

Abstract

A series of imidazo[1,2-a]pyridine derivatives against c-Met was designed by means of bioisosteric replacement. In this study, a selective, potent c-Met inhibitor, 22e was identified, with IC50 values of 3.9 nM against c-Met kinase and 45.0 nM against c-Met-addicted EBC-1 cell proliferation, respectively. Compound 22e inhibited c-Met phosphorylation and downstream signaling across different oncogenic forms in c-Met overactivated cancer cells and model cells. Compound 22e significantly inhibited tumor growth (TGI = 75%) with good oral bioavailability (F = 29%) and no significant hERG inhibition. On the basis of systematic metabolic study, the pathway of all possible metabolites of 22e in liver microsomes of different species has been proposed, and a major NADPH-dependent metabolite 33 was generated by liver microsomes. To block the metabolic site, 42 was designed and synthesized for further evaluation. Taken together, the imidazo[1,2-a]pyridine scaffold showed promising pharmacological inhibition of c-Met and warrants further investigation.
Keywords: Receptor tyrosine kinase; c-Met inhibitor; imidazo[1,2-a]pyridine; metabolic stability
Receptor tyrosine kinases (RTKs) have a critical role in the development and progression of many types of cancer.1 The receptor tyrosine kinase c-Met is expressed mainly by epithelial cells of many organs.2 Activation of c-Met is regulated upon binding with its natural ligand, hepatocyte growth factor (HGF),3 and interaction with other membrane receptors.4 The normal functions of HGF/c-Met pathway are largely restricted to embryogenesis, tissue injury repair, and regeneration in adults.5 Dysregulation of the HGF/c-Met pathway (through, e.g., c-Met transcriptional upregulation, MET gene amplification or rearrangement, and activating mutations of MET gene) can induce cell proliferation, invasion, migration, and apoptosis avoidance, leading to tumor growth, angiogenesis, and metastasis.3 Importantly, aberrant c-Met activation observed frequently in many human solid tumors and hematological malignancies is associated with poor clinical outcomes.6 Furthermore, overactivation of c-Met causes therapeutic resistance.7 For these reasons, c-Met has become an attractive target for cancer therapy.
Recently, a numerous number of small molecule c-Met inhibitors have been reported (Figure 1). Compound 1 (Crizotinib)8 developed by Pfizer is a potent c-Met/ALK dual inhibitor, which demonstrated marked efficacy for a subset of nonsmall cell lung cancer (NSCLC) patients with an EML4-ALK fusion gene. Phase I clinical trial of inhibitor 2 (JNJ38877605)9 has been terminated because of an increase in serum creatinine levels. Inhibitor 3 (SGX-523) is no longer in clinical development because of acute renal failure.10 Without any safety concerns, the clinical study of inhibitor 4 (PF-04217903)11 was prematurely discontinued. In addition, compounds 5 (AMG-337)12 and 6 (INC-280)13 were reported and have entered phase II clinical trials.
Figure 1.

Representative c-Met inhibitors.
Among the known c-Met inhibitors, bicyclic triazole-based or triazine-based inhibitors demonstrated high c-Met inhibitory potency and excellent selectivity against other kinases. However, many projects of these inhibitors in clinical or discovery development were stopped due to toxicity or unknown reasons.9,10,14 Thus, there is still a need to discover new classes of inhibitors against c-Met for the treatment of cancer patients. The publicly available cocrystal structure of PF-04217903 within c-Met (PDB 3ZXZ) disclosed the binding mode of this kind of inhibitors.11 Generally, the quinoline nitrogen atom participates in a hydrogen bond with the hinge region residue Met-1160. The N-3 nitrogen of triazolopyrazine backbone formed a hydrogen bond with Asp-1222. Meanwhile, triazolopyrazine backbone plays an important role in both c-Met activity and kinase selectivity via the unique face-to-face π-stacking interaction with the activation loop residue Tyr-1230, and there is a positive correlation between electron deficiency of bicyclic aromatic rings and the strength of π–π interaction with Tyr-1230.11 The substituted aryl/heteroaryl (e.g., 1-methylpyrazole) on the backbone reaches out into the solvent (Figure 2). Bioisosteric replacement is a powerful method for the identification of novel chemical series in medicinal chemistry. On the basis of the previous work by Merck, 8-fluoroimidazo[1,2-a]pyridine has been established as a physicochemical mimic of imidazo[1,2-a]pyrimidine, using both in silico and traditional techniques.15 To the best of our knowledge, there are still no examples of using this strategy to discover novel c-Met inhibitors. Hence, we tried to replace the 8-position N atom with a C–F bond to mimic the properties of N-8 atom, including electrostatic surface and lipophilicity to keep the electron deficiency of bicyclic aromatic rings. Meanwhile, we tried to introduce Cl, CN, and CF3 on the imidazo[1,2-a]pyridine to evaluate the effluence of the electron density of bicyclic aromatic rings on c-Met inhibition. On the basis of the cocrystal structural information and principles of bioisosterism, we designed a novel imidazo[1,2-a]pyridine scaffold as c-Met inhibitors. Herein, we describe our recent effort on the synthesis and SARs of this series of compounds.
