

Abstract

Deregulated kinase activities of tropomyosin receptor kinase (TRK) family members have been shown to be associated with tumorigenesis and poor prognosis in a variety of cancer types. In particular, several chromosomal rearrangements involving TRKA have been reported in colorectal, papillary thyroid, glioblastoma, melanoma, and lung tissue that are believed to be the key oncogenic driver in these tumors. By screening the Novartis compound collection, a novel imidazopyridazine TRK inhibitor was identified that served as a launching point for drug optimization. Structure guided drug design led to the identification of (R)-2-phenylpyrrolidine substituted imidazopyridazines as a series of potent, selective, orally bioavailable pan-TRK inhibitors achieving tumor regression in rats bearing KM12 xenografts. From this work the (R)-2-phenylpyrrolidine has emerged as an ideal moiety to incorporate in bicyclic TRK inhibitors by virtue of its shape complementarity to the hydrophobic pocket of TRKs.
Keywords: Neurotrophins, tropomyosin receptor kinase, TRK, (R)-2-phenylpyrrolidine, imidazopyridazines, GNF-8625
The tropomyosin receptor kinase (TRK) receptors, TRKA, TRKB, and TRKC (also known as NTRK1, NTRK2 and NTRK3) and their respective neurotrophin ligands (NGF, BDNF, and NT-3) are implicated in proliferation, survival, differentiation, and functional regulation of different neuronal populations in the nervous system, especially during embryogenesis.1 In the past decade, the dysregulation of neurotrophin signaling has been linked to the development and progression of cancer.2−9 Several different mechanisms have been proposed involving oncogenic activation of TRK receptors, expression of alternative and constitutively active splice variants under hypoxia conditions, point mutations, and genomic rearrangements.2 TRKA rearrangement leading to constitutively active and ligand-independent fused protein TPM3-TRKA was originally identified in a colorectal carcinoma biopsy10,11 and was later found to be associated with a subset of papillary thyroid carcinomas (PTC).12−15 Recently the TPM3-TRKA fusion has also been described to be harbored in KM12 colorectal cells.16 Other TRKA fusions with TGF and TPR have been found in colorectal and thyroid cancers,10−15 and recently, TRKA fusions with MPRIP and CD74 have been identified in a subset of patients (3.3%) with lung adenocarcinoma that did not contain other common genetic alterations.16 TRKC fusion protein Tel-TRKC has been identified as the causative event in rare cancers such as secretory breast carcinoma (SBC),17 congenital fibrosarcoma (CFS),18 and congenital mesoblastic nephroma (CMN).19 In addition, there may be a role for small molecule inhibitors in other cancer settings. For example, BDNF levels have been associated with unfavorable pathological parameters and adverse clinical outcome in breast cancer, and TRKB has been reported as a strong predictor of aggressive tumor growth in neuroblastoma.20−23 Selective TRK inhibitors, AZ623 and GNF-4256, have been shown to inhibit the growth of human neuroblastoma xenograft tumors and, in combination with chemotherapy, have prolonged inhibition of tumor regrowth.24,25 Moreover, high and increased expression of TRK receptors in pancreatic cancer correlates with tumor progression, perineural invasion, pain, and metastasis.7,26−28
Given the growing evidence for a role of TRK in cancer there is interest in discovering developable selective TRK inhibitors for evaluation in the clinic. Here we describe the discovery of (R)-2-phenylpyrrolidine substituted imidazopyridazines as a novel class of selective pan-TRK inhibitors with efficacy in a KM12 rat tumor model.
The novel benzonitrile substituted imidazopyridazine 1 was identified as an inhibitor of TRKB (IC50 = 83 nM) from screening using a TRKB biochemical inhibition assay (Scintillation Proximity Assay). Submicromolar cellular activity across the three TRK isoforms was shown in Ba/F3 assays where Ba/F3 cells were rendered TRKA, TRKB, and TRKC dependent and IL-3 independent by respectively overexpressing the constitutively active Tel-TRKA, Tel-TRKB, and Tel-TRKC fusions (Table 1). In this assay setting, 1 showed differential cytotoxicity as compared to parental wild-type (WT) Ba/F3 cells grown in the presence of IL-3. The cellular kinase selectivity profile of 1 was determined in a Ba/F3 panel using Ba/F3 cells rendered IL-3 independent by stable transduction with 29 selected kinases activated by fusion with a dimerizing protein partner (e.g. BCR or Tel).29 In this panel, 1 showed reasonable selectivity with submicromolar inhibitory activity for FLT3 and ROS (Scheme 1, Table 3).
