

Abstract
AIM: To investigate the role of reactive oxygen species in the ulcer-aggravating effect of lead in albino rats.
METHODS: Albino Wistar rats were randomly divided into three groups and treated orally with 100 mg/L (low dose) or 5000 mg/L (high dose) of lead acetate for 15 wk. A third group received saline and served as control. At the end of wk 15, colorimetric assays were applied to determine the concentrations of total protein and nitrite, the activities of the oxidative enzymes catalase and superoxide dismutase, and lipid peroxidation in homogenized gastric mucosal samples.
RESULTS: Exposure of rats to lead significantly increased the gastric mucosal damage caused by acidified ethanol. Although the basal gastric acid secretory rate was not significantly altered, the maximal response of the stomach to histamine was significantly higher in the lead-exposed animals than in the unexposed control group. Exposure to low and high levels of lead significantly increased gastric lipid peroxidation to 183.2% ± 12.7% and 226.1% ± 6.8% of control values respectively (P < 0.0). On the other hand, lead exposure significantly decreased catalase and superoxide dismutase (SOD) activities and the amount of nitrite in gastric mucosal samples.
CONCLUSION: Lead increases the formation of gastric ulcers by interfering with the oxidative metabolism in the stomach.
Keywords: Lead, Ulcer, Lipid peroxidation, Catalase, Gastric acid
INTRODUCTION
Reactive oxygen species (ROS) have been shown to be to be involved in the etiology of many inflammatory disorders of the gastrointestinal system[1,2]. This is evidenced by the increased oxidative stress by pro-ulcerative factors in the gut such as H pylori[3,4], use of non-steroidal anti-inflammatory drugs[5], smoking[6], psychological stress, corticosteroid use[7], and loss of sleep[8]. Lipid peroxidation (LPO), a result of the reaction of oxyradicals and polyunsaturated acids, has been suggested as an attack factor in the gastric mucosa[9]. Also GSH, an endogenous sulfhydryl compound, is an important substance in the cellular defense system[10].
Nitric oxide (NO) depletion also plays significant roles in the pathogenesis of gastric ulcers. NO along with superoxide (02.-) and the products of their interaction initiate a wide range of toxic oxidative reactions causing tissue injury[11]. Large amounts of NO, generated primarily by iNOS, can be toxic and pro-inflammatory. Likewise, neutrophils too produce oxidants and release granular constituents comprising lytic enzymes performing an important role in inflammatory injury[12].
Although lead acetate does not initiate any excess generation of reactive oxygen species in a cerebral synaptosomal suspension, it has a marked ability to enhance the pro-oxidant properties of ferrous iron in the same system[13]. It also amplifies oxidative stress induced by l-glutamate[14]. Recent studies suggest that accumulated lead exposure is related to several chronic disorders of aging including disorders that have been associated with oxidative stress[15-17]. Sass[19] first reported that lead poisoning in dogs is associated with perforating gastrointestinal ulcers. Bercowitz and Laufer[19] observed that gastrointestinal ulcer patients had accumulated lead in their teeth, thus suggesting a relationship between lead and ulcer. In a previous study, we reported that long term exposure of rats to lead predisposes the stomach to higher ulcerogenic effects of indomethacin and acidified ethanol[20]. However, the exact mechanism(s) by which lead promotes gastric ulceration is not yet understood. The objective of the present study thus was to investigate the involvement of ROS in the aggravation of ulceration in the stomach of lead-exposed rats.
MATERIALS AND METHODS
Chemicals and animals
Lead acetate and urethane were obtained from BDH chemicals Ltd, Poole. England. Epinephrine, 5,5-dithio-bis-2-nitrobenzoic acid, hydrogen peroxide, sodium pentobarbitone, and thiobarbituric acid (TBA) were purchased from Sigma (St Loius, MO USA). All other reagents were of analytical grade and were obtained from the British Drug Houses, Poole, UK.