Figure 2.
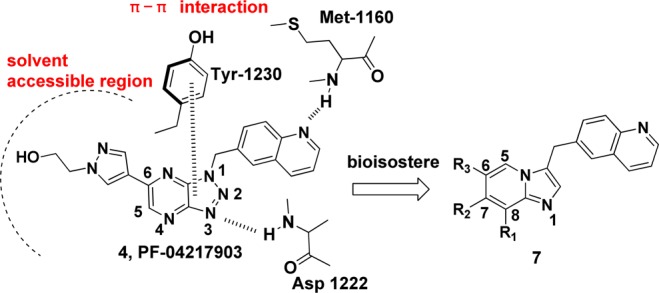
Design of the imidazo[1,2-a]pyridine scaffold.
To achieve a structure–activity relationship (SAR) exploration, efficient synthetic routes were employed for preparation of the analogues (Schemes 1 and 2 in Supporting Information).
These novel imidazo[1,2-a]pyridine derivatives against c-Met have not been previously synthesized and evaluated; therefore, the initial goal was to identify the optimal substituent. To identify potent c-Met inhibitors efficiently, we initially selected 1-methylpyrazole as a substituent at the C-6 position while varying the C-7 and C-8 substituents (Table 1). The initial biochemical assay found that compound 15a exhibited moderate activity with an enzymatic IC50 of 2.18 μM against c-Met. The activity did not significantly change with the introduction of different substituents at the C-7 or C-8 position (15b–15f). To our delight, compound 15g exhibited c-Met inhibition with an enzymatic IC50 of 7.8 nM and an EBC-1 cell IC50 of 0.27 μM, respectively. These encouraging results prompted us to investigate the SAR for substituents on the pyrazole while keeping the fluorine substitute at the C-8 position. The incorporation of polar groups, such as an ethanolic group (15h) and a piperidine group (15i) on the pyrazole of 15g resulted in 1.2-fold and 5.4-fold loss of cellular potency, respectively.
Table 1. SAR of Substituents on the Imidazo[1,2-a]pyridine Scaffold.
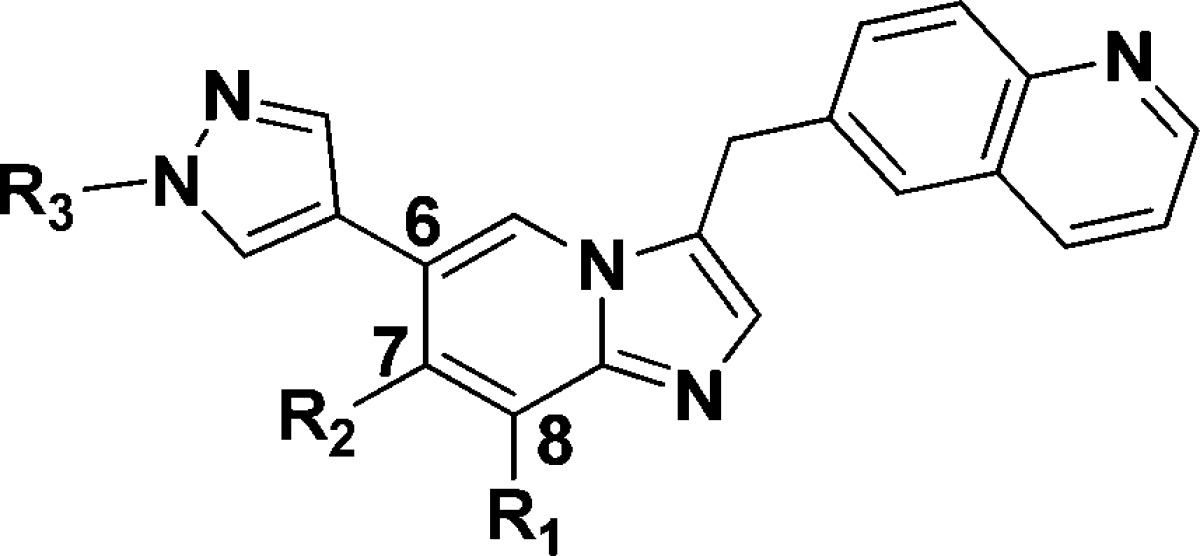
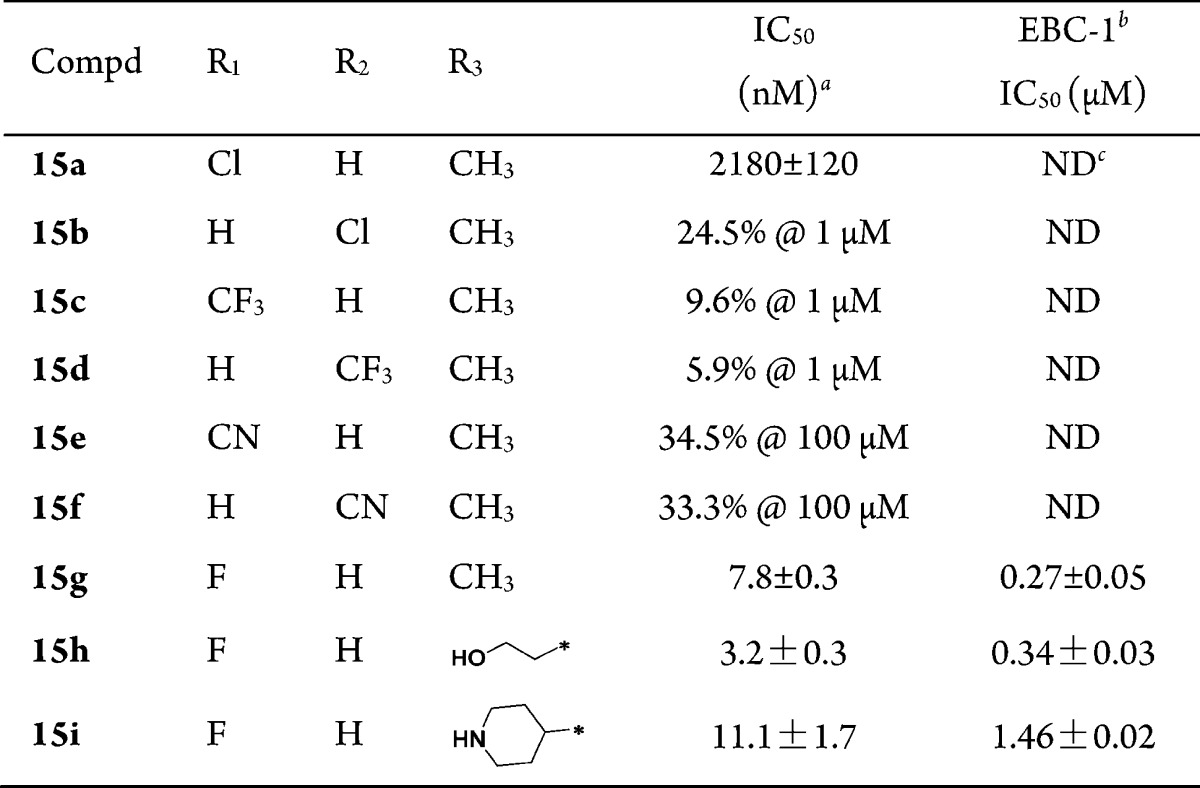
IC50 values were calculated by the Logit method from the results of at least two independent tests with eight concentrations each and expressed as mean ± SD; c-Met IC50 of Crizotinib = 2.4 ± 0.1 nM; c-Met IC50 of JNJ38877605 = 1.8 ± 0.3 nM.