Table 1. Cellular Activities of Imidazopyridazines.
| Cellular
Ba/F3 assaysa |
||||
|---|---|---|---|---|
| compd | TRKA | TRKB | TRKC | WTb |
| 1 | 0.85 | 0.27 | 0.10 | 7.13 |
| 2 | 0.74 | 0.20 | 0.23 | >10 |
| 3 | 0.050 | 0.021 | 0.006 | 3.92 |
| 4 | 0.17 | 0.12 | 0.075 | 1.01 |
| 5 | 0.076 | 0.027 | 0.040 | 1.92 |
| 6 | 0.006 | 0.007 | 0.003 | 3.00 |
| 7 | 0.027 | 0.013 | 0.004 | 2.53 |
| 8 | 0.52 | 0.13 | 0.020 | 5.89 |
| 9 | 0.17 | 0.050 | 0.011 | >10 |
| 10 | 0.075 | 0.047 | 0.019 | >10 |
| 11 | 0.068 | 0.029 | 0.008 | >10 |
| 12 | 3.13 | 1.97 | 0.98 | 9.23 |
| 13 | 0.021 | 0.011 | 0.003 | >10 |
| 14 | 0.003 | 0.001 | 0.001 | >10 |
| 15 | 0.005 | 0.004 | 0.002 | >10 |
| 16 | 0.001 | 0.001 | <0.001 | 4.79 |
| 17 | 0.004 | 0.004 | 0.002 | 5.91 |
Proliferation assays with Tel-TRK fusions; IC50, μM.
Proliferation assay using parental Ba/F3 cells.
Scheme 1. Imidazopyridazines SAR Progression and Numbering Assignment.

Table 3. Ba/F3 Cellular Kinase Panel (IC50, μM).
| Ba/F3a | 1 | 14 | 16 | 17 |
|---|---|---|---|---|
| ABLb | 3.8 | NA | 2.4 | 5.2 |
| ALKc | 4.8 | 6.3 | 1.2 | 3.0 |
| ALK | 3.1 | NA | 2.2 | 3.9 |
| BRAF | 2.5 | NA | 6.2 | 5.9 |
| BMX | 9.7 | >10 | 2.7 | 4.3 |
| FGFR3 | 3.9 | 9.5 | 4.9 | 5.0 |
| FGFR4 | 5.9 | 9.2 | 5.3 | 4.4 |
| FGR | 6.4 | 8.7 | 2.8 | 5.9 |
| FLT1 | 3.0 | NA | 2.6 | 5.3 |
| FLT3 | 0.72 | 8.1 | 3.3 | 4.3 |
| FMS | 3.4 | >10 | 2.5 | 5.3 |
| IGF1R | 4.0 | NA | 5.1 | 5.4 |
| INSR | 8.6 | 8.8 | 5.3 | 6.0 |
| JAK2 | 4.7 | 6.7 | 2.7 | 5.6 |
| KDR | 4.1 | 6.5 | 1.0 | 5.0 |
| KIT | 2.2 | 5.5 | 0.79 | 7.0 |
| LCK | 2.0 | NA | 3.0 | 5.2 |
| LYN | 7.5 | 6.4 | 3.4 | 4.9 |
| MER | 5.4 | NA | 4.7 | 5.1 |
| MET | 5.1 | 7.7 | 4.3 | 5.7 |
| PDGFRβ | 2.0 | NA | 2.2 | 4.6 |
| RET | 4.9 | 8.0 | 3.7 | 6.0 |
| RON | 6.1 | NA | 5.0 | 4.8 |
| ROS | 0.30 | 0.6 | 0.06 | 0.21 |
| SRC | 4.6 | NA | 2.5 | 4.6 |
| SYK | 4.0 | NA | 4.4 | 7.9 |
| TIE1 | 5.5 | 6.5 | 4.3 | 5.5 |
| TYRO3 | NA | NA | 5.2 | 5.0 |
| ZAP70 | 6.4 | NA | 5.2 | 5.5 |
Ba/F3 cells rendered IL-3 independent by stable transduction with the indicated kinase fused with a Tel dimerization partner unless otherwise specified.
BCR-ABL.
NMP-ALK.
The X-ray cocrystal structure of 1 showed binding to the inactivated (DFG-out) kinase domain of TRKC and forming a key hydrogen bond between N1 of the imidazopyridazine and the hinge amide NH of Met620 (Figure 1).30,31 The benzonitrile ring was wedged between phenyl rings from the gatekeeper Phe617, Phe698 from the DFG motif, and the phenyl ring of the benzylamino group of compound 1. This phenyl ring sits under the glycine-rich loop and faces the solvent exposed front of the binding pocket, lending itself to immediate structure–activity relationship (SAR) investigations.
Figure 1.
