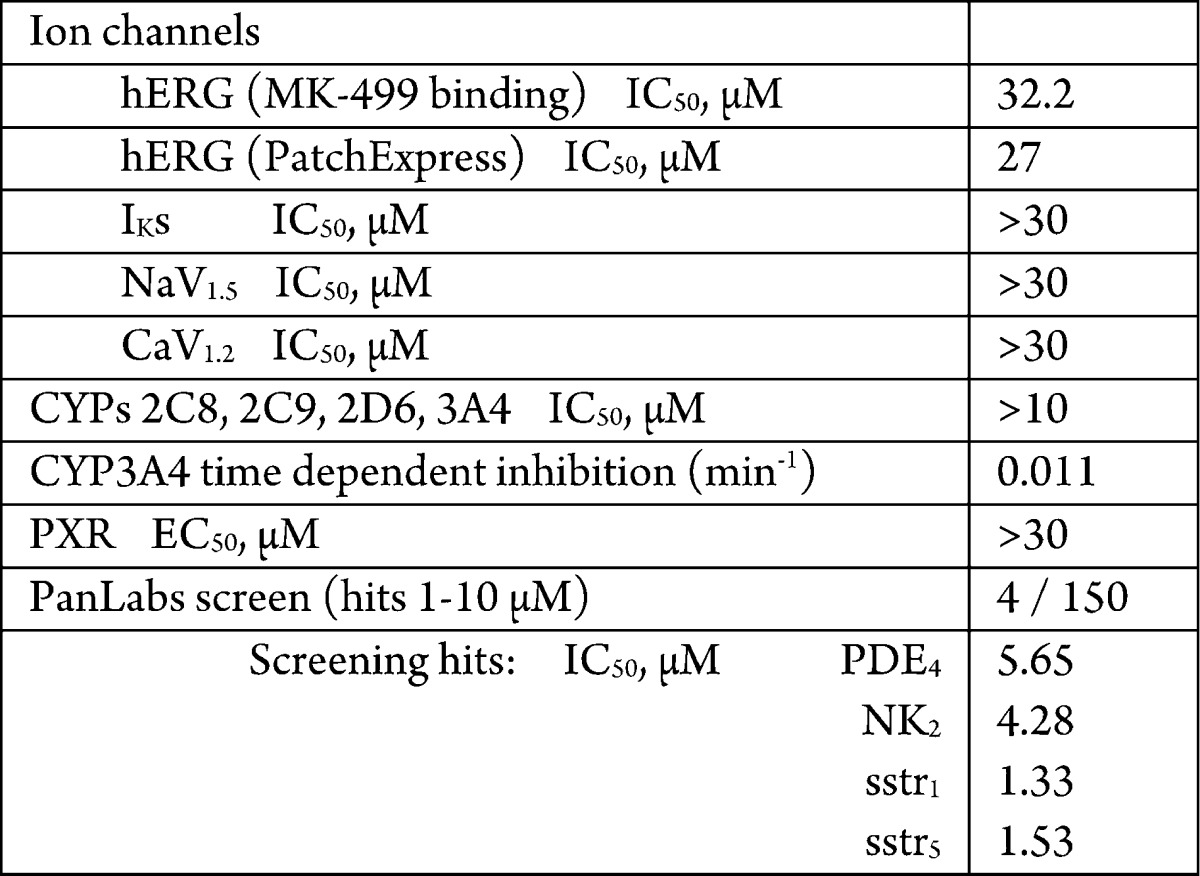
Additional evaluation of off-target hits is shown in Table 5. Selectivity vs ion channels, CYPs, and a screening panel of 150 enzyme and receptor targets was excellent as was the lack of activity against PXR activation. Acceptable time-dependent inhibition of CYP3A4 was noted.
Table 5. Off-Target Profile of 17e.