EBC-1: human nonsmall-cell lung cancer cell line that expresses elevated levels of constitutively active c-Met.
Not detected.
To gain structural information for further optimization, the 3D proposed binding modes of representative compounds 15a, 15c, 15e, and 15g were generated by docking simulation (Figure 3). For each compound, the best pose with the lowest binding energy was selected for further analysis with Glide16 and PyMOL.17 The binding mode indicated that compound 15g was bound to the active site of c-Met, similar to PF-04217903 (Figure 3A). The nitrogen of quinoline H-bonds well with the hinge region Met-1160, whereas the N-1 nitrogen of the imidazo[1,2-a]pyridine scaffold formed a hydrogen bond with Asp-1222. In addition, imidazo[1,2-a]pyridine core kept a π–π interaction with electron rich Tyr-1230, which is critical for c-Met inhibition. Compared with 15g, the binding pose of 15c (Figure 3B) was entirely different since the bulky C-8 CF3 substituent could not enter the inside pocket, which could be the main reason for the loss in inhibitory activity. Compared with 8-fluoroimidazo[1,2-a]pyridine core (15g), the 8-chloroimidazo[1,2-a]pyridine is more electron-rich, leading to a decreased interaction with Tyr-1230. Taken together, these results could give an explanation to the different inhibition of these compounds.
Figure 3.
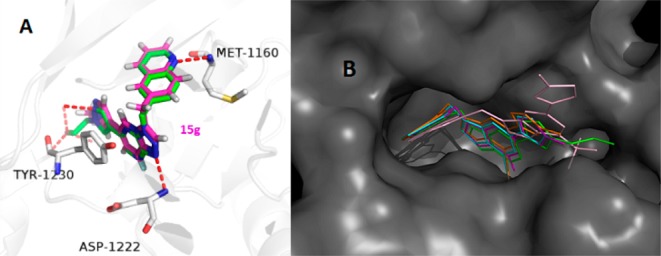
Binding model comparison of designed compounds with PF-04217903. (A) Binding pose of compound 15g (purple) and PF-04217903 (green) with c-Met. (B) Overlay of 15a (blue), 15c (pink), 15e (orange), and 15g (purple) in the same cavity along with PF-04217903 (green) as the reference molecule.