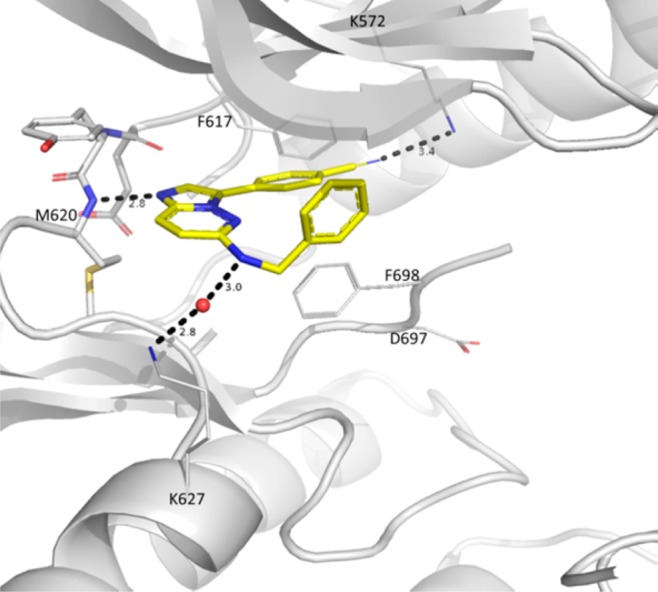
X-ray cocrystal structure of 1 (carbon in yellow) binding to the active site of TRKC kinase. Hydrogen bonds are depicted as dashed lines. Nitrogen atoms are shown in blue, oxygen atoms in red, and a water molecule depicted as a red sphere.
Chemistry optimization was initiated with the goal of identifying compounds with improved potency and pharmacokinetic properties suitable for in vivo profiling. New compounds were screened for antiproliferative effects in Ba/F3-Tel-TRKA, TRKB, and TRKC assays, and cytotoxicity effects were compared with parental wild-type (WT) Ba/F3 cells. Initial efforts focused on exploring the SAR around the benzyl amine moiety (R1, R2, R4) and the phenyl group linked to the imidazopyrimidine core (R3) (Scheme 1 and Table 1).
From this work, it appeared that nitrile substitutions in the phenyl group (R3) at the 3 or 4 ring-position were equally tolerated (1 vs 2). It was also observed that 3-substitution at the benzylamine phenyl ring (R1) led to a 10-fold increase in potency relative to the unsubstituted 1, especially with EWG groups such as fluorine (3 vs 4). Shimming the phenyl ring (R3) with the potency boosting 3-F substitution at R1 furnished many tolerated functional groups that could either increase solubility or further drive potency such as hydroxymethyl derivative 5, amide 6, and sulfonamide 7.
Methylation of the benzylic amine (R4) led to no significant change in potency (8 vs 1) suggesting that the benzylic amine is not involved in a critical proton donating interaction.
Docking models using the cocrystal structure of 1 with TRKC supported that potency could be further increased by structurally rigidifying the benzyl amine moiety in a cyclic fashion by reducing conformational entropy. Cyclic piperdine, morpholine, and pyrrolidine analogues were synthesized as racemates (9, 10, and 11) and revealed a potency preference toward the five-membered heterocycle. A cocrystal structure between TRKA and 10 indicated a preference for the R-enantiomer and induced protein conformation change from the DFG-out to the DFG-in state, which allowed the fluorophenyl to bind near the ribose binding pocket (Figure 2).30 Moreover, the DFG-in conformation prevented a clash between Phe669 and the compound’s fluorophenyl group. Chiral resolution of the pyrrolidine derivative 11 into its two enantiomers, 12 and 13, indicated the R-enantiomer (13) as the more potent isomer, which also demonstrated a 2-fold activity improvement relative to acyclic compound 3.
Figure 2.
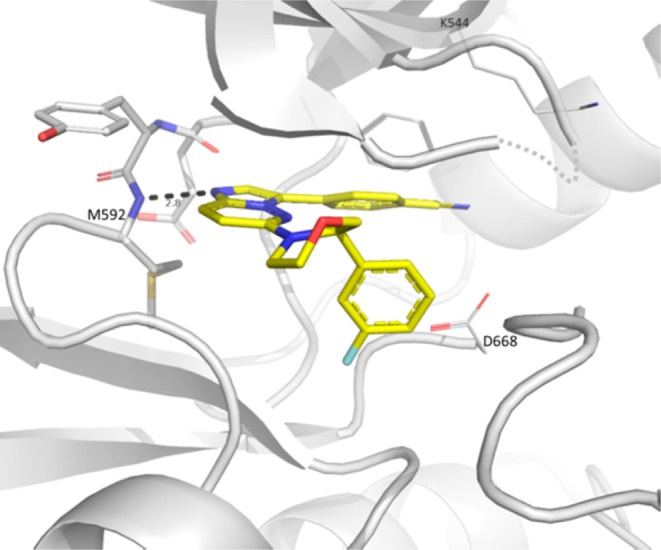
X-ray cocrystal structure of TRKA with 10. The fluorophenyl points toward the pocket usually occupied by the ribose from ATP. The protein adopts a DFG-in conformation to allow for the fluorophenyl to bind. Although the 10 racemate was used for crystallization, only the R-enantiomer binds to TRKA. Color coding according to Figure 1.
