

Abstract
Background
Preclinical testing of potential therapies for Duchenne muscular dystrophy (DMD) is conducted predominantly of the mdx mouse. But lack of a detailed quantitative description of the pathology of this animal limits our ability to evaluate the effectiveness of putative therapies or their relevance to DMD.
Methods
Accordingly, we have measured the main cellular components of muscle growth and regeneration over the period of postnatal growth and early pathology in mdx and wild-type (WT) mice; phalloidin binding is used as a measure of fibre size, myonuclear counts and BrdU labelling as records of myogenic activity.
Results
We confirm a two-phase postnatal growth pattern in WT muscle: first, increase in myonuclear number over weeks 1 to 3, then expansion of myonuclear domain. Mdx muscle growth lags behind that of WT prior to overt signs of pathology. Fibres are smaller, with fewer myonuclei and smaller myonuclear domains. Moreover, satellite cells are more readily detached from mdx than WT muscle fibres. At 3 weeks, mdx muscles enter a phase of florid myonecrosis, accompanied by concurrent regeneration of an intensity that results in complete replacement of pre-existing muscle over the succeeding 3 to 4 weeks.
Both WT and mdx muscles attain maximum size by 12 to 14 weeks, mdx muscle fibres being up to 50% larger than those of WT as they become increasingly branched. Mdx muscle fibres also become hypernucleated, containing twice as many myonuclei per sarcoplasmic volume, as those of WT, the excess corresponding to the number of centrally placed myonuclei.
Conclusions
The best-known consequence of lack of dystrophin that is common to DMD and the mdx mouse is the conspicuous necrosis and regeneration of muscle fibres. We present protocols for measuring this in terms both of loss of muscle nuclei previously labelled with BrdU and of the intensity of myonuclear labelling with BrdU administered during the regeneration period. Both measurements can be used to assess the efficacy of putative antinecrotic agents. We also show that lack of dystrophin is associated with a number of previously unsuspected abnormalities of muscle fibre structure and function that do not appear to be directly associated with myonecrosis.
Keywords: Muscular dystrophy, Muscle regeneration, Hypertrophy, Hypotrophy, Hyperplasia
Background
From shortly after its discovery in the early 1980s [1], the mdx mouse fell into disrepute and was widely dismissed as a model of human muscular dystrophy [2-5]; that its muscles are hypertrophic and of similar absolute strength to those of the C57Bl/10 wild-type (WT) mouse did not fit well with the muscle-wasting phenotype of Duchenne muscular dystrophy (DMD) boys. Its eventual revelation as a genetic homologue of DMD [6] subsequently established it as the most-used preclinical model of DMD but with the common proviso that the mdx pathology is much less severe. This comparison is based mainly on the progression of muscle fibrosis and, importantly, of clinical incapacity expressed as a proportion of the lifespans of man and mouse, a debatable basis for comparison of pathological severity between two such different species [7]. Persistence of such opinions is abetted by a lack of well-authenticated quantitative criteria in either species by which such comparisons might validly be made. While current technologies permit us to gather detailed quantitative information on gene expression and proteomic profiles in DMD and mdx dystrophies [8-10], at the level of cellular pathology, we lack equivalent data to permit accurate translation of molecular events into pathological processes.
To realize the full potential utility and to accommodate the limitations of the mdx mouse as a preclinical model requires a deeper understanding of its pathology by comparison with that of DMD in man. A full quantitative description of the processes involved in generating pathological changes over the course of disease, especially of the dynamics of those processes, could provide a basis for determining the role of each pathological feature of the mdx dystrophy in the development of the disease. Such information is required to fully evaluate the applicability, or otherwise, to DMD of data emerging from this animal model and to better interpret results from tests of potentially therapeutic preclinical interventions.
To understand those features that are common to the two species and those that differ between them, we need, at a minimum, to develop quantitative methods in the mouse for measuring and comparing the benefits to pathology and function of any putative therapeutic treatment. At present, we have only a hazy understanding of the relationship between the pathological targets that we are aiming to modify with a given treatment and the outcome criteria that are commonly assessed in such investigations. For example, variation in fibre size, the frequency of centrally placed myonuclei or the deposition of fibrous connective tissue are all recognized as consequent to dystrophic pathological processes in the mdx mouse, but their relationships to the primary pathology unleashed by the lack of dystrophin are indirect and obscure. Likewise, although the molecular mechanisms by which putative treatments are thought to modify these processes are often identified, the cellular mechanisms that translate these molecular changes into structural and functional improvements remain largely unresolved. As a first approach to gaining a better understanding of these various relationships, we have made a detailed examination of the mdx mouse muscle during its phase of postnatal growth, driven by conspicuous satellite cell activity, and of its transition, at the end of this period, into a florid myopathic stage that is accompanied by even more intense satellite cell activity.
We have assessed these events by three main methods. First, we have monitored the accumulation of myonuclei within muscle fibres as a measure of the contribution of myogenic cell fusion to fibre growth. Second, the content of fibrous actin has been measured as an index of the amount of contractile material within each muscle fibre. Third, we have followed the incorporation of the thymidine analogue BrdU into muscle fibre nuclei as a measure of myogenic activity and loss of these labelled nuclei as an indicator of myonuclear turnover in the period of growth and at the onset of myopathology in mdx mice.
We demonstrate a number of previously unrecognized consequences of absence of dystrophin from skeletal muscle. In particular, muscle growth in mdx mice is markedly compromised prior to the onset of overt myonecrosis; growth of mdx fibres lags behind that of WT fibres, with fewer myonuclei that have smaller sarcoplasmic domains. This hypotrophy is accompanied by feeble attachment of satellite cells to the mdx fibres but appears to originate pre- or peri-natally. At 3 weeks, WT muscle growth involving satellite cell activity ceases, further enlargement being accomplished by expansion of the myonuclear domain. In contrast, mdx mice at this age enter a sudden myonecrotic phase and myogenic activity becomes yet more conspicuous, coping with both the hypertrophic growth and the repair of extensively destroyed muscle fibres. At maturity, mdx muscles and their component myofibres are larger than those of WT and are hypernuclear, with more than double the number of myonuclei per fibre. Thus, although hypertrophic in terms of absolute bulk, muscles of the mdx mouse can be considered hypotrophic in terms of myonuclear domain, which falls to half the WT value. We propose that the mdx mouse differs greatly from DMD in having no problem of repair and regeneration. This brings into question its value as a model of this aspect of dystrophinopathy. However, the basic similarity of myonecrotic events commends it as a model for testing therapies aimed at modifying this aspect of DMD.