Subsequently, the SAR at the 6-position of the imidazo[1,2-a]pyridine scaffold was investigated (Table 2). Introduction of heteroaryl substituents (16a–c) displayed good c-Met inhibition. The pyridinyl analogue 16b, which incorporated a cyano group, was more effective, and the EBC-1 cell IC50 reached to 188.5 nM. Among the phenyl derivatives (16d–g), benzonitrile analogues 16d and 16f demonstrated improved c-Met inhibition, with an EBC-1 cell IC50 of 106.7 and 145.0 nM, respectively. These observations indicated that incorporation of polar groups on 6-phenyl had a significant influence on cellular activity. Encouraged by these results, analogues incorporated by polar amide groups on 6-phenyl were prepared for SAR exploration. 6-Benzamide analogue 22a displayed 4-fold loss in cellular activity compared with 16d. Derivatives bearing 4-fluoro-3-N-methylbenzamide (22b) and 4-chloro-3-N-methylbenzamide (22c) groups displayed less potent c-Met inhibition than 22a. However, derivatives bearing 3-methoxy-4-N-methylbenzamide (22d) and 3-fluoro-4-N-methylbenzamide (22e) groups exhibited remarkably improved cellular activity. In particular, compound 22e showed an enzymatic IC50 of 3.9 nM and good EBC-1 cell IC50 of 45.0 nM. These results indicated that the 3-F and 3-OMe might be important for c-Met inhibition.
Table 2. SAR at the 6-Position of the Imidazo[1,2-a]pyridine Scaffold.
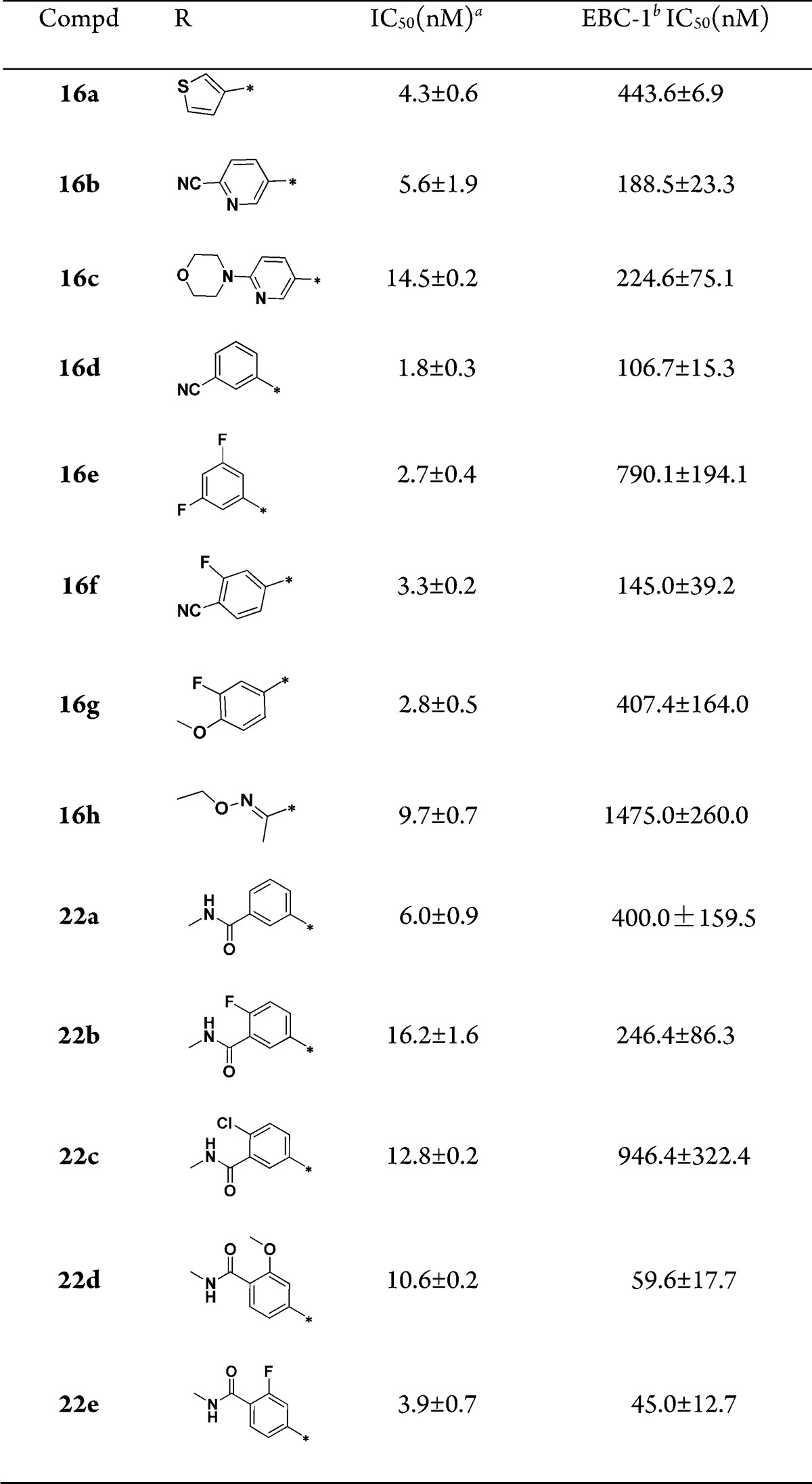
IC50 values were calculated by the Logit method from the results of at least two independent tests with eight concentrations each and expressed as mean ± SD; c-Met IC50 of Crizotinib = 2.4 ± 0.1 nM; c-Met IC50 of JNJ38877605 = 1.8 ± 0.3 nM.