By maintaining the 3-F-phenylpyrrolidine in 11, a subsequent round of optimization at R3 was performed. This effort showed that replacing the 3-CN-phenyl moiety with a 2-pyridine (14) led to an 8–20-fold potency improvement across all three TRK isoforms.
The cocrystal structure of 14 with TRKA revealed an unanticipated flip of the compound around the imidazopyridazine core while maintaining the same key hinge interaction (Figure 3).30 Binding could only be observed for the R-enantiomer where the 3-F-phenyl is optimally positioned in the hydrophobic pocket originally occupied by Phe669 and provides excellent shape complementary, likely to contribute significantly to the observed potency improvement. Molecular modeling indicated that two distinct binding modes (i.e. core flipping) are indeed possible for the DFG-in conformation. The preferred binding mode probably depends upon the substitution on the 6-position of the imidazopyridazine. When this substitution is the (R)-phenylpyrrolidine, the “flipped” orientation appears to be preferred as the pyrrolidine anchors the phenyl group in the hydrophobic pocket.
Figure 3.
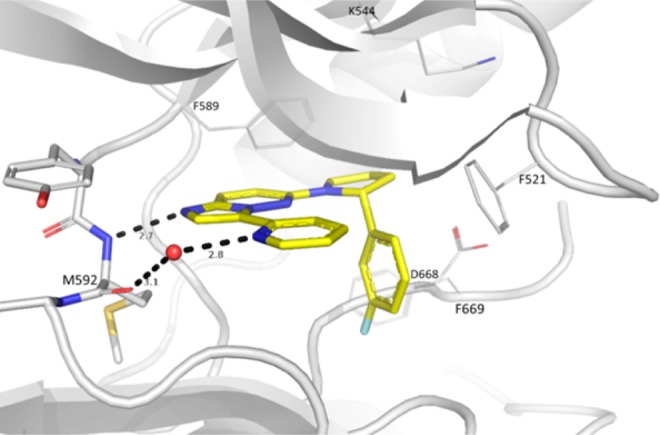
X-ray cocrystal structure of TRKA with the R-enantiomer of 14. The protein adopts the DFG-in conformation. Compound 14 maintains the key hydrogen bond between the hinge NH of Met592 and N1 of imidazopyridazine. Surprisingly, the imidazopyridazine is now flipped. This allows for the fluorophenyl group to better fit the ribose binding pocket compared to 10. Color coding according to Figure 1.
It is worth noting that since our initial disclosure of the (R)-2-phenylpyrrolidine in 2008,32 this moiety has become a privileged TRK structure and was incorporated effectively in closely related scaffolds33−38 and on unrelated bicycle scaffolds.39
The cocrystal structure of 14 indicated that the pyridine ring at the 3-position faces the solvent exposed front of the binding pocket, suggesting that polar groups in this area should be tolerated. Indeed, decorating the 3-position (R5) of the pyridine maintained very good potency across the TRK isoforms (15, 16, and 17).
To determine the in vivo PK properties, 15, 16, and 17 were administered intravenously to Wistar or Sprague–Dawley rats. High plasma clearance (CL), moderate volume distribution (Vss), and short half-life (T1/2) were observed (Table 2). The plasma clearances of these compounds were about 82% (16 and 17) and 100% (15) of rat hepatic blood flow (55 mL/min/kg). Compound 17, with an additional heterocyclic group between the pyridine and the solubilizing group, led to a higher volume of distribution (4.9 vs 2.3 L/kg), which resulted in a longer half-life (1.6 vs 0.9 h) compared to 15 and 16. When administered orally by gavage, 15 showed low bioavailability (10%), while 16 and 17 had 2.6-fold higher oral bioavailability (Table 2). In mouse brain tissue distribution studies, the AUC and Cmax ratios of brain to plasma were lower for 17 (∼0.2) compared to 16 (∼0.8) (Supporting Information).
Table 2. In Vivo Rat PK Properties after Dosing of 3 mg/kg IV and 10 mg/kg PO.
| rat PKa | 15b | 16b | 17c |
|---|---|---|---|
| CL (mL/min/kg) | 56.9 | 45.5 | 45.2 |
| Vss (L/kg) | 2.3 | 2.4 | 4.9 |
| T1/2 (h) | 0.83 | 0.9 | 1.6 |
| AUC PO (h·nM) | 659 | 1897 | 1879 |
| Cmax PO (nM) | 110 | 250 | 215 |
| F(%) PO | 10 | 26 | 27 |
Route: Intravenous (3 mg/kg) and oral administrations (10 mg/kg). Formulation as a solution in 75% PEG300/25% D5W.