Methods
Animals and BrdU and EdU administration
C57Bl10/ScSn mice and mdx C57Bl10/ScSn mice were managed and handled according to protocols approved ethically and scientifically by the local animal care and use committee guidelines of the Children’s National Medical Center, Washington, DC. For some experiments, mice were injected subcutaneously with BrdU (30 mg/kg) morning and evening for the first postnatal week. Administration of BrdU to post-weaning mice was by inclusion in their drinking water (0.8 mg/ml) for a period of 1 week. In some experiments, pre-weaning mice were injected twice daily with EdU, which is more easily visualized but too expensive to be administered in the drinking water and is more toxic than BrdU in longer term administration protocols.
Mice aged 1, 2, 3, 4, 6, 14 and 28 weeks were euthanased by CO2 flooding followed by cervical dislocation, and tibialis anterior (TA), gastrocnemius and extensor digitorum longus (EDL) muscles were dissected from both hindlimbs.
Isolation of myofibres
Reagents were from Invitrogen (Thermo Fisher Scientific, Grand Island, NY, USA) unless otherwise stated. Single myofibres were isolated as described previously [11]. Briefly, right limb EDL muscles were carefully dissected immediately after euthanasia of the mouse and incubated in 0.2% Collagenase Type 1 (Sigma-Aldrich, St. Louis, MO, USA) in DMEM for 1 to 2 h, depending on muscle size and age, to digest the connective tissue. Single myofibres were liberated by gentle trituration with fire-smoothed wide-mouthed Pasteur pipettes in DMEM in dishes pre-coated with horse serum. Liberated myofibres were washed by transfer through four such dishes. TA and gastrocnemius muscles of mice that had been given BrdU were frozen on corks in isopentane held at freezing point in liquid nitrogen for subsequent cryostat sectioning.
Immunostaining
EDL muscles from 16-day-old mdx and WT mice (n = 4) were frozen in liquid-nitrogen-cooled isopentane. Transverse sections cut at −20°C were incubated for 1 h at RT in permeabilization/blocking buffer (TBS-T, 0.5% Triton X-100, 2% BSA 2% (w:v), 20% goat serum) and then overnight at RT in primary antibodies diluted in permeabilization and blocking buffer. Anti-laminin-2 (clone 4H8-2, 1:400, Axxora, Lausen, Switzerland) was used to define fibre margins and LAMP1 (dilution 1:300, H-228, Santa Cruz Biotechnology, Santa Cruz, CA, USA) to visualize lysosomal structures. Slides were then washed three times in TBS-T 10 min at RT prior to incubation in secondary antibodies (goat anti-rat Alexa Fluor 488, 1:400, Invitrogen; donkey anti-rabbit Alexa Fluor 595, 1:400, Invitrogen) diluted in permeabilization and blocking buffer. After three 10-min washes in TBS-T at RT, the slides were counter-stained with DAPI, 1 μg/ml for 2 min, rinsed two times and mounted with Gel Mount (Sigma). Four images of 1-μm-thick optical sections were gathered from each section, using Zeiss LSM 510 Meta NLO on an Axiovert 200 M microscope (Carl Zeiss, Jena, Germany). Images were acquired using AxioVision software (Carl Zeiss).
BrdU staining with biotin-conjugated anti-BrdU (Life Technologies B35138) 1:100 overnight, followed by Alexa 488 streptavidin (Invitrogen S32354) 1:500, was performed according to the supplier’s instructions. Laminin co-staining was performed by overnight incubation at 4°C with anti-laminin antibody (Sigma L9393) 1:400, followed by Marina Blue goat-anti-rabbit IgG (Invitrogen M10992) 1:500 for 1 h at RT to reveal the primary antibody localization.
EdU-treated mice were euthanased on postnatal day 18 and EDL muscles were harvested. Single myofibres were isolated as described above. EdU detection was performed on fixed myofibres using the Click-iT Assay kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. DAPI was used to visualize nuclei.
Isolation, immunostaining and counting of Pax7-positive cells
Myofibres were isolated as described above, fixed with 4% paraformaldehyde and immunostained with anti-Pax7 antibody (Developmental Studies Hybridoma Bank, Iowa City, IA, USA) as previously described [12].
Fixation and phalloidin staining of myofibres
Batches of myofibres harvested at each age were transferred to a 2-ml Eppendorf vial containing 1.5 ml 3.65% formaldehyde and fixed at 37°C for 15 min. The vial was centrifuged at 700 g for 10 min, to loosely pellet the myofibres. The formaldehyde supernatant was carefully removed, and 1 ml of 30% sucrose was added to the pellet which was resuspended by a 3-s vortex and stored at −80°C pending analysis.
For staining, the vials were thawed at room temperature and the contents deposited into a Petri dish together with the products of a rinse with TBS-Tween (Tween 0.1% in TBS). Using a stereo microscope and watchmaker’s forceps, myofibres were carefully transferred into a 2-ml Eppendorf vial containing permeabilization and blocking buffer (0.5% Triton, 0.1% Tween, 2% BSA and 20% goat serum) in which they were incubated overnight at 4°C. After centrifugation for 10 min at 15,000 g, the buffer was removed and the fibres were rinsed three times with alternate centrifugations in 1 ml of rinse solution (0.1% Tween in TBS).
The myofibres were then re-suspended in Alexa Fluor 594-conjugated phalloidin at a dilution of 1:40 for 20 min at room temperature, as previously described [11], rinsed three times and left in the rinse solution overnight at 4°C to maximize elution of unbound phalloidin. After centrifugation for 10 min at 15,000 g, myofibres were re-suspended in a 0.75 μg/ml solution of DAPI and immediately centrifuged for a further 10 min at 15,000 g, then rinsed once more. The contents of the vial were deposited into a Petri dish and transferred gently, using a stereo microscope and forceps, and mounted onto a microscope slide.