The pharmacokinetic profile in four preclinical species is listed in Table 6. Bioavailability was poor in rodent species but much better in higher species. This compound is an efficient substrate for the transporter P-glycoprotein (PGP) in several species (BA/AB: mouse 19; rat 16; human 31).11 As a consequence, brain penetration is very low (brain/plasma concentration ratio b/p = 0.02; see Supporting Information).11 The compound also exhibits poor permeability with a permeability coefficient Papp < 10 cm·10–6/sec. The combination of PGP transport and low permeability may explain the poor bioavailability in rodents. Plasma protein binding is comparable among all species.
Table 6. Pharmacokinetic Profile and Plasma Protein Binding (PPB) of 17e in Preclincal Speciesa.
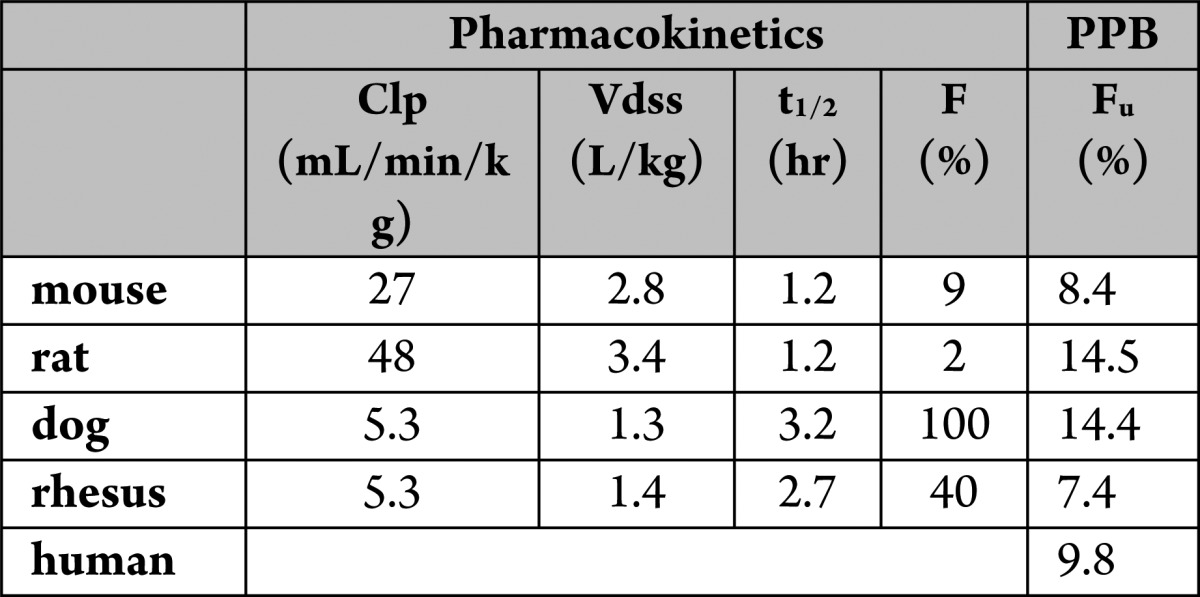
Clp = plasma clearance; Vdss = volume of distribution at steady state; t1/2 = plasma half-life after iv dosing; F = oral bioavailability; Fu = unbound fraction determined in vitro.
Compound 17e was assessed for its ability to improve glucose tolerance in lean C57BL/6N mice. Oral administration of 17e 1 h prior to dextrose challenge in an oral glucose tolerance test (oGTT) significantly reduced blood glucose excursion in a dose-dependent manner from 0.03 to 3 mg/kg (Figure 1). The cumulative results from separate titrations demonstrated that at 3 mg/kg inhibition was near 100%. The plasma levels of 17e determined at the completion of the oGTT study (∼2.5 h post dose) were 1, 2, 4, 12, and 105 nM at 0.03, 0.1, 0.3, 1, and 3 mg/kg po, respectively. The ED50 for 17e-induced suppression of oGTT glucose levels in mouse was 0.16 mg/kg, and the MEDmax (minimal efficacious dose for maximal suppression of blood glucose during oGTT) was 3 mg/kg po. The effect of 17e on insulin release in the presence of low and high glucose concentrations in isolated human islets was examined. As shown in Figure 2, compound 17e (5 μM) augmented insulin secretion at 16 mM glucose, but not at 2 mM glucose during a 60 min static incubation of human islets from two nondiabetic donors. Compound 17e did not affect the release of glucagon nor somatostatin (SS-14) at either glucose concentrations (data not shown). GLP-1 (50 nM) and linoleic acid (100 μM) were used as positive controls for insulin/somatostatin and glucagon release, respectively.