Male Sprague–Dawley rat.
Male Wistar rat.
Compounds 14, 16, and 17 were tested in the Ba/F3 panel and showed much improved selectivity across all the kinases when compared to 1 (Table 3; see Table 1 for TRK activity).
The main kinase off-target activity remained ROS but with an improved selectivity index (SI) (SI < 3 for 1 and SI >50 for 14, 16, and 17). This demonstrated that (R)-2-fluorophenylpyrrolidine-containing analogues were able to achieve higher potency as well as increased selectivity due to their high specificity at the hydrophobic pocket.
In addition, the anti-TRKA activity for compounds 16 and 17 was further demonstrated in two other cellular systems, Ba/F3 and KM12. In Ba/F3 cells engineered to express both TRKA and NGF, both compounds demonstrated potent antiproliferation activity with IC50 of 0.001 and 0.003 μM, respectively. In KM12 cells, derived from a colon cancer cell line that harbors the TPM3-TRKA fusion and is dependent on TRKA kinase for proliferation and survival, the compounds were also very potent with IC50 of 0.001 and 0.01 μM, respectively.
By virtue of its pan-TRK in vitro potency, good selectivity, relatively low brain exposure, and overall acceptable PK properties, 17 (GNF-8625) was selected for in vivo efficacy studies (synthesis in Supporting Information). In a tumor xenograft model derived from the KM12 cell line,4017 demonstrated in vivo antitumor efficacy when administered at ascending doses twice daily (bid) for 14 days in rats (Figure 4).
Figure 4.
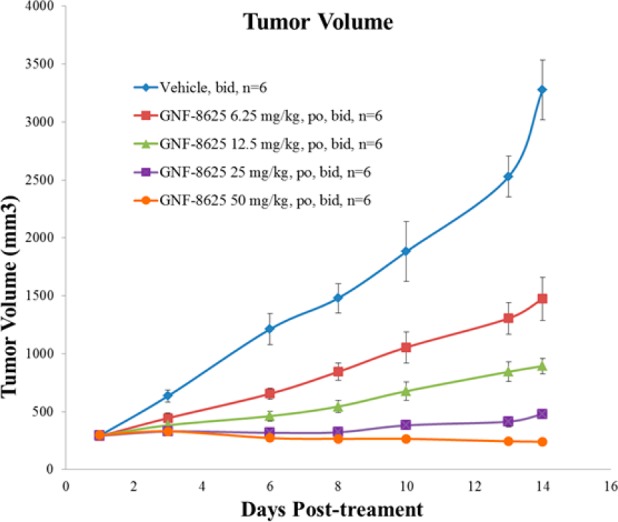
KM12 efficacy model in female CRL RNU nude rats with compound 17 (GNF-8625). Animals were transplanted with KM12 tumor tissues, and dosing began 10 days postimplant. Vehicle and 17 were dosed twice a day (bid) for 14 days. Efficacy measured as tumor volume.
In this study, 17 induced 20% tumor regression at 50 mg/kg bid and achieved partial tumor growth inhibition at 6.25, 12.5, and 25 mg/kg bid, in a dose-dependent manner. A dose proportional increase in exposure of compound 17 was observed, and at the highest doses, a slight increase in body weight was observed (Supporting Information).
In summary, using a TRK/ligand cocrystal structural guided approach, we have developed a series of potent, selective, and efficacious (R)-2-phenylpyrrolidine substituted imidazopyridazine TRK inhibitors. This work has unveiled the (R)-2-phenylpyrrolidine as a privileged moiety for TRK inhibitors due to its optimal shape complementarity to the TRK protein’s hydrophobic pocket available in the “DFG-in” conformation.
Acknowledgments
We thank Daniel Raymond and Thomas H. Marsilje for help in proof reading.
Glossary
ABBREVIATIONS
- TRK
tropomyosin receptor kinase
- NGF
nerve growth factor
- BDNF
brain-derived neurotrophic factor
Supporting Information Available
Experimental procedures, characterization of compounds, kinase selectivity, ADMET data for compound 17, and details of in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Novartis Pharma AG, Novartis Institutes for Biomed. Research, Postfach, CH-4002 Basel, Switzerland.
Author Present Address
‡ Loma Linda University Medical Center, Rehabilitation Institute, 25333 Barton Road, Loma Linda, California 92354, United States.
Author Present Address
§ Sanofi Oncology, 640 Memorial Drive, Cambridge, Massachusetts 02139, United States.
Author Present Address
∥ Parallel Computing Laboratories, Inc., 3525 Del Mar Heights Road, #288, San Diego, California 92130, United States.