Imaging and software analysis
Single fibre imaging and quantification of F-actin was performed as previously described [11]. Briefly, stained myofibres were imaged on a Nikon Eclipse E500 epifluorescence microscope with Spot Camera (Nikon Corporation, Chiyoda-ku, Japan). Overlapping segments of each myofibre were recorded under a Nikon PlanApo × 10 objective. Between two to six images were required, depending on the length of the myofibre. Exposure time was set to fall within a previously validated linear response range of signal intensities [11] and was kept constant across all samples within a comparison. Images of each myofibre were stitched together using the CS2 version ‘Interactive Layout’ mode of the Photomerge feature of Adobe Photoshop in Adobe Photoshop Elements.
For measurement of volume, confocal microscopy z-stacks were acquired on a Zeiss LSM 510 Meta NLO system with an Axiovert 200 M microscope. Depths ranged from 35.3 to 61.8 μm, depending on myofibre thickness. Slices of 2.2 μm thick composed of 512 × 512 pixels were collected with × 20 objective. Between two to six non-adjacent z-stacks were collected per myofibre, depending on myofibre length. DAPI-stained nuclei were counted for each z-stack by eye from three-dimensional representation using AxioVision software (Zeiss) and added together. Volume data of each segment are expressed per myonucleus within that segment.
Integrated density of signal from fluorophore-conjugated phalloidin for each slice was calculated in ImageJ and added together to give total signal per z-stack. Signals per z-stack were added for each myofibre and divided by the number of nuclei to give signal per nucleus.
Measurement of growth of muscle fibres
To follow growth of muscle fibres from WT and mdx mice, we recorded the two main contributory factors: the numbers of nuclei within the muscle fibre (myonuclei) and the amount of filamentous actin (F-actin). Since most fibrous actin within the fibre is associated with the myofibrillar structures, this is an index of the amount of contractile apparatus. These methods were developed and analysed for consistency and reproducibility in a previous study [11]. EDL muscles were dissected from WT and mdx mice aged from 1 to 28 weeks of age (three animals per time point) and dissociated in type 1 collagenase as described above. Batches of fibres obtained from mdx and WT mice were thawed in age-matched pairs, subjected to the staining regime, and mounted for microscopic examination.
Validation of fluorescent phalloidin signal as an index of volume
To determine the relationship between fibrous actin content and sarcoplasmic volume, we compared the phalloidin signal from segments of individual muscle fibres extracted from EDL muscle fibres of 2-, 6- and 28-week mdx and WT mice with the calculated volumes of the same segments from z-stacks of confocal images as described previously [11]. These two measures were normalized to the number of myonuclei in these same segments and plotted against one another (Figure 1). The strong correlation between these two parameters across the two strains and spanning the entire age range validates the use of the fluorescent phalloidin signal as a reliable index of sarcoplasmic volume within these bounds, irrespective of age or strain. Thus, we apply the term ‘myonuclear domain’ as a descriptor of both the amount of contractile material associated with each myonucleus as measured by the fluorescent phalloidin signal and the volume of sarcoplasm calculated from the regression relationship.
Figure 1.
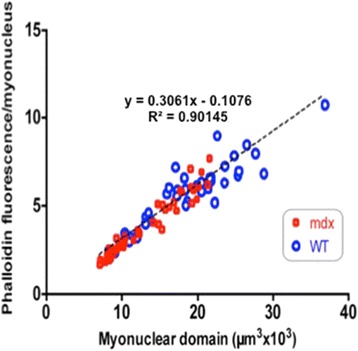
Plot of fluorescent phalloidin signal per myonucleus against estimates of fibre volume per myonucleus from stacked confocal images visualized in segments of muscle fibres isolated from mdx and WT muscles at ages of 2, 6 and 28 weeks. The strong linear relationship between the two parameters across the age range and the two mouse strains validates the use of phalloidin fluorescence as a reliable index of fibre volume within these limits.
Statistics
For myofibres processed by the PhAct method on a standard fluorescent microscope, statistical tests for significant differences between WT and mdx myofibres in terms of nuclear counts (myonuclei, peripheral, centralized or Pax7-positive) and actin signal (per fibre or normalized to the number of myonuclei) were analysed within an ANOVA framework. Some parameters were transformed to achieve normal distributions (square-root transformation: Pax7-positive nuclei per total nuclei and actin signal; log transformation: actin signal per myonucleus, total nuclei and myonuclei), then two-way ANOVA was performed for each age. Factors included into the model were the murine strain and the experimental batch of the myofibres.
For myofibres imaged using confocal microscopy, for which volume per nucleus and actin per volume were measured, because of the non-normality of dependent variables, non-parametric tests were used to compare medians. For the analysis at each time point, the Wilcoxon rank-sum test was used to compare strains.
Results
Postnatal fibre growth differs substantially between mdx and WT muscle fibres
Simple visual comparison of age-matched isolated fibre preparations from WT and mdx muscles indicated that for the first 4 weeks, mdx fibres tended to be smaller but, beyond 6 weeks, appeared larger than those of WT and showed branching that was not observed in those of WT (Figure 2). To quantify myofibre growth, we analysed myofibres isolated from WT and mdx mice by two main criteria: the number of myonuclei and the amount of fibrous actin they contained as plotted in Figure 3A,B. These data confirmed the visual impression that mdx myofibres lagged behind those of WT in both respects for the first 4 postnatal weeks but overtook them in terms of absolute size (phalloidin fluorescent yield) in mature animals.
Figure 2.