Young male albino Wistar rats (80-90 g) were obtained from the small animal house, College of Medicine, University of Ibadan, Nigeria. They were randomly divided into three groups with adequate matching of weight. They were kept in wire meshed cages and fed with commercial rat pellets (Ladokun Feeds Ltd, Ibadan, Nigeria) and allowed to take water ad libitum.
Lead treatment
Animals were exposed to lead as previously described[20]. The high lead group (HiPb) was given 5000 mg/L lead acetate in drinking water daily while the low lead group (LoPb) received 100 mg/L lead acetate in drinking water. The control animals received only drinking water. At the end of a 15 wk exposure, blood samples were collected from the rats for analysis of the Packed Cell Volume (PCV), red and white blood cell counts, and neutrophile numbers by standard methods. Afterwards, ulcer was induced as follows.
Induction of experimental ulcer
Acidified ethanol was used to induce experimental gastric ulcers. Thirty six hours fasted rats were given 1.0 mL of HCl/ethanol mixture containing 0.15 mol/L HCl in 70% mL/L ethanol[21]. Four hours after administering the ulcerogen and under sodium pentobarbitone anaesthesia (60 mg/kg, ip), the animals were sacrificed. The stomachs were removed, inflated with 10 mL of 2% formaldehyde for 10 min to fix the tissue walls and opened along the greater curvature. The hemorrhagic lesions were stretched out on a glass plate and their sizes were estimated using an underlying graph paper with a 1 mm2 grid. Lesion areas were summed up per stomach and expressed as % of total mucosal area.
Measurement of gastric acid secretion
Gastric acid secretion was studied in the 3 groups using a modification of the method originally described by Ghosh[22]. The animals were fasted for 24 h at the end of the 15 wk lead treatment. Under urethane anaesthesia [250 g/L urethane (6 mL/kg body, ip)], the animals were surgically prepared for in situ stomach perfusion. The pylorus was semi-transected at its junction with the duodenum and a pyloric cannula inserted and tied into place. An oesophageal cannula for infusion from a flow meter (Watson-Marlow Inc., USA) was passed through the mouth to the stomach. Ten minute effluent samples (10.0 ± 0.1 mL each) were collected via a pyloric cannula and titrated against 0.01 mol/L NaOH using phenolphthalein as indicator. Acidity was expressed in mol/L/L per min life. After obtaining a consistent basal acid output, each animal was injected via a femoral vein cannula with either 9 g/L NaCl or histamine acid phosphate.
Biochemical analysis
At the end of chronic lead exposure, the animals were killed under deep ether anaesthesia and the stomachs were removed. Each stomach was cut open through the greater curvature, rinsed in normal saline, weighed, and homogenized in 10 mL KCl (pH = 7.4). The homogenate was subsequently used for total protein estimation. The protein content of the gastric mucosa was estimated by the method of Lowry et al[23] using bovine serum albumin as a standard.
Determination of lipid peroxidation. Lipid peroxidation was assayed by measuring the thiobarbituric acid reactive (TBAR) products using the procedure of Walls et al[24]. Briefly, the homogenate was supplemented with 0.75 g/L TBA in 0.1 mol/L HCl. The reactants were then supplemented with 5 mL n-butanol-pyridine mixture, shaken vigorously for 1 min and centrifuged for 10 min at 4000 r/min. Absorbance was then read at 532 nm and the results expressed as nmol TBA per 100 mg wet tissue.
Determination of catalase activity. Activity of catalase in gastric mucosa was determined according to the procedure of Sinha[25]. This method is based on the reduction of dichromate in acetic acid to chromic acetate when heated in the presence of H2O2, with the formation of perchromic acid as an unstable intermediate. The chromic acetate so produced is measured calorimetrically at 530 nm.
Determination of SOD activity. For determination of SOD activity, samples of gastric mucosal homogenates were taken as described above. A method originally described by Misra and Fridovich[26] as reported by Magwere et al[27] was employed. This method is based on the ability of superoxide dismutase to inhibit the autoxidation of epinephrine caused by O2 generated by xanthine oxidase reaction.