Figure 1.

Titration of 17e in mouse oGTT.
Figure 2.

Insulin secretion from human islets induced by 17e, GLP-1, and linoleic acid.
In light of the cardiovascular issues associated with compound 1, the potential of 17e to elicit any CV effects was evaluated. Compound 17e was administered by iv infusion to anesthesized vagus-intact dogs at various doses to achieve plasma concentrations (Cmax) of 17, 43, and 140 μM.12 There were no changes to any electrophysiologic parameters, including no prolongation of the QTc interval. Subsequent studies with 17e in oral, telemetrized cardiovascular dogs revealed no significant changes (Cmax = 5.8 μM).13 As a result, 17e was chosen as an early development candidate and designated as MK-1421.
In summary, we disclose the discovery and profile of a potent, selective sstr3 antagonist (17e, MK-1421) that showed excellent efficacy in a mouse model of glucose excursion (oGTT) and demonstrated glucose-dependent release of insulin from relevant human tissue. This compound has an acceptable preclinical pharmacokinetic profile. Unlike a previous candidate, 17e demonstrated little potential for cardiovascular effects. It was chosen for further development as a potential treatment for type 2 diabetes mellitus. Further information regarding its clinical profile will be reported in due course.
Acknowledgments
The authors thank Li-Kang Zhang at the Merck Research Laboratories for measuring the high resolution mass of 17e.
Supporting Information Available
Synthetic methods and characterization of key compounds are described along with protocols for biological assays. Table S1 is also presented. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- He S.; Ye Z.; Truong Q.; Shah S.; Du W.; Guo L.; Dobbelaar P. H.; Lai Z.; Liu J.; Jian T.; Qi H.; Bakshi R. K.; Hong Q.; Dellureficio J.; Pasternak A.; Feng Z.; deJesus R.; Yang L.; Reibarkh M.; Bradley S. A.; Holmes M. A.; Ball R. G.; Ruck R. T.; Huffman M. A.; Wong F.; Samuel K.; Reddy V. B.; Mitelman S.; Tong S.; Chicchi G.; Tsao K.-L.; Trusca D.; Wu M.; Shao Q.; Trujillo M. E.; Eiermann G. J.; Li C.; Zhang B.; Howard A. D.; Zhou Y.-P.; Nargund R. P.; Hagmann W. K. The discovery of MK-4256, a potent SSTR3 antagonist as a potential treatment of type-2 diabetes. ACS Med. Chem. Lett. 2012, 3, 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S.; Lai Z.; Ye Z.; Dobbelaar P.; Shah S. K.; Truong Q.; Du W.; Guo L.; Liu J.; Jian T.; Qi H.; Bakshi R. K.; Hong Q.; Dellureficio J.; Reibarkh M.; Samuel K.; Reddy V. B.; Mitelman S.; Tong S. X.; Chicchi G. G.; Tsao K.-L.; Trusca D.; Wu M.; Shao Q.; Trujillo M. E.; Fernandez G.; Nelson D.; Bunting P.; Kerr J.; Fitzgerald P.; Morissette P.; Volksdorf S.; Eiermann G. J.; Li C.; Zhang B.; Howard A. D.; Zhou Y.-P.; Nargund R. P.; Hagmann W. K. Investigation of cardiovascular effects of tetrahydro-β-carboline sstr3 antagonists. ACS Med. Chem. Lett. 2014, 5, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli A.; Poluzzi E.; Fabrizio De Ponti F.; Recanatini M. Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K+ channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M. C.; Mitcheson J. S. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol. Sci. 2005, 26 (3), 119–124. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal chemistry of hERG optimizations: highlights and hang-ups. J. Med. Chem. 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- Poitout L.; Roubert P.; Contour-Galcera M.; Moinet C.; Lannoy J.; Pommier J.; Plas P.; Bigg D.; Thurieau C. Identification of potent non-peptide somatostatin antagonists with sst3 selectivity. J. Med. Chem. 2001, 44, 2990–3000. [DOI] [PubMed] [Google Scholar]
- Pasternak A.; Feng Z.; deJesus R.; Ye Z.; He S.; Dobbelaar P. H.; Bradley S. A.; Chicchi G. G.; Tsao K.; Trusca D.; Eiermann G. J.; Feng Y.; Wu M.; Shao Q.; Zhang B.; Nargund R. P.; Mills S. G.; Howard A. D.; Yang L.; Zhou Y.-P. Stimulation of glucose-dependent insulin secretion by a potent, selective sst3 antagonist. ACS Med. Chem. Lett. 2012, 3, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum C. A.; Zhang X.; De Lombaert S. Design, synthesis, and biological evaluation of substituted 2-cyclohexyl-4-phenyl-1H-imidazoles: potent and selective neuropeptide Y Y5-receptor antagonists. J. Med. Chem. 2004, 47, 2318–2325. [DOI] [PubMed] [Google Scholar]
- QTc interval prolongation may be observed in the guinea pig when free plasma concentrations are 3-fold the hERG IC20. This Letter also shows that the GP results translate well to non-rodents and to the clinic.Morissette P.; Nishida M.; Trepakova E.; Imredy J.; Lagrutta A.; Chaves A.; Hoagland K.; Lei Hoe C.-M.; Zrada M. M.; Travis J. J.; Zingaro G. J.; Gerenser P.; Friedrichs G.; Salata J. J. The anesthetized guinea pig: An effective early cardiovascular derisking and lead optimization model. J. Pharmacol. Toxicol. Methods 2013, 68, 137–149. [DOI] [PubMed] [Google Scholar]
- Ernst R. R.; Bodenhausen B.; Wokaun A.. Principles of Nuclear Magnetic Resonances in One or Two Dimensions; Oxford University Press: Oxford, U.K., 1992. [Google Scholar]
- Neuhaus D.; Williamson M. P.. The Nuclear Overhauser Effect in Structural and Conformational Analysis. In Methods in Stereochemical Analysis, 2nd ed.; Marchand A. P., Ed.; John A. Wiley and Sons: New York, 2000. [Google Scholar]
- He H.; Lyons K. A.; Yao Z.; Bleasby K.; Chan G.; Hafey M.; Li X.; Salituro G. M.; Cohen L. H.; Tang W. Utility of unbound plasma drug levels and P-glycoprotein transport data in prediction of central nervous system exposure. Xenobiotica 2009, 39, 687–693. [DOI] [PubMed] [Google Scholar]
- Tashibu H.; Miyazaki H.; Aoki K.; Akie1 Y.; Yamamoto K. QT PRODACT: in vivo QT assay in anesthetized dog for detecting the potential for QT interval prolongation by human pharmaceuticals. J. Pharmacol. Sci. 2005, 99, 473–486. [DOI] [PubMed] [Google Scholar]
- Toyoshima S.; Kanno A.; Kitayama T.; Sekiya K.; Nakai K.; Haruna M.; Mino T.; Miyazaki H.; Koji Yano K.; Yamamoto K. QT PRODACT: in vivo QT assay in the conscious dog for assessing the potential for QT interval prolongation by human pharmaceuticals. J. Pharmacol. Sci. 2005, 99, 459–471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.