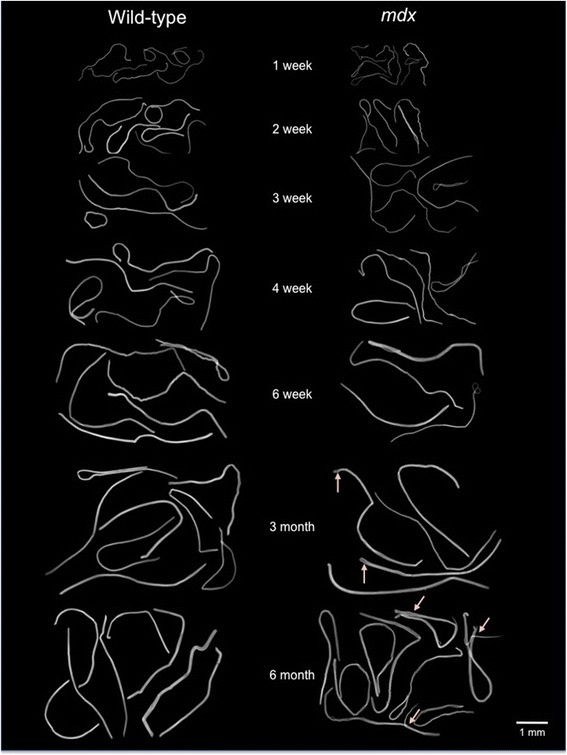
Individual fibres isolated from muscles of mdx and WT mice aged from 1 to 28 weeks. The visual impression that muscle fibres of mdx mice are thinner than those of age-matched WT controls is borne out by analyses shown in subsequent figures. Branching is evident in the older mdx fibres.
Figure 3.

Growth of mdx and WT muscle fibres up to 28 weeks of age, showing the total phalloidin fluorescence per myofibre, as an indicator of fibre volume, plotted against the number of myonuclei in that myofibre, displayed as individual data points and linear best fits across the range of ages as identified by colour. (A) Growth of WT fibres. Myonuclear number, plotted along the X-axis, increases up to week 3 but not thereafter, all further growth being accomplished by increase in fibrous actin content, along the Y-axis, as indicated too by the increasing slope of the best fit lines. (B) The equivalent plot to (A) of mdx muscle fibres using the same scales, to show the marked difference between growth patterns of the two strains. Up to week 4, mdx growth is broadly similar to that of WT but, beyond this point, continues both by a great increase in number of myonuclei per fibre and by increase in F-actin content. The total F-actin per fibre becomes larger than that of WT, in association with fibre branching, but the ratio of fibrous actin per myonucleus is low, as indicated by the shallow slopes of the best fit lines.
Myofibre growth in WT mice
As expected from previous studies [13], myonuclear number per WT fibre increased steadily over the first 3 weeks, when it ceased abruptly (Figure 3A). All further growth occurred by increase in the amount of F-actin per fibre, as indicated by the progressive rise along the Y-axis of the individual data points in Figure 3A, without further increase along the X-axis. The increasing slope of the correlation lines within the WT fibre population also indicates an increase in the F-actin (that is, sarcoplasmic volume) per myonucleus up to 14 weeks of age, with no further increase by 28 weeks.
Growing mdx muscle is hypotrophic prior to the onset of disease
For the first 3 to 4 weeks, muscle fibres of mdx mice followed a largely similar pattern of increase in myonuclear number per fibre accompanied by a small increase in fibrous actin content (Figure 3B). But, thereafter, the myonuclear number per mdx fibre increased progressively along the X-axis to more than twice that seen in WT muscles of equivalent age. The F-actin content also increased to higher average levels than that of WT but were highly variable and not in proportion to myonuclear number.
The difference between patterns of myogenesis in WT and mdx mice is depicted more succinctly as plots of the average fluorescent phalloidin signal per muscle fibre (Figure 4A) and per myonucleus (Figure 4B) across the range of ages.
Figure 4.
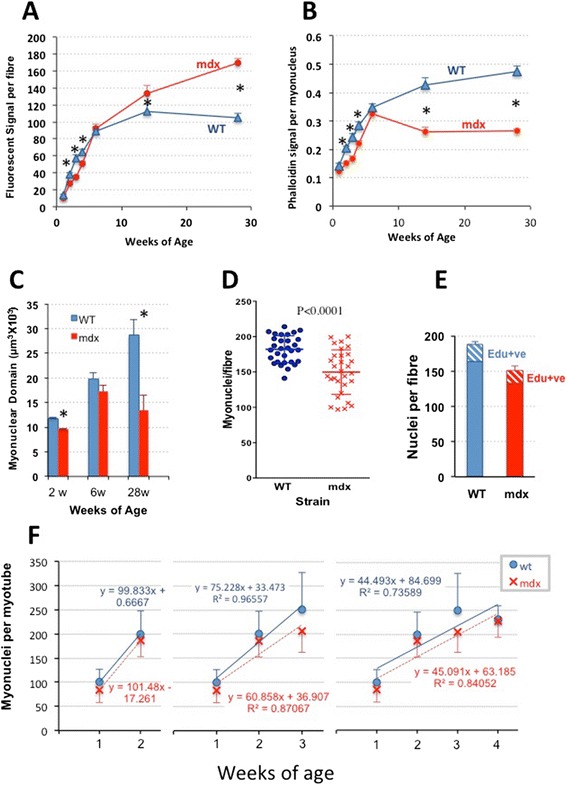
Difference between patterns of myogenesis in WT and mdx mice. (A) Fluorescent phalloidin signal per myofibre (mean ± SEM) plotted against age, illustrating the lag in mdx fibre growth in up to 6 weeks but progressive hypertrophy beyond this point (*P < 0.05). (B) The fluorescent phalloidin signal per nucleus (mean ± SEM) also increases rapidly in both mouse strains up to 6 weeks, but with mdx lagging significantly (*P < 0.05) behind WT. The subsequent continuing rise in WT fibres contrasts with the fall to half of the WT values in mdx. (C) Column plot of the estimated myonuclear domains in muscle fibres isolated from muscles of 2-, 6- and 28-week WT and mdx mice (mean ± SEM, *P < 0.05). (D) Scatterplots, together with depiction of mean ± SD, of myonuclear number per myofibre isolated from 18-day-old WT and mdx mouse EDL muscles. (E) EDL fibres from mice labelled with EdU at 14, 15 and 16 days of age and analysed on day 18 showing similar labelling frequencies of fibre-associated nuclei (new-formed myonuclei plus Pax7-positive satellite cells). (F) Nuclear number/fibre (mean ± SD) of fibres isolated from muscles of mdx and WT mice at weeks 1 to 4. The successive incremental plots show a progressive fall in slope from 100 nuclei/fibre between weeks 1 and 2 to a net rise of approximately 45 nuclei per fibre averaged across weeks 1 to 4. Cessation of WT growth is evident beyond week 3. Myonuclei numbers in mdx fibres fall consistently below those of WT fibres, but the plots run largely parallel suggesting that the intensity of myogenesis in the two strains is closely similar and that the mdx fibres had fallen behind WT during prenatal development.