Determination of nitrite in gastric mucosa. The gastric mucosa was cooled in ice-cold distilled water before homogenization (100 g/L). The crude homogenate was centrifuged at 21.000 g for 20 min at 4°C. Aliquots of the supernatants were taken to determine nitrite levels. The amounts of nitrite were measured in the gastric mucosa by performing the Griess reaction. 100 mL of sample were incubated with 100 mL of Griess reagent (Sigma) at room temperature for 20 min. Nitrite level was determined by measuring the absorbance at 550 nm using a spectrophotometer (DU 640B, Beckman, Fullerton, California, USA)[28,29].
Determination of gastric mucous. Adherent gastric glandular mucous was measured by the method of Corne et al[30]. Excised gastric glandular portion of the stomachs were transferred for 2 h to 0.1% Alcian blue dissolved in buffer solution containing 0.1 mol/L sucrose and 0.05 mol/L sodium acetate (pH adjusted to 5.8 with hydrochloric acid). After washing the stomach twice in 0.25 mol/L sucrose (15 min and 45 min), the dye complexed with mucous was eluted by immersion in 10 mL aliquots of 0.5 mol/L MgCl2 for 2 h. The resulting blue solution was shaken with equal volumes of diethyl ether and the optical density of the aqueous phase was measured spectrophotometerically at 605 nm.
Statistical analysis
All values presented in tables are expressed as mean ± SEM. The appropriate comparisons between groups were made using Student’ t-test. The difference between the groups is taken to be significant at P < 0.05.
RESULTS
Development of gastric lesions
Administration of acidified ethanol caused severe damage to the rat stomachs under investigation. The site of the formation of lesions is the corpus mucosa. The mean ulcer score in the control animals was 6.94% ± 0.42%. The severity of lesions was markedly increased by chronic exposure to both low and high lead concentrations (Table 1, P < 0.05). Also shown in Table 1 is the result of the total protein and adherent mucous content of the glandular stomach. Long term exposure to lead significantly decreased both gastric mucous and protein contents, the inhibition being greatest in the high lead treated group.
Table 1.
Effects of lead exposure on acidified ethanol-induced gastric injury (mean ± SEM, n = 8)
| Treatment | Mass gain (% of initial mass) | Mucosal damage score (% of total mucosal area) | Total protein (mg/g) | Gastric mucus (mg/g) |
| Control | 43.3 ± 4.4 | 6.94 ± 0.42 | 9.80 ± 0.01 | 31.0 ± 0.31 |
| Low lead | 17.4 ± 6.3a | 9.72 ± 0.65a | 7.50 ± 0.14 | 28.0 ± 0.27a |
| High lead | 6.3 ± 5.1a | 12.50 ± 0.51a | 6.20 ± 0.01 | 23.0 ± 0.24a |
P < 0.05 vs control.
Basal rate of gastric acid secretion was not significantly affected by lead exposure. However, the maximal response of the stomach of lead-exposed animals to histamine was significantly higher than those of the unexposed control group (Figure 1).
Figure 1.
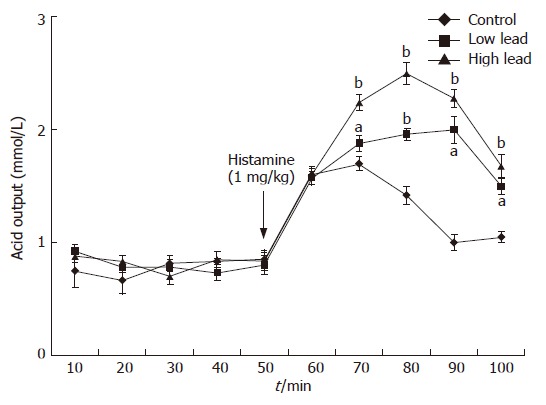
Gastric acid secretory output after administration of histamine (1 mg/kg) in rats (aP < 0.05, bP < 0.01 vs control).