EBC-1: human nonsmall-cell lung cancer cell line that expresses elevated levels of constitutively active c-Met.
To further modulate physical chemical properties of these inhibitors, substituted amides were introduced, especially amides with polar groups such as basic amines on their N-terminus (Table 3). First, the N,N′-disubstituted amide analogues were evaluated. Compounds 22f and 22g showed an EBC-1 cell IC50 of 405.5 and 868.9 nM, respectively, suggesting that N-monosubstituents were preferred here. Next, we investigated the influence of monosubstitution of the N-terminus of the amide on c-Met inhibition. For example, compound 22h, which contained a morpholinoethyl group on the N-terminus of the amide, demonstrated c-Met inhibition by 2-fold compared with 22e, and there was no significant differences between their EBC-1 activity. Replacement of the morpholinoethyl group with a noncyclic dimethylaminoethyl group (compound 22i, enzymatic IC50 = 2.4 nM and EBC-1 cell IC50 = 49 nM) slightly improved the cell inhibitory activity. Compound 22j, with a longer C-chain of the amide, reduced EBC-1 cell inhibitory activity by 3-fold. We also investigated the SARs of the compounds by restricting the side-chain conformation at the N-terminus of the amides (compounds 22k, 22l, and 22o). When the N-terminus of the amide was replaced with a piperidin-2-ylmethyl group (compound 22k, EBC cell IC50 = 276.8 nM) or a piperidin-3-yl group (compound 22l, EBC cell IC50 = 137.5 nM), c-Met inhibition was 5.5- and 4.3-fold more potent than 22e, respectively. In addition, we investigated compounds that contained an aryl group or a heteroaryl group on the N-terminal side of the amides. Compound 22m showed sharply decreased c-Met cell inhibition, which could be caused by an intramolecular hydrogen bond interaction between the hydrogen atom of the amide and the pyridine nitrogen. The potency of compound 22n against EBC-1 cell proliferation is comparable to that of optimal compound 22e.
Table 3. SAR of Substituents on the Amides.
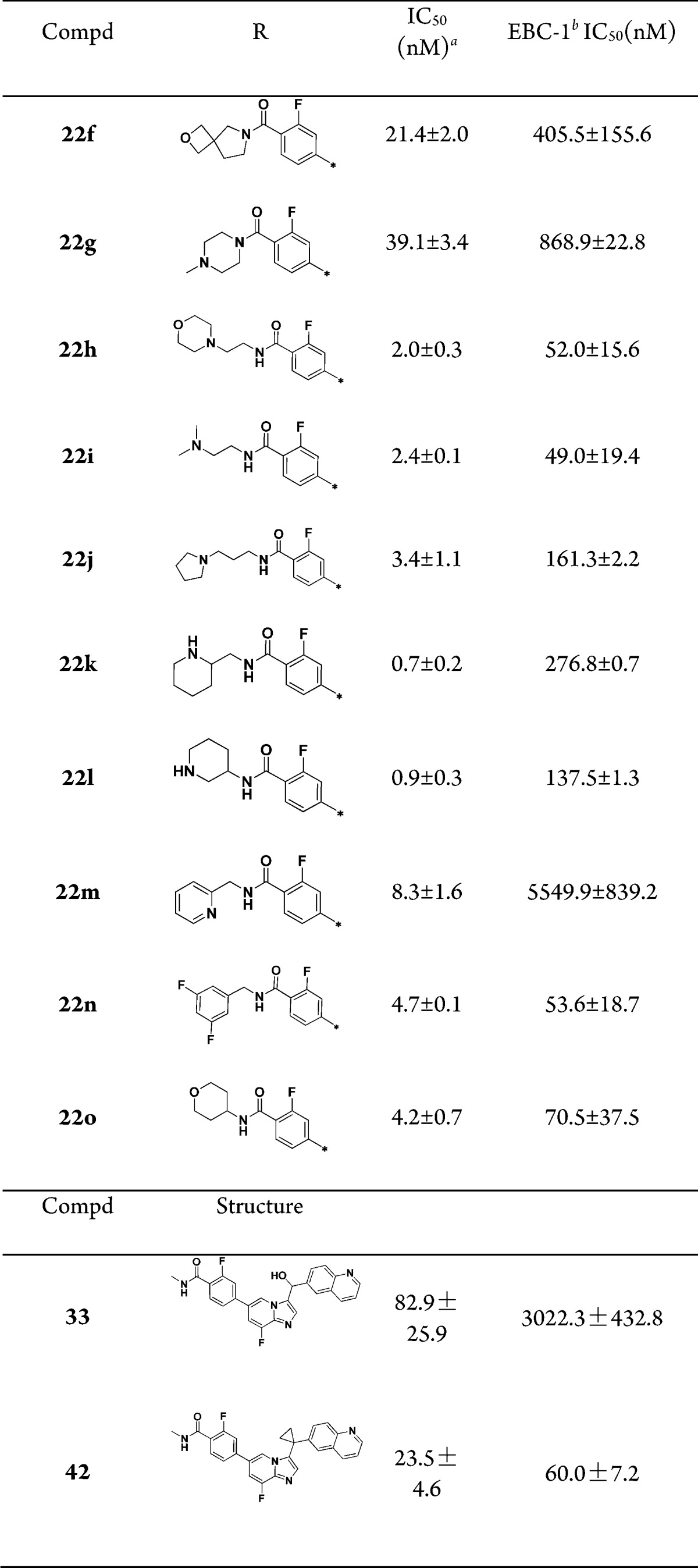
IC50 values were calculated by the Logit method from the results of at least two independent tests with eight concentrations each and expressed as mean ± SD; c-Met IC50 of Crizotinib = 2.4 ± 0.1 nM; c-Met IC50 of JNJ38877605 = 1.8 ± 0.3 nM.