In WT myofibres, the fluorescent signals per myofibre and per myonucleus both increase continuously up to 14 weeks of age, where they stabilize.
In mdx myofibres, by contrast, both relationships are biphasic, following two distinct courses. Up to the onset of frank myonecrotic disease at 3 to 4 weeks, the fluorescent phalloidin signal, both per myofibre and per myonucleus, lags significantly behind that of the age-matched WT. This confirms the visual impression in Figure 2 that individual mdx fibres are smaller in these young mice (Figure 4A). Calculation of the myonuclear domain from the fluorescent phalloidin yield (Figure 4C) confirms that this too is significantly smaller in mdx than in WT fibres in these young growing muscles as well as later in the adult mice.
Hypotrophic growing mdx muscle fibres contain fewer myonuclei than WT fibres
Up to week 4, fewer myonuclei were routinely seen in mdx than in the age-matched WT myofibres but not significantly so at any given time point. We therefore conducted a larger experiment to determine whether the difference was real and whether myogenic activity was lower in mdx mice during this postnatal growth period. Three WT and three mdx mice were each injected twice per day with EdU (0.5 mg/g) from p13 to p15, prior to euthanasia at 18 days. Analysis of ten isolated EDL fibres per mouse showed that mdx myofibres contained significantly fewer myonuclei than age-matched WT fibres (Figure 4D). However, the proportion marked by a 3-day administration of EdU over this period, which would comprise both myogenic cells in mitosis over the labelling period and myonuclei derived from them, did not differ significantly between WT and mdx (Figure 4E).
We conclude that for the first 3 to 4 postnatal weeks, mdx muscle fibres are hypotrophic, in terms of all parameters we have measured; that is, they are of significantly smaller volume and contain significantly fewer myonuclei with significantly smaller sarcoplasmic domains than the age-matched WT fibres.
Detailed plots of the myonuclear numbers in fibres over weeks 1 to 4 (Figure 4F) confirm that the lower myonuclear numbers in mdx myofibres are not attributable to differences in rate of proliferation of myogenic cells. They show that the accumulation of myonuclei in mdx and WT fibres rose in parallel over this growth period but with the mdx lagging slightly behind the WT; both showed an increase of 100 myonuclei per fibre between weeks 1 and 2, dropping to a mean increase of 60/75 myonuclei per fibre by week 3 and further, to approximately 45 when averaged over the first 4 weeks. We conclude that lack of myonuclei over the postnatal growth period does not reflect a lower rate of myogenic cell proliferation and fusion during this time and that it must have arisen at or before birth.
In the context of continuously degenerating/regenerating muscle beyond week 3, the low F-actin content per myonucleus is unsurprising [14] but the seeming lag in growth of this factor prior to this time was unexpected. The small myonuclear domain in these young mdx is not attributable, as it is during the subsequent myopathic stage, to excessive myonuclear numbers and must therefore reflect a diminished maintenance of sarcoplasmic protein per myonucleus during this growth phase.
Thus, the postnatal hypotrophy of mdx EDL muscle arises from two factors: a smaller myonuclear domain in combination with fewer myonuclei per fibre than WT. In previous tissue culture studies, we had observed a similar hypotrophic pattern to that seen here in vivo, that is, hyponucleated myofibres in the context of equivalent overall myogenicity and small myonuclear domain; this in association with elevated lysosomal content as well as with excess secretion of protein in lysosomal vesicles [15]. As shown in Figure 5A, the hypotrophic mdx fibres in pre-necrotic mdx muscle in vivo are also heavily loaded with lysosomes, suggesting that similar aberrations of protein handling and secretion may contribute to the hypotrophic status of these fibres.
Figure 5.
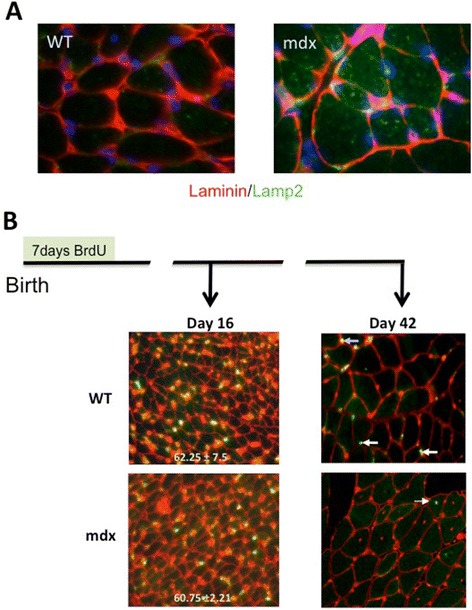
Mdx muscles show excess lysosomal activity prior to disease and massive turnover after onset. (A) Cryostat sections of TA muscles from 2-week-old mdx and WT muscles immunostained for laminin (red) to show fibre outlines and Lamp1 (green) and DAPI (blue) to show the strong representation of lysosomes within mdx fibres as a possible contributory factor to the hypotrophic condition of these fibres. (B) Immunostains for BrdU (green) and laminin (red) counter-stained with propidium iodide (red) of the section of TA muscles from mice injected twice daily with BrdU during the first week of life and euthanased at 16 or 42 days of age. At 16 days, prior to the onset of myonecrosis, muscle from both strains showed similar frequencies of labelled nuclei. N = 4 muscles. Data are shown as mean ± SD. In the WT muscle, the BrdU label had been retained at 42-day muscle, both by identifiable myonuclei (arrowed) and by interstitial cells but had been lost, apart from the occasional rare (1 to 2 per whole section) centrally placed labelled myonucleus in the mdx muscles.