Lipid peroxidation and gastric antioxidant enzymes
Lipid peroxidation (measured as the amount of TBA reactants in the gastric mucosa) in the unexposed control animals was 595 ± 82 mmol/g tissue. Exposure of rats to low and high lead levels significantly increased gastric TBA reactant level to 183.2% ± 12.7% and 226.1% ± 6.8% of the control value, respectively (Figure 2A). On the other hand, SOD activity in the intact gastric mucosa (control group) averaged 334 ± 22 nkat/g. Exposure of rats to low and high lead levels significantly decreased SOD activity to 78% ± 3% and 66% ± 6% of the control, respectively (Figure 2B). Similarly, lead exposure decreases gastric mucosal catalase activity to 88.22% ± 3.21% and 55.29% ± 2.15% of the control, respectively (Figure 2C).
Figure 2.
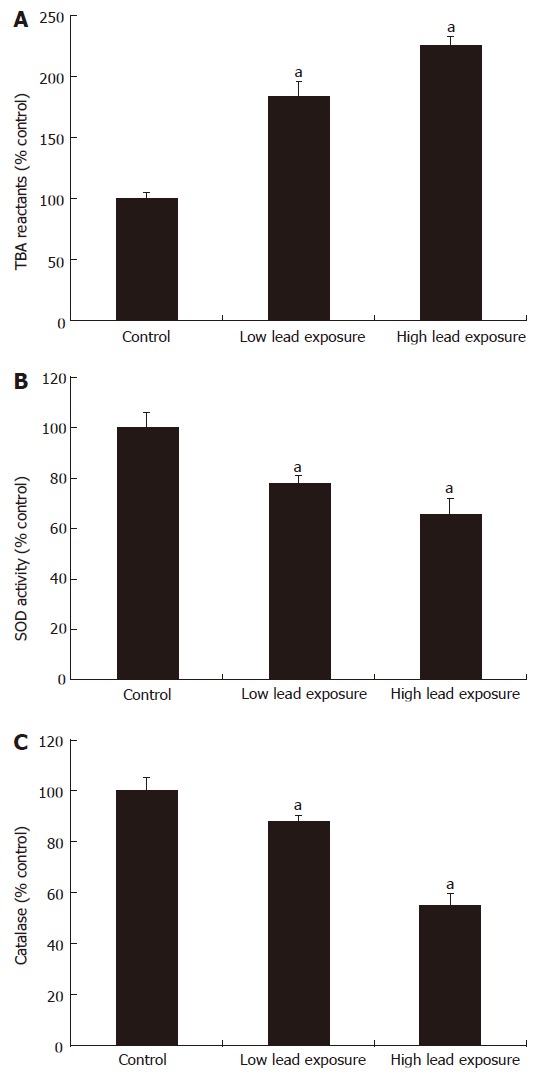
TBA reactants, superoxide dismutase and catalase activities in gastric mucosa of rats exposed to chronic lead treatments (mean ± SEM, n = 8, aP < 0.05 vs control).
Gastric mucosal nitrite concentration
The control gastric mucosal nitrite level was 62.4 ± 4.2 nmol/g tissue (Figure 3). Chronic exposure of rats to lead for 15 wk depleted the gastric nitrite level to 51.6 ± 3.1 nmol/g tissue (P < 0.05) and 49.7 ± 2.5 nmol/g tissue (P < 0.01).
Figure 3.
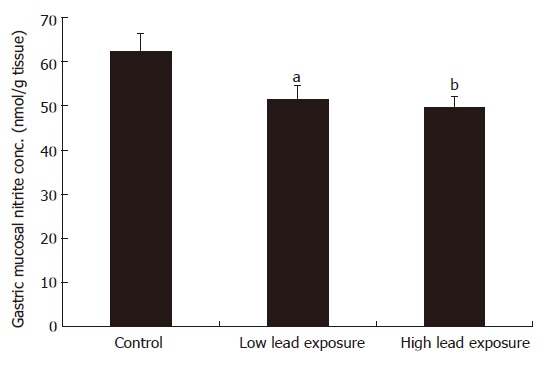
Gastric mucosal nitrite concentration in lead exposed rats (mean ± SEM, n = 8 rats, aP < 0.05, bP < 0.01 vs control).