EBC-1: human nonsmall-cell lung cancer cell line that expresses elevated levels of constitutively active c-Met.
Given its promising enzymatic and cellular potency, 22e was selected as the lead compound for subsequent evaluation. In contrast to its high potency against c-Met, 22e had barely any inhibitory effect on the other 21 tested kinases, including c-Met family member RON and highly homologous kinases Axl, c-Mer, and Tyro-3 (IC50 > 10 μM), indicating that 22e was a selective c-Met inhibitor (Table S1 in Supporting Information).
As shown in Figure S1 (See Supporting Information), 22e inhibited the phosphorylation of c-Met and its key downstream molecules Akt and Erk in a dose-dependent manner in representative cancer or model cell lines (EBC-1, MKN45, and BaF3/TPR-Met cells). These results suggested that 22e suppressed c-Met signaling across different oncogenic forms in c-Met overactivated cells.
Compound 22e significantly inhibited cell proliferation of EBC-1, MKN-45, SNU-5, and BaF3/TPR-Met cells, which are characterized by a c-Met-dependent cell growth, with an IC50 value of 45.0 to 203.2 nM (Table 4); however, 22e barely inhibited these other cell lines, of which had Met low expression or activation (IC50 > 50 μM) (Figure S2A in Supporting Information). c-Met inhibition blocks cell proliferation via arresting cells in G1/S phase.18 Compound 22e induced a G1/S phase arrest in the EBC-1 cells, with 81.84% of the cell population in G1 phase in the presence of 1 μM 22e (versus 52.95% in the vehicle control group) (Figure S2B,C in Supporting Information).
Table 4. Effects of 22e on Cell Proliferation.
| IC50 (nM)a | 22e | JNJ38877605 | Crizotinib |
|---|---|---|---|
| EBC-1 | 45.0 ± 12.7 | 9.5 ± 2.0 | 21.8 ± 6.5 |
| MKN-45 | 90.9 ± 17.4 | 10.9 ± 0.4 | 38.1 ± 8.8 |
| SNU-5 | 70.2 ± 19.6 | 15.8 ± 3.2 | 20.4 ± 14.4 |
| BaF3/TPR-Met | 203.2 ± 4.4 | 17.6 ± 2.2 | 127.4 ± 8.1 |
The IC50 values are shown as the mean ± SD (nM) from three separate experiments.
The HGF/c-Met axis activation promotes cell invasion and migration to allow cancer metastasis.19 Compound 22e inhibited the migration of HGF-induced NCI-H441 cells, and almost completely blocked the cell migration phenotype at a dose of 500 nM (Figures S3A,C in Supporting Information). Further, 22e strongly suppressed the invasion of HGF-induced NCI-H441 cell (Figures S3B,D in Supporting Information).
Cell scattering induced by HGF/c-Met activation is a hallmark of cancer invasiveness and metastasis.20 Upon HGF stimulation, epithelial cells undergo colony dispersal and become migratory, fibroblast-like cells.21 Compound 22e treatment reduced HGF-induced cell scattering of MDCK canine kidney epithelial cells in a dose-dependent manner (Figures S4 in Supporting Information). Collectively, these results indicated that 22e inhibited the metastasis and invasiveness phenotype evoked by the HGF/c-Met axis in cancer.
Upon HGF stimulation, c-Met induces several biological responses that collectively give rise to a program known as invasive growth, which is pivotal to drive cancer cell invasion and metastasis.22 Thus, we investigated whether 22e inhibited c-Met-mediated invasive growth. Exposure to 22e inhibited branching morphogenesis in MDCK cells (Figures S5 in Supporting Information), indicating the 22e inhibited HGF-induced c-MET-mediated invasive growth.
The pharmacokinetic (PK) profiles of the selected compound 22e were assessed in Sprague–Dawley (SD) rats (Table S2 in Supporting Information). Compound 22e showed a high area under the curve (AUC0-∞) when dosed orally. The half-life and the absolute oral bioavailability of compound 22e were 1.17 h and 29.4%, respectively.