Onset of myopathy in mdx mice involves a switch from hypotrophy to a hyperplasia-driven hypertrophy
Beyond 3 to 4 weeks of age, the phalloidin signal per mdx myofibre increases more rapidly than WT with age. This reflects an increase to a very large size of mature mdx myofibres (Figures 3B and 4A) accompanied, at 14 and 28 weeks, by an even greater increase in myonuclear number with the result that sarcoplasmic volume per myonucleus, that is the myonuclear domain, was reduced to half of that of WT fibres at these ages (Figure 4B,C).
Onset of mdx myopathy involves massive turnover of muscle fibres
To examine the dynamics of the initial growth and transition from growth into disease, we injected a cohort each of newborn mdx and WT mice with BrdU subcutaneously twice per day (morning and evening) on postnatal days 1 to 7. At 16 days of age, counts of labelled muscle fibre nuclei per unit area of muscle from mdx and WT mice (Figure 5B, n = 4 mice) showed no difference between the two strains, adding to the preceding evidence that the lower myonuclear content in mdx compared to that in WT muscle fibres over the first 3 weeks is not attributable to diminished myoblast proliferation during this period.
By 42 days of age, muscle sections from four mice from each of these same labelled cohorts showed a stark divergence between the two strains in retention of labelled nuclei. In WT muscles, numerous BrdU-labelled nuclei were identified both in myonuclear positions within the myofibres and in the interstitium between fibres. By contrast, in mdx muscles at this age, only a very rare isolated labelled nucleus per whole muscle section was found in either position. Since myonuclei do not divide, loss of labelled myonuclei from mdx muscle must reflect loss of the nuclei themselves, presumably associated with death and replacement of the fibre, rather than by the main alternative, namely, mitotic dilution of the BrdU label to subliminal levels. For the interstitial nuclei, either explanation is possible. Overall, the loss of BrdU-stained myonuclei indicates that the majority of muscle fibres had been lost and replaced over the 3 weeks between the onset of the myopathy and time of examination. Thus, muscle precursor cell activity over this 3- to 6-week period had not only replaced the complete muscle mass but, as shown explicitly in Figure 3B and by implication from the halving of the myonuclear domain in Figure 4B,C, had augmented the total myonuclear content by approximately 100% over the number in the WT mouse.
The changing relationship between satellite cells and muscle fibres during postnatal growth and regeneration in mdx muscles
Counts of Pax7-positive satellite cells on WT fibres over the first 3 weeks corresponded well to those found by White et al. [13], but muscle fibres isolated from mdx muscles carried far fewer of these cells than their age-matched WT equivalents (Figure 6A,B). This reconciles poorly with the similar rates of incorporation of new myonuclei into growing fibres of the two strains over this period (Figure 4F) or the similar frequencies of incorporation of EdU and BrdU labels into nuclei during postnatal weeks 1 to 3 (Figures 4E and 5B). Suspecting that satellite cells were being lost preferentially from these young mdx muscle fibres during the isolation process, we counted Pax7-positive cells, per fibre profile, in cryostat sections of EDL muscles at 14 days of age and found no significant difference between mdx and WT muscles (Figure 6C). Thus, Pax7-positive cells were present with equal frequency in situ in these young mdx and WT EDL muscles but were lost preferentially from mdx fibres during the isolation procedure, suggesting that their adhesiveness to these young growing fibres was reduced in the absence of dystrophin.
Figure 6.

Numbers of Pax7-positive satellite cells and of fibre branches in mdx and WT muscles. (A) Numbers of satellite cells on myofibres extracted from EDL muscles of 1- to 28-week-old WT and mdx mice, shown as Pax7-positive cells per fibre (mean ± SEM). Up to week 4, mdx fibres carried markedly fewer satellite cells than those of WT. Beyond 6 weeks of age, the numbers of satellite cells on mdx fibres increased progressively and significantly exceeded the WT number at 14 and 28 weeks.(B) Pax7-positive cells on fibres extracted from EDL muscles of 1- to 28-week-old WT and mdx mice, normalized to total myofibre nuclei (mean ± SEM). Again, during the first 4 weeks, the percentage of satellite cells on mdx fibres is markedly lower than that on WT fibres but, at 6 weeks and beyond, the numbers are closely similar, suggesting a link between myonuclear density and satellite cell number in mature muscle. (C) Numbers of Pax7-positive cells per myofibre profile in frozen sections of muscle from 2-week-old mdx and WT mice (three individual mice of each strain). In the sectioned muscle, Pax7-positive cells are equally frequent in WT and mdx mice at this age. (D) Frequency of fibres bearing one to four branches in muscles of mdx mice at 6, 14 and 28 weeks of age. No branches were seen on fibres extracted from three batches of mdx mice younger than 6 weeks or from WT mice at any age examined, but the frequency and complexity of branching increased progressively with age of the mdx mouse.
By 6 weeks of age, mdx muscle fibres carried equivalent numbers of satellite cells to those of WT muscle fibres and the frequencies of satellite cells per mdx fibre rose consistently over the remaining period of observation to higher numbers than those of satellite cells per WT fibre (Figure 6A). Interestingly, the satellite cell number standardized to myonuclear number per fibre was constant over the entire period from 6 to 28 weeks (Figure 6B). Maintenance of so similar a proportionality in such dissimilar conditions as the WT and mdx muscles implies some hitherto unremarked interaction between the myonuclear population and satellite cell number.
Fibre splitting increases progressively with age in mdx muscle
Beyond 6 weeks of age, we show a significant increase in mdx myofibre volume, as indicated by fluorescent phalloidin signal, that is not proportionate to the more than doubling of myonuclei. The reported hypertrophy of mdx mouse muscles is not matched by increased fibre diameter [16] but, as shown in Figures 2, 6D and 7A, is accompanied by a progressive increase in the frequency of branched or split fibres [17-19], which has recently been shown to play a major role in mdx muscle hypertrophy [19].
Figure 7.