DISCUSSION
The present study investigated the role of oxidative stress in the ulcer-promoting action of prolonged lead exposure in rats. The doses and the duration of lead treatment used in our study have been shown to cause significant high blood lead levels in previous studies[20,31]. Thus, there is no doubt that the animals had high blood lead levels. The acidified ethanol model has been used widely to produce gastric mucosal damage[21]. We observed that long-term exposure of rats to lead given through drinking water significantly increased the incidence of gastric ulcer produced by the ulcerogen. This agrees with our previous report in which lead was shown to aggravate gastric ulcers induced by indomethacin and acidified ethanol[20].
It is well established that gastric acid secretion plays a role in gastric ulcer. Moreover, many anti-ulcerogenic drugs act by reducing the acid secretion[32]. The present result suggests that basal gastric acid secretory rate was not significantly affected by prolonged lead treatment. However, maximal stimulation by the established secretagogue, histamine, was markedly increased in the lead exposed rats. We had earlier reported that acid output from pylorus ligated stomach of lead exposed rats was increased by lead treatment. Further studies may therefore be required using different acid secretory models before a conclusion can be made on the exact effect of lead exposure on gastric acid secretion.
Lipid peroxidation has been implicated in the etiology of damage to subcellular membranes and then injury in the cell. In the present study, lipid peroxidation, as measured by the amount of TBA reactants, was increased by lead exposure. The implication of this is that lead causes an increase in the formation of free radicals, which, if not mopped up by free radical scavengers as SOD, CAT, or glutathione, will expose the stomach to inflammation.
Wapnir et al[33] reported that juvenile rats fed a diet containing 1% lead acetate for 7 wk suffered from malabsorption of certain amino acids, as the intestinal absorption of glycine, lycine, and phenylalanine were decreased. Furthermore, administration of HCl and ethanol has been shown to produce ulcerative lesions and increased lipid peroxidation in the gastric mucosa with depletion of endogenous antioxidants[21]. The result of this study showed a significant decrease in the protein glandular protein level of the rats given low and high doses of lead acetate. This may also explain the significant low levels of the antioxidizing enzymes SOD and CAT observed in the treated groups.
In gastroprotection, the first line of antioxidative enzyme is SOD which catalyses the dismutation of superoxide radical anion (O2) into less noxious hydrogen peroxide (H2O2). H2O2 is then inactivated by the degradation into water by catalase or glutathione peroxidase[35,36]. Depletion of these enzymes, as evident by the results of this study, therefore predisposes the stomach to a greater impact of the free radicals produced via increased lipid peroxidation, hence increased ulcer formation following lead exposure.
Apart from the free radical scavenging enzymes, another agent that has been widely believed to be involved in the regulation of various gastric functions and in the modulation of gastric mucosal integrity is nitric oxide (NO). This is evidenced by reports showing that suppression of NO production by NO synthase inhibitor worsens gastric lesions induced by ethanol in rats[37,38]. NO has also been suggested to prevent infiltration by neutrophils, which are a source of superoxide radical anions[39]. The reduction in the gastric tissue nitrite in the lead treated groups in the present study is therefore indicative for a reduced protective capacity of endogenous NO.
In summary, chronic exposure of rats to lead intensifies HCl/ethanol-induced gastric mucosal damage and this may be related to reduced gastric contents of endogenous NO and the antioxidative enzymes SOD and CAT.
COMMENTS
Background
A number of humans are occupationally exposed to high levels of lead through paints, car exhausts battery fumes, etc. Lead exposure also occurs through ingestion with food and via inhalation. Previous studies in dogs and rats have shown that long-term exposure to high level lead in drinking water predisposes the stomach to ulcer. The mechanism of this effect of lead is not understood.