Author Present Address
⊥ Novartis Institutes for BioMedical Research, Inc. 250 Massachusetts Avenue, Cambridge, Massachusetts 02139, United States.
Author Present Address
# Novartis Pharmaceuticals Corporation, One Health Plaza, East Hanover, New Jersey 07936, United States.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang E. J.; Reichardt L. F. TRK Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [DOI] [PubMed] [Google Scholar]
- Pierotti M. A.; Greco A. Oncogenic Rearrangements of the NTRK1/NGF Receptor. Cancer. Lett. 2006, 232, 90–98. [DOI] [PubMed] [Google Scholar]
- Lei L.; Parada L. F. Transcriptional Regulation of TRK Family Neurotrophin Receptors. Cell. Mol. Life Sci. 2007, 64, 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadec C.; Meignan S.; Adriaenssens E.; Foveau B.; Vanhecke E.; Romon R.; Toillon R. A.; Oxombre B.; Hondermarck H.; Le Bourhis X. TRKA Overexpression Enhances Growth and Metastasis of Breast Cancer Cells. Oncogene 2009, 28, 1960–1970. [DOI] [PubMed] [Google Scholar]
- Borrello M. G.; Bongarzone I.; Pierotti M. A.; Luksch R.; Gasparini M.; Collini P.; Pilotti S.; Rizzetti M. G.; Mondellini P.; De Bernardi B.; Di Martino D.; Garaventa A.; Brisigotti M.; Tonini G. P. TRK and ret Proto-Oncogene Expression in Human Neuroblastoma Specimens: High Frequency of TRK Expression in Non-Advanced Stages. Int. J. Cancer 1993, 54, 540–545. [DOI] [PubMed] [Google Scholar]
- Ma J.; Jiang Y.; Jiang Y.; Sun Y.; Zhao X. Expression of Nerve Growth Factor and Tyrosine Kinase Receptor A and Correlation with Perineural Invasion in Pancreatic Cancer. J. Gastroenterol. Hepatol. 2008, 23, 1852–1859. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Friess H.; diMola F. F.; Zimmermann A.; Graber H. U.; Korc M.; Buchler M. W. Nerve Growth Factor Expression Correlates with Perineural Invasion and Pain in Human Pancreatic Cancer. J. Clin. Oncol. 1999, 17, 2419–2428. [DOI] [PubMed] [Google Scholar]
- Festuccia C.; Muzi P.; Gravina G. L.; Millimaggi D.; Speca S.; Dolo V.; Ricevuto E.; Vicentini C.; Bologna M. Tyrosine Kinase Inhibitor CEP-701 Blocks the NTRK1/NGF Receptor and Limits the Invasive Capability of Prostate Cancer Cells in vitro. Int. J. Oncol. 2007, 30, 193–200. [PubMed] [Google Scholar]
- Davidson B.; Reich R.; Lazarovici P.; Nesland J. M.; Skrede M.; Risberg B.; Trope C. G.; Florenes V. A. Expression and Activation of the Nerve Growth Factor Receptor TRKA in Serous Ovarian Carcinoma. Clin. Cancer. Res. 2003, 9, 2248–2259. [PubMed] [Google Scholar]
- Martin-Zanca D.; Hughes S. H.; Barbacid M. A Human Oncogene Formed by the Fusion of Truncated Tropomyosin and Protein Tyrosine Kinase Sequences. Nature 1986, 319, 743–748. [DOI] [PubMed] [Google Scholar]
- Butti M. G.; Bongarzone I.; Ferraresi G.; Mondellini P.; Borrello M. G.; Pierotti M. A. A Sequence Analysis of the Genomic Regions Involved in the Rearrangements Between TPM3 and NTRK1 Genes Producing TRK Oncogenes in Papillary Thyroid Carcinomas. Genomics 1995, 28, 15–24. [DOI] [PubMed] [Google Scholar]
- Greco A.; Mariani C.; Miranda C.; Pagliardini S.; Pierotti M. A. Characterization of the NTRK1 Genomic Region Involved in Chromosomal Rearrangements Generating TRK Oncogenes. Genomics 1993, 18, 397–400. [DOI] [PubMed] [Google Scholar]
- Bounacer A.; Schlumberger M.; Wicker R.; Du-Villard J. A.; Caillou B.; Sarasin A.; Suarez H. G. Search for NTRK1 Proto-Oncogene Rearrangements in Human Thyroid Tumours Originated After Therapeutic Radiation. Br. J. Cancer 2000, 82, 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beimfohr C.; Klugbauer S.; Demidchik E. P.; Lengfelder E.; Rabes H. M. NTRK1 Re-Arrangement In Papillary Thyroid Carcinomas of Children After the Chernobyl Reactor Accident. Int. J. Cancer 1999, 80, 842–847. [DOI] [PubMed] [Google Scholar]