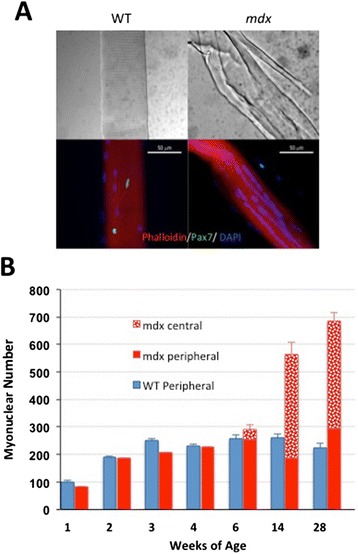
Increase with age in the proportion of ‘central nuclei’ and of fibre branching in mdx mice. (A) Interference contrast and fluorescent micrographs of single fibres isolated from 1-year-old mdx and WT mice and immunostained for Pax7. These not only bear readily identifiable satellite cells (green) but also illustrate the extensive branching of fibres in older mdx but not in WT mice together with the tendency for myonuclei to be arranged in linear ‘central strings’. (B) Counts of myonuclei classified into peripheral location and ‘central’ location, the latter being classified on the basis of their distribution in linear arrays. These ‘central’ myonuclei are first seen in 6-week-old mdx mice. At 14 and 28 weeks, however, it is notable that the numbers of peripherally located myonuclei per fibre are not substantially different from those of the WT and that the centrally located nuclei account for all of the excess myonuclei found in older mdx muscles.
‘Central’ nuclei account for the myonuclear excess in mdx over WT
A noted feature of mdx mouse muscle and of regenerated muscle in the adult mouse in general [20] is the large proportion of nuclei distributed in centrally placed strings [20,21] (Figure 7A). In our counts, we designate myonuclei in aligned strings as ‘central nuclei’ (that is, not in the normal juxta-sarcolemmal position) in distinction from those not in this configuration which we classify as peripheral myonuclei. In this investigation, we find no central strings of nuclei in the WT fibres at any age. In mdx fibres, they are first seen at 6 weeks of age and increase rapidly by 14 and 28 weeks. Counts reveal that these ‘central’ myonuclei account fully for the higher total myonuclear number in mdx muscle fibres (Figure 7B). In fact, the numbers of myonuclei located peripherally in the regenerated myofibres remain constant beyond 4 weeks of age in mdx mouse muscle and do not differ significantly from the numbers of total myonuclei per fibre in age-matched WT muscles. Although the supernumerary myonuclei in older mdx muscle correspond numerically to the central nuclei, myonuclei in both positions are marked by 1 week of BrdU labelling and we are unable, at present, to identify either with any specific aspect of the regenerative process.
Phasing of muscle growth and regeneration is not limited to the EDL
Our picture of a cessation of satellite cell-based growth of WT muscle at 3 weeks of age is based on investigation of fibres from the EDL muscle. To determine whether this was more broadly true, we gave BrdU to mice in their drinking water from weeks 4 to 5, looking for signs of continuing proliferation of myogenic cells in a range of muscles. As outlined in Figure 8A, mice were euthanased after a further week on water not laced with BrdU to give any myogenic cells labelled during week 4 ample time to differentiate and enter a growing or regenerating muscle fibre. Gastrocnemius and TA muscles of mdx mice subjected to this protocol showed conspicuous labelling of a significant proportion of myonuclei within focal areas of regeneration, in which virtually all myonuclei were labelled with BrdU (Figure 8B), reflecting synchrony of regeneration within such spontaneous lesions and implying that the proliferation of myogenic cells had not diluted the label to subliminal levels within this protocol. Muscles of WT mice subjected to this same regimen showed only occasional BrdU labelling of interstitial cells and no labelling of myonuclei (Figure 8C).
Figure 8.

Assessment of myogenic activity in muscles of mdx and WT mice. (A) Protocol for labelling cell participation in muscle regeneration. Mice are given drinking water containing 0.8 mg/ml BrdU for 7 days and maintained on normal drinking water for a further 7 days before being euthanased for analysis. (B) Section from a gastrocnemius muscle taken from a mdx mouse subjected to the regime in (A). Seven days after the end of BrdU administration, it shows extensive labelling of both interstitial cells and centrally located myonuclei in a spontaneous mdx muscle lesion (green or yellow). Virtually all of the centrally placed myonuclei are labelled, showing that the BrdU had not been diluted to subliminal levels by proliferation of the myogenic cells that had repaired this particular lesion. (C) Gastrocnemius of a WT mouse subjected to this same labelling regimen. An occasional interstitial nucleus is stained, but there are no centrally placed myonuclei and no identifiable myonucleus is stained. Thus, the cessation of myonuclear-based growth at 3 weeks, identified in the isolated fibres of the WT EDL muscles, is a generalized phenomenon extending to the gastrocnemius muscle and to other limb muscles examined.
Discussion
It is of practical concern that, although the mdx mouse has been the animal model of first resort for preclinical testing of potential therapeutic ideas and agents for treatment of DMD, we lack a coherent pathological picture of it. The common assertion that the mdx dystrophy is milder than DMD remains unsupported by quantitative data in either species [7]. Here, we have gathered detailed quantitative information outlining the main features that distinguish the mdx mouse from the WT mouse during its postnatal growth and the transition to severe pathology. Although comparison with DMD is constrained by the scanty human data, we do identify several similarities and differences between mdx and WT that would invite further investigation in man as well as highlighting a number of features of dystrophinopathy that are not apparent from more casual observation of either species.
Of practical interest, we delineate a set of readily measurable criteria for use in assessment of the beneficial effects of putative therapeutic strategies. We also identify several features that make this animal less than ideal, sometimes misleading, as a model of DMD. Recognition of such issues is important if we are to avoid developing therapeutic strategies based on misconception.