Research frontiers
Oxidative stress has been shown to be a major causative factor for many diseases, including gastrointestinal ulcers. The hotspot of this study is to examine if the increased susceptibility of the stomach to ulcer after prolonged lead exposure can be explained (partly or totally) by an increased oxidative stress in the stomach.
Innovations and breakthroughs
The results of this study show that chronic exposure of rats to lead intensifies HCl/ethanol-induced gastric mucosal damage and this may be related to reduced gastric contents of endogenous nitric oxide and antioxidative enzymes.
Applications
The study suggests that prolonged exposure to lead may be a factor in the etiology of gastrointestinal ulcers.
Terminology
Oxidative stress: a condition of increased oxidant production in animal cells characterized by the release of free radicals and resulting in cellular degeneration. Free radicals damage components of the cells’ membranes, proteins, or genetic material by “oxidizing” them-the same chemical reaction that causes iron to rust; Lead: a soft heavy toxic malleable metallic element; bluish white when freshly cut but tarnishes readily to dull grey; Lead poisoning: Lead can come into the body in a number of ways: through water that goes through lead pipes, through badly canned food and through small pieces of paint. Victims of lead poisoning may get headaches, dizziness, confusion, and problems seeing. They may also become slowly paralyzed, starting with the hands. In very serious cases, it can cause death.
Peer review
This is an interesting and generally well written paper with some new information.
Footnotes
Supported by the Senate, University of Ibadan, Nigeria partly through SRG grant to SBO UI/SRG/COM/2000/10A
S- Editor Ma N L- Editor Mihm S E- Editor Lu W
References
- 1.Perry MA, Wadhwa S, Parks DA, Pickard W, Granger DN. Role of oxygen radicals in ischemia-induced lesions in the cat stomach. Gastroenterology. 1986;90:362–367. doi: 10.1016/0016-5085(86)90933-9. [DOI] [PubMed] [Google Scholar]
- 2.Schmassmann A, Stettler C, Poulsom R, Tarasova N, Hirschi C, Flogerzi B, Matsumoto K, Nakamura T, Halter F. Roles of hepatocyte growth factor and its receptor Met during gastric ulcer healing in rats. Gastroenterology. 1997;113:1858–1872. doi: 10.1016/s0016-5085(97)70005-2. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi N, Kakizoe T. Synergistic interaction between Helicobacter pylori gastritis and diet in gastric cancer. Lancet Oncol. 2001;2:88–94. doi: 10.1016/S1470-2045(00)00225-4. [DOI] [PubMed] [Google Scholar]
- 4.Janulaityte-Günther D, Günther T, Pavilonis A, Kupcinskas L. What Bizzozero never could imagine - Helicobacter pylori today and tomorrow. Medicina (Kaunas) 2003;39:542–549. [PubMed] [Google Scholar]
- 5.Rostom A, Wells G, Tugwell P, Welch V, Dube C, McGowan J. Prevention of chronic NSAID induced upper gastrointestinal toxicity. Cochrane Database Syst Rev. 2000:CD002296. doi: 10.1002/14651858.CD002296. [DOI] [PubMed] [Google Scholar]
- 6.Ma L, Wang WP, Chow JY, Yuen ST, Cho CH. Reduction of EGF is associated with the delay of ulcer healing by cigarette smoking. Am J Physiol Gastrointest Liver Physiol. 2000;278:G10–G17. doi: 10.1152/ajpgi.2000.278.1.G10. [DOI] [PubMed] [Google Scholar]