We used the ultraperformance liquid chromatography quadrupole time-of-flight mass spectrometry (UPLC-qTOF-MS) method to identify the possible metabolites of 22e in liver microsomes of different species. We have proposed the metabolism pathway and all the metabolites of 22e (Scheme 3a in Supporting Information). In view of the result that the major oxidant metabolism take place in the benzyl position of this scaffold, we speculated that metabolite 33 was the major metabolite of 22e after incubation in mice, dog, monkey, and human liver microsomes, except rat liver microsomes (Table S3 in Supporting Information).
The IC50 value of compound 22e on hERG was 93.56 μM using a FluxOR thallium assay. Thus, 22e did not show significant hERG inhibition.
To assess the in vivo antitumor efficacy of 22e, an EBC-1 xenograft model specifically driven by MET amplification was chosen. After 21 days of 22e (methanesulfonic salt) oral administration, dose-dependent tumor growth inhibition was observed in 22e treated groups, with an inhibitory rate of 75.0% (P < 0.05) and 62.9% (P < 0.05) at doses of 100 and 50 mg/kg, respectively (Figure 4). No significant weight losses were observed (data not shown).
Figure 4.
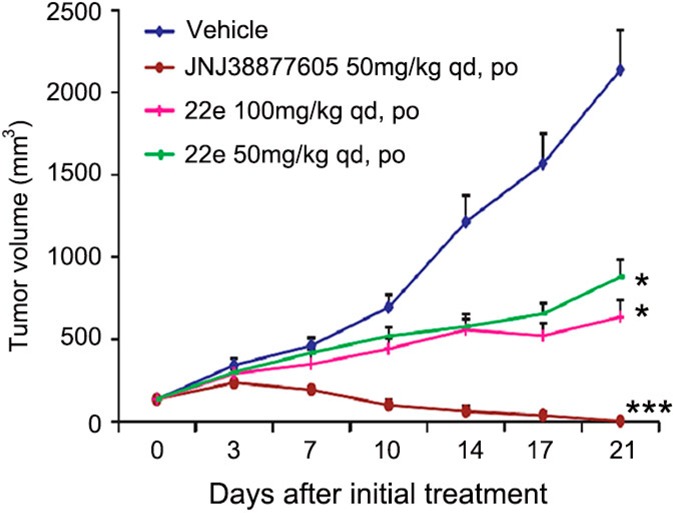
Compound 22e inhibits tumor growth in EBC-1 xenografts. Compound 22e in the methanesulfonic acid salt form or JNJ38877605 was administered orally once daily for 3 weeks after the tumor volume reached 100 to 150 mm3. The results are expressed as the mean ± SEM (drug-treated group, n = 6; vehicle group, n = 12). *p < 0.05, ***p < 0.001 vs control group, determined using Student’s t test.
Furthermore, we synthesized the major metabolite 33 for investigation (Scheme 3b in Supporting Information); however, it had no significant inhibitory effect on c-Met-dependent cell proliferation (enzymatic IC50 = 82.9 ± 25.9 nM and EBC-1 cell IC50 = 3022.3 nM). Since we have identified metabolic hot spots of 22e, we designed and synthesized compound 42 (Scheme 4 in Supporting Information), which bears a cyclopropyl at the linker of this scaffold that might block the metabolic site.23 Even though 42 demonstrated potent inhibitory effect on c-Met kinase and c-Met-dependent cell viability (enzymatic IC50 = 23.5 nM and EBC-1 cell IC50 = 60.0 nM), the in vivo antitumor efficacy of 42 was disappointing. Further optimization to improve the metabolic stability and in vivo antitumor efficacy are ongoing in our laboratory.
In conclusion, a series of novel imidazo[1,2-a]pyridine derivatives were designed, synthesized, and evaluated as c-Met inhibitors by means of bioisosteric replacement. SAR exploration led to the identification of a potent, selective c-Met inhibitor 22e. Compound 22e showed strong potency against c-Met kinase and inhibited c-Met phosphorylation and downstream signaling across different oncogenic forms in c-Met overactivated cancer cells and model cells. Furthermore, 22e treatment resulted in significant antitumor activity in c-Met-driven EBC-1 xenografts. Compound 42 was designed by means of metabolite identification. Further optimizations are currently under investigation.
Glossary
ABBREVIATIONS
- c-Met
mesenchymal-epithelial transition factor
- ALK
anaplastic lymphoma kinase
- SARs
structure–activity relationships
- AUC
area under curve
Supporting Information Available
Synthetic procedures and analytical data for compounds reported in this letter and procedures for in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ C.L., J.A., and D.Z. contributed equally to this work.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grants 91229204 and 81220108025), Major Project of Chinese National Programs for Fundamental Research and Development (2015CB910304), National High Technology Research and Development Program of China (2012AA020302), National Basic Research Program of China (2012CB518005), and National ST Major Projects (2012ZX09103101-072, 2014ZX09507002-001, and 2013ZX09507-001).
The authors declare no competing financial interest.