- Alberti L.; Carniti C.; Miranda C.; Roccato E.; Pierotti M. A. RET and NTRK1 Proto-Oncogenes in Human Diseases. J. Cell. Phyisiol. 2003, 195, 168–186. [DOI] [PubMed] [Google Scholar]
- Vaishnavi A.; Capelletti M.; Le A. T.; Kako S.; Butaney M.; Ercan D.; Mahale S.; Davies K. D.; Aisner D. L.; Pilling A. B.; Berge E. B.; Kim J.; Sasaki H.; Park S.; Kryukov G.; Garraway L. A.; Hammerman P. S.; Haas J.; Andrews S. W.; Lipson D.; Stephens P. J.; Miller V. A.; Varella-Garcia M.; Jänne P. A.; Doebele R. C. Oncogenic and Drug-Sensitive NTRK1 Rearrangements in Lung Cancer. Nat. Med. 2013, 19, 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognon C.; Knezevich S. R.; Huntsman D.; Roskelley C. D.; Melnyk N.; Mathers J. A.; Becker L.; Carneiro F.; MacPherson N.; Horsman D.; Poremba C.; Sorensen P. H. B. Expression of the ETV6-NTRK3 Gene Fusion as a Primary Event in Human Secretory Breast Carcinoma. Cancer Cell 2002, 2, 367–376. [DOI] [PubMed] [Google Scholar]
- Lannon C. L.; Sorensen P. H. B. ETV6-NTRK3: a Chimeric Protein Tyrosine Kinase with Transformation Activity in Multiple Cell Lineages. Semin. Cancer Biol. 2005, 15, 215–223. [DOI] [PubMed] [Google Scholar]
- Watanabe N.; Kobayashi H.; Hirama T.; Kikuta A.; Koizumi S.; Tsuru T.; Kaneko Y. Cryptic t(12;15)(p13;q26) Producing the ETV6-NTRK3 Fusion Gene and No Loss of IGF2 Imprinting in Congenital Mesoblastic Nephroma with Trisomy 11 Fluorescence in situ Hybridization and IGF2 Allelic Expression Analysis. Cancer Genet. Cytogenet. 2002, 136, 10–16. [DOI] [PubMed] [Google Scholar]
- Patani N.; Jiang W. G.; Mokbel K. Brain derived Neurotrophic Factor Expression Predicts Adverse Pathological and Clinical Outcomes in Human Breast Cancer. Cancer Cell. Int. 2011, 11, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesler R.; Brunetto de Farias C.; Abujamra A. L.; Brunetto A. L.; Schwartsmann G. BDNF/TrkB Signaling as an Anti-Tumor Target. Expert Rev. Anticancer Ther. 2011, 11, 1473–1475. [DOI] [PubMed] [Google Scholar]
- Izbicka E.; Izbicki T. Therapeutic Strategies for the Treatment of Neuroblastoma. Curr. Opin. Investig. Drugs 2005, 6, 1200–1214. [PubMed] [Google Scholar]
- Douma S.; Van Laar T.; Zevenhoven J.; Meuwissen R.; Van Garderen E.; Peeper D. S. Suppression of Anoikis and Induction of Metastasis by the Neurotrophic Receptor TRKB. Nature 2004, 430, 1034–1039. [DOI] [PubMed] [Google Scholar]
- Zage P. E.; Graham T. C.; Zeng L.; Fang W.; Pien C.; Thress K.; Omer C.; Brown J. L.; Zweidler-McKay P. A. The Selective Trk Inhibitor AZ623 Inhibits Brain-Derived Neurotrophic Factor-Mediated Neuroblastoma Cell Proliferation and Signaling and is Synergistic with Topotecan. Cancer 2011, 117, 1321–1329. [DOI] [PubMed] [Google Scholar]
- Croucher J. L.; Iyer R.; Li N.; Molteni V.; Loren J.; Gordon W. P.; Tuntland T.; Liu B.; Brodeur G. M. TrkB Inhibition by GNF-4256 Slows Growth and Enhances Chemotherapeutic Efficacy in Neuroblastoma Xenografts. Cancer Chemother. Pharmacol. 2015, 75, 131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miknyoczki S. J.; Chang H.; Klein-Szanto A.; Dionne C. A.; Ruggeri B. A. The TRK Tyrosine Kinase Inhibitor CEP-701 (KT-5555) Exhibits Significant Antitumor Efficacy in Preclinical Xenograft Models of Human Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 1999, 5, 2205–2212. [PubMed] [Google Scholar]