- 7.Levenstein S. Peptic ulcer at the end of the 20th century: biological and psychological risk factors. Can J Gastroenterol. 1999;13:753–759. doi: 10.1155/1999/521393. [DOI] [PubMed] [Google Scholar]
- 8.Bercovitz K, Laufer D. Lead accumulation in teeth of patients suffering from gastrointestinal ulcers. Sci Total Environ. 1991;101:229–234. doi: 10.1016/0048-9697(91)90036-e. [DOI] [PubMed] [Google Scholar]
- 9.Guo JS, Chau JF, Cho CH, Koo MW. Partial sleep deprivation compromises gastric mucosal integrity in rats. Life Sci. 2005;77:220–229. doi: 10.1016/j.lfs.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 10.Hung CR. Importance of histamine, glutathione and oxyradicals in modulating gastric haemorrhagic ulcer in septic rats. Clin Exp Pharmacol Physiol. 2000;27:306–312. doi: 10.1046/j.1440-1681.2000.03241.x. [DOI] [PubMed] [Google Scholar]
- 11.Poli G, Albano E, Dianzani MU. Free radicals: From Basic Science to Medicine. Basel: Birkhauser Verlag AG; 1993. pp. 18–30. [Google Scholar]
- 12.Hogg N. Free radicals in disease. Semin Reprod Endocrinol. 1998;16:241–248. doi: 10.1055/s-2007-1016284. [DOI] [PubMed] [Google Scholar]
- 13.Yoshikawa T, Naito Y. The role of neutrophils and inflammation in gastric mucosal injury. Free Radic Res. 2000;33:785–794. doi: 10.1080/10715760000301301. [DOI] [PubMed] [Google Scholar]
- 14.Bondy SC, Guo SX. Lead potentiates iron-induced formation of reactive oxygen species. Toxicol Lett. 1996;87:109–112. doi: 10.1016/0378-4274(96)03766-6. [DOI] [PubMed] [Google Scholar]
- 15.Naarala JT, Loikkanen JJ, Ruotsalainen MH, Savolainen KM. Lead amplifies glutamate-induced oxidative stress. Free Radic Biol Med. 1995;19:689–693. doi: 10.1016/0891-5849(95)00067-8. [DOI] [PubMed] [Google Scholar]
- 16.Romero-Alvira D, Roche E. High blood pressure, oxygen radicals and antioxidants: etiological relationships. Med Hypotheses. 1996;46:414–420. doi: 10.1016/s0306-9877(96)90196-6. [DOI] [PubMed] [Google Scholar]
- 17.Thylefors B, Négrel AD, Pararajasegaram R, Dadzie KY. Available data on blindness (update 1994) Ophthalmic Epidemiol. 1995;2:5–39. doi: 10.3109/09286589509071448. [DOI] [PubMed] [Google Scholar]
- 18.Mecocci P, Mariani E, Cornacchiola V, Polidori MC. Antioxidants for the treatment of mild cognitive impairment. Neurol Res. 2004;26:598–602. doi: 10.1179/016164104225017659. [DOI] [PubMed] [Google Scholar]
- 19.Sass B. Perforating gastric ulcer associated with lead poisoning in a dog. J Am Vet Med Assoc. 1970;157:76–78. [PubMed] [Google Scholar]
- 20.Olaleye SB, Raji Y, Onasanwo SA, Erigbali P, Oyesola SO, Odukanmi A, Omotosho IO, Elegbe RA. Potentiation of Gastric Ulceration By Experimental Lead Exposure In Rats. J Biol Sci. 2006;6:480–484. [Google Scholar]
- 21.Anadan R, Rekha RD, Saravanan N, Devaki T. Protective effects of Picrorrhiza kurroa against HCl/ethanol induced ulceration in rats. Fitoterapia. 1999;70:498–503. [Google Scholar]
- 22.Ghosh MN, Schild HO. Continuous recording of acid gastric secretion in the rat. Br J Pharmacol Chemother. 1958;13:54–61. doi: 10.1111/j.1476-5381.1958.tb00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 24.Walls R, Kumar KS, Hochstein P. Aging human erythrocytes. Differential sensitivity of young and old erythrocytes to hemolysis induced by peroxide in the presence of thyroxine. Arch Biochem Biophys. 1976;174:463–468. doi: 10.1016/0003-9861(76)90374-x. [DOI] [PubMed] [Google Scholar]
- 25.Sinha AK. Colorimetric assay of catalase. Anal Biochem. 1972;47:389–394. doi: 10.1016/0003-2697(72)90132-7. [DOI] [PubMed] [Google Scholar]
- 26.Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- 27.Magwere T, Naik YS, Hasler JA. Effects of chloroquine treatment on antioxidant enzymes in rat liver and kidney. Free Radic Biol Med. 1997;22:321–327. doi: 10.1016/s0891-5849(96)00285-7. [DOI] [PubMed] [Google Scholar]
- 28.Green LC, Ruiz de Luzuriaga K, Wagner DA, Rand W, Istfan N, Young VR, Tannenbaum SR. Nitrate biosynthesis in man. Proc Natl Acad Sci USA. 1981;78:7764–7768. doi: 10.1073/pnas.78.12.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crespo E, Macías M, Pozo D, Escames G, Martín M, Vives F, Guerrero JM, Acuña-Castroviejo D. Melatonin inhibits expression of the inducible NO synthase II in liver and lung and prevents endotoxemia in lipopolysaccharide-induced multiple organ dysfunction syndrome in rats. FASEB J. 1999;13:1537–1546. [PubMed] [Google Scholar]
- 30.Corne SJ, Morrissey SM, Woods RJ. Proceedings: A method for the quantitative estimation of gastric barrier mucus. J Physiol. 1974;242:116P–117P. [PubMed] [Google Scholar]
- 31.Gruber HE, Gonick HC, Khalil-Manesh F, Sanchez TV, Motsinger S, Meyer M, Sharp CF. Osteopenia induced by long-term, low- and high-level exposure of the adult rat to lead. Miner Electrolyte Metab. 1997;23:65–73. [PubMed] [Google Scholar]
- 32.Schmassmann A. Mechanisms of ulcer healing and effects of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104:43S–51S; discussion 79S-80S. doi: 10.1016/s0002-9343(97)00211-8. [DOI] [PubMed] [Google Scholar]
- 33.Wapnir RA, Exeni RA, McVicar M, Lipshitz F. Experimental lead poisoning and intestinal transport of glucose, amino acids, and sodium. Pediatr Res. 1977;11:153–157. doi: 10.1203/00006450-197703000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Blum J, Fridovich I. Inactivation of glutathione peroxidase by superoxide radical. Arch Biochem Biophys. 1985;240:500–508. doi: 10.1016/0003-9861(85)90056-6. [DOI] [PubMed] [Google Scholar]
- 35.Kwiecień S, Brzozowski T, Konturek PC, Pawlik MW, Pawlik WW, Kwiecień N, Konturek SJ. Gastroprotection by pentoxyfilline against stress-induced gastric damage. Role of lipid peroxidation, antioxidizing enzymes and proinflammatory cytokines. J Physiol Pharmacol. 2004;55:337–355. [PubMed] [Google Scholar]
- 36.Masuda E, Kawano S, Nagano K, Tsuji S, Takei Y, Tsujii M, Oshita M, Michida T, Kobayashi I, Nakama A. Endogenous nitric oxide modulates ethanol-induced gastric mucosal injury in rats. Gastroenterology. 1995;108:58–64. doi: 10.1016/0016-5085(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 37.Konturek SK, Konturek PC. Role of nitric oxide in the digestive system. Digestion. 1995;56:1–13. doi: 10.1159/000201214. [DOI] [PubMed] [Google Scholar]
- 38.Gaboury J, Woodman RC, Granger DN, Reinhardt P, Kubes P. Nitric oxide prevents leukocyte adherence: role of superoxide. Am J Physiol. 1993;265:H862–H867. doi: 10.1152/ajpheart.1993.265.3.H862. [DOI] [PubMed] [Google Scholar]
- 39.Okcu N, Onuk MD, Yilmaz A, Gundogdu M, Baran T. The effects of omeprazole and ranitidine on the gastric ulcer healing. Doga Trop J Med Sci. 1992;16:657–658. [Google Scholar]