Hypotrophy in growing mdx muscles
This is the first report of a lag in growth of mdx EDL muscle fibres behind that of WT during the early postnatal growth period. It is a hypotrophy; small fibre size arises from a combination of fewer myonuclei, each with a smaller myonuclear domain. We dismiss the idea that lower myonuclear numbers in isolated mdx fibres during this period are attributable to the low numbers of satellite cells, since this is revealed as an artefact by the comparable satellite cell frequency at 2 weeks in histological sections of mdx and WT muscles (Figure 6C). This bespeaks excess satellite cell loss during fibre isolation from young mdx compared with WT mice, implying a looser association of satellite cells with mdx than with WT fibres that might itself predispose to slower growth. But other evidence argues against this. Even at week 1, mdx fibres contained fewer myonuclei than WT fibres, while the actual rate of growth across the 3 weeks was indistinguishable between the two (Figure 4F), as were the rates of incorporation of EdU (Figure 4E) and BrdU (Figure 5B). These data converge on the idea that the difference had arisen at or before birth and merely persisted over the postnatal myogenic growth period. A reported paucity of Pax7-positive cells in early development in the mdx mouse [22] might be held responsible for this, but the need for sequestration of satellite cells beneath the basement membrane during late prenatal growth [23] provides an alternative mechanism potentially susceptible to disturbance of the dystrophin-dependent link between the basement membrane and the fibre plasmalemma [24]. A hangover of this disturbance would also account for the excessive loss of satellite cells during isolation of fibres from mdx muscle during early postnatal growth [25].
The features of pre-myonecrotic mdx muscle fibres paint a picture of a more complex role of dystrophin in muscle fibre function than simply sustaining resilience of the surface. Thus, the mechanisms that bind satellite cells tightly to the fibre surface are defective, and the small mdx fibre size is associated with a small myonuclear domain. Both accord well with our previous in vitro studies where mdx myotubes were atrophic, hyponuclear, highly enriched in lysosomal structures and chronically exported large amounts of intact protein in aberrant lysosomal vesicles [15]. We show here that this is reflected in vivo by hypotrophy and excess lysosomal content of pre-necrotic muscle fibres (Figure 5A). Taken together, these data portray a broad and pervasive derangement of the metabolic and protein degradation processes, which precede myonecrosis [25-27], as integral features of dystrophinopathy.
The early phase of the mdx mouse myopathy is severe
We show that myonuclei labelled during the first postnatal week are largely lost by 7 weeks, 3 to 4 weeks after the onset of the myopathy (Figure 5B). This indicates massive myonecrosis and regenerative replacement, as echoed in the heavy BrdU labelling of myonuclei during weeks 4 to 5 in newly regenerated foci within all muscles examined (Figure 8B). Thus, the lag in growth in mdx muscle up to week 4 is fully compensated by week 6 by active myofibre regeneration (Figure 4A), even in the face of massive contemporaneous myonecrosis; this finding fits badly with the idea of distinct and separate phases of early myonecrosis followed subsequently by repair [28].
Overall, these findings argue against attribution of the mild clinical deficiency in mdx mouse to a less aggressive primary pathology than DMD. Indeed, far from being milder than DMD, it is doubtful whether the intensity of myonecrosis seen in 4- to 6-week mdx mice would be survivable by a DMD boy. Instead, our data show that the mdx mouse endures severe early myonecrotic pathology but is better able than man to cope with this by virtue of a more effective and harmonious regenerative response, as epitomized in Figure 8B. Differences in scale between mouse and man may play some part in this [7], but a more fundamental distinction between the mdx and DMD pathologies is the different timing of the phases of growth and regeneration in the two species. Satellite cell-dependent growth in the mouse ceases at 3 weeks, coincident with the onset of myonecrosis and regeneration, with no overlap between the separate programmes followed by satellite cells undertaking these two activities [29]. By contrast, in growing boys, myogenic cell proliferation continues throughout the juvenile and pubertal periods [30,31], presenting every prospect of adverse interference in DMD boys between the separate growth and regeneration programmes. Thus, some benefit might be achieved by blockade of discrepant signals between these two different satellite cell functions.
Quantitative evaluation of disease severity
One objective of this investigation was to develop robust quantifiable criteria by which to evaluate the severity of the mdx myopathy and any benefits of putative therapies. Because the spontaneous mdx lesions are focal, episodic and fleeting [32], there is considerable experimental noise between individual small samples such as histological sections, so we have broadened the sample space by adoption of two strategies based on labelling proliferating 1-week cohorts with BrdU.
Twice-daily injection of BrdU for the first postnatal week labels a neonatal cohort of muscle nuclei whose loss provides a dramatic signal against which to test any therapeutic agent. However, the intensive administration is difficult to standardize and the rapidity of loss of this labelled cohort limits the period of testing of therapeutic actions to the initial myopathic period.
We therefore favour the simpler and readily standardized protocol depicted in Figure 8A, whereby BrdU is added to the drinking water for 1 week to deliver a chronic supply of the label to the cohort of cells proliferating during that week, giving any labelled cells that are destined to participate in myogenesis ample time to do so during the subsequent BrdU-free week. As seen in Figure 8B, virtually every identifiable myonucleus is labelled by BrdU in regions of intense regeneration triggered by the spontaneous focal myopathy. Thus, this protocol fully samples events where myogenic proliferation falls completely within the week of BrdU administration as well as partially sampling those that overlap this week. It also shows that the serial cell divisions within any single regenerative event do not dilute the label to below its subliminal threshold. We can thus measure the number and size of focal lesions occurring within a given week and the intensity of labelling within each lesion. In our hands, this regimen labels myonuclei in the mdx mouse up to at least 18 months of age and is currently being used to compare regeneration across the mdx mouse lifespan and to investigate the effectiveness of exon-skipping treatments in suppressing the mdx myopathy.
Muscle hypertrophy and hyperplasia of myogenic cells
Muscle hypertrophy is an ill-understood feature of dystrophinopathies [33]. Calf muscle hypertrophy is pathognomonic of DMD, although the gradual replacement of muscle with fibro-fatty tissue converts this to ‘pseudohypertrophy’ in older boys [34]. In the GRMD dog, the gluteus, cranial sartorius and tongue show conspicuous hypertrophy [35,36], and in the mdx mouse, many muscles are routinely heavier than the equivalent WT [37]. Here, we make new observations pertinent to the role of myogenic proliferation in the hypertrophy.
It remains an enigma that, despite conspicuous myonecrosis, muscle of the mdx actually increases in weight beyond 3 to 4 weeks more rapidly than that of the WT and maintains normal levels of absolute strength [14,38,39]. With the possible exception of the diaphragm, there is little evidence that diminished regeneration plays any significant part in the histopathological or physiological decline of mdx muscle. Here, we show that hypertrophic regenerated mdx