Supplementary Material
References
- Zwick E.; Bange J.; Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr. Relat. Cancer 2001, 8, 161–173. [DOI] [PubMed] [Google Scholar]
- Trusolino L.; Comoglio P. M. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [DOI] [PubMed] [Google Scholar]
- Bottaro D. P.; Rubin J. S.; Faletto D. L.; Chan A. M.; Kmiecik T. E.; Vande Woude G. F.; Aaronson S. A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [DOI] [PubMed] [Google Scholar]
- Garouniatis A.; Zizi-Sermpetzoglou A.; Rizos S.; Kostakis A.; Nikiteas N.; Papavassiliou A. G. Vascular endothelial growth factor receptors 1,3 and caveolin-1 are implicated in colorectal cancer aggressiveness and prognosis--correlations with epidermal growth factor receptor, CD44v6, focal adhesion kinase, and c-Met. Tumour Biol. 2013, 34, 2109–2117. [DOI] [PubMed] [Google Scholar]
- Porter J. Small molecule c-Met kinase inhibitors: a review of recent patents. Expert Opin. Ther. Pat. 2010, 20, 159–177. [DOI] [PubMed] [Google Scholar]
- Parr C.; Watkins G.; Mansel R. E.; Jiang W. G. The hepatocyte growth factor regulatory factors in human breast cancer. Clin. Cancer Res. 2004, 10, 202–211. [DOI] [PubMed] [Google Scholar]
- Engelman J. A.; Zejnullahu K.; Mitsudomi T.; Song Y.; Hyland C.; Park J. O.; Lindeman N.; Gale C. M.; Zhao X.; Christensen J.; Kosaka T.; Holmes A. J.; Rogers A. M.; Cappuzzo F.; Mok T.; Lee C.; Johnson B. E.; Cantley L. C.; Janne P. A. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dube M.; Shen H.; Nambu M.; Kung P. P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [DOI] [PubMed] [Google Scholar]
- Clinical trial data: https://www.clinicaltrials.gov/ct2/show/NCT00651365.
- Diamond S.; Boer J.; Maduskuie T. P. Jr.; Falahatpisheh N.; Li Y.; Yeleswaram S. Species-specific metabolism of SGX523 by aldehyde oxidase and the toxicological implications. Drug Metab. Dispos. 2010, 38, 1277–1285. [DOI] [PubMed] [Google Scholar]
- Cui J. J.; McTigue M.; Nambu M.; Tran-Dube M.; Pairish M.; Shen H.; Jia L.; Cheng H.; Hoffman J.; Le P.; Jalaie M.; Goetz G. H.; Ryan K.; Grodsky N.; Deng Y. L.; Parker M.; Timofeevski S.; Murray B. W.; Yamazaki S.; Aguirre S.; Li Q.; Zou H.; Christensen J. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1 -yl)ethanol (PF-04217903) for the treatment of cancer. J. Med. Chem. 2012, 55, 8091–8109. [DOI] [PubMed] [Google Scholar]
- Clinical trial data: https://www.clinicaltrials.gov/ct2/show/NCT02016534.
- Clinical trial data: https://www.clinicaltrials.gov/ct2/show/NCT01737827.
- Clinical trial data: https://www.clinicaltrials.gov/ct2/show/NCT00706355.
- Humphries A. C.; Gancia E.; Gilligan M. T.; Goodacre S.; Hallett D.; Merchant K. J.; Thomas S. R. 8-Fluoroimidazo[1,2-a]pyridine: synthesis, physicochemical properties and evaluation as a bioisosteric replacement for imidazo[1,2-a]pyrimidine in an allosteric modulator ligand of the GABA A receptor. Bioorg. Med. Chem. Lett. 2006, 16, 1518–1522. [DOI] [PubMed] [Google Scholar]
- Glide, version 5.5, Schrödinger, LLC, New York, NY, 2009. [Google Scholar]
- PyMOL, Version 1.4.1, Schrödinger, LLC, New York, NY, 2011. [Google Scholar]
- Bertotti A.; Burbridge M. F.; Gastaldi S.; Galimi F.; Torti D.; Medico E.; Giordano S.; Corso S.; Rolland-Valognes G.; Lockhart B. P.; Hickman J. A.; Comoglio P. M.; Trusolino L. Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci. Signaling 2009, 2, ra80. [DOI] [PubMed] [Google Scholar]
- Jeffers M.; Rong S.; Vande Woude G. F. Hepatocyte growth factor/scatter factor-Met signaling in tumorigenicity and invasion/metastasis. J. Mol. Med. 1996, 74, 505–513. [DOI] [PubMed] [Google Scholar]
- Stoker M.; Gherardi E.; Perryman M.; Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [DOI] [PubMed] [Google Scholar]
- Thiery J. P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–54. [DOI] [PubMed] [Google Scholar]
- Gherardi E.; Birchmeier W.; Birchmeier C.; Vande Woude G.; Targeting M. E. T. in cancer: rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Metcalf B.; Xu M.; He C.; Zhang C.; Qian D.; Burns D. M.; Li Y.; Yao W.. Imidazotriazines and Imidazopyrimidines as kinase inhibitors. PCT Int. Appl. WO 2008/064157A1, 2008.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.