- Ketterer K.; Rao S.; Friess H.; Weiss J.; Buechler M. W.; Korc M. Reverse Transcription-PCR Analysis of Laser-Captured Cells Points to Potential Paracrine and Autocrine Actions of Neurotrophins in Pancreatic Cancer. Clin. Cancer Res. 2003, 9, 5127–5136. [PubMed] [Google Scholar]
- Sclabas G. M.; Fujioka S.; Schmidt C.; Li Z.; Frederick W. A. I.; Yang W.; Yokoi K.; Evans D. B.; Abbruzzese J. L.; Hess K. R.; Zhang W.; Fidler I. J.; Chiao P. Overexpression of Tropomysin-Related Kinase B in Metastatic Human Pancreatic Cancer Cells. Clin. Cancer Res. 2005, 11, 440–449. [PubMed] [Google Scholar]
- Melnick J. S.; Janes J.; Kim S.; Chang J. Y.; Sipes D. G.; Gunderson D.; Jarnes L.; Matzen J. T.; Garcia M. E.; Hood T. L.; Beigi R.; Xia G.; Harig R. A.; Asatryan H.; Yan S. F.; Zhou Y.; Gu X. J.; Saadat A.; Zhou V.; King F. J.; Shaw C. M.; Su A. I.; Downs R.; Gray N. S.; Schultz P. G.; Warmuth M.; Caldwell J. S. An Efficient Rapid System for Profiling the Cellular Activities of Molecular Libraries. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 3153–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structure coordinates have been deposited into the PDB: 4YMJ; 4YNE; 4YPS.
- Zuccotto F.; Ardini E.; Casale E.; Angiolini M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2010, 53, 2681–2694. [DOI] [PubMed] [Google Scholar]
- Ferrand S.; Glickman F.; Leblanc C.; Ritchie C.; Shaw D.; Stiefl N. J.; Furet P.; Imbach P.; Stauffer F.; Capraro H. G.; Gessier F.; Gaul C.; Albaugh P. Chopiuk G.. Heterocyclic Compounds as Antiinflammatory Agents. WO/2008/052734; International Application No. PCT/EP2007/009382; May 8, 2008.
- Andrews S. W.; Haas J.; Jiang Y.; Zhang G.. Substituted Imidazo[1,2b]pyridazine Compounds as TRK Kinase Inhibitors. WO/2010/033941; International Application No. PCT/US2009/057729; March 25, 2010.
- Haas J.; Andrews S. W.; Jiang Y.; Zhang G.. Substituted Pyrazolo[1,5-a]pyrimidine Compounds as TRK Kinase Inhibitors. WO/2010/048314 A1; International Application No. PCT/US2009/061519; April 29, 2010.
- Allen S.; Andrews S. S.; Condroski K. R.; Haas J.; Huang L.; Jiang Y.; Kercher T.; Seo J.. Substituted Pyrazolo[1,5-a]pyrimidine Compounds as TRK Kinase Inhibitors. WO/2011/006074 A1; International Application No. PCT/US2010/041538; January 11, 2011.
- Andrews S. W.; Condroski K. R.; Haas J.; Jiang Y.; Kolakowski G. R.; Seo J.; Yang H. W.; Zhao Q.. Macrocyclic Compounds as Trk Kinase Inhibitors. WO/2011/146336 A1; International Application No. PCT/US2011/036452; November 24, 2011.
- Sasmal P. K.; Ahmed S.; Tehim A.; Pradkar V.; Dattatreya P. M.; Mavinahalli N. J.. Substituted Pyrazolo[1,5-a]pyridine as Tropomyosin Receptor Kinase (Trk) Inhibitors. WO/2013/088256 A1; International Application No. PCT/IB2012/003012; June 20, 2013.
- Sasmal P. K.; Ahmed S.; Prabhu G.; Tehim A.; Pradkar V.; Dattatreya M. P.; Mavinahalli N. J.. Substituted Heterocyclic Compounds as Tropomyosin Receptor Kinase A (TRKA) Inhibitors. WO/2013/088257 A1; International Application No. PCT/IB2012/003022; June 20, 2013.
- Stachel S. J.; Sanders J. M.; Henze D. A.; Rudd M. T.; Su H. P.; Li Y.; Nanda K. K.; Egbertson M. S.; Manley P. J.; Jones K. L. G.; Brnardic E. J.; Green A.; Grobler J. A.; Hanney B.; Leitl M.; Lai M. T.; Munshi V.; Murphy D.; Rickert K.; Riley D.; Krasowska-Zoladek A.; Daley C.; Zuck P.; Kane S. A.; Bilodeau M. T. Maximizing Diversity from a Kinase Screen: Identification of Novel and Selective pan-Trk Inhibitors for Chronic Pain. J. Med. Chem. 2014, 57, 5800–5816. [DOI] [PubMed] [Google Scholar]
- Unpublished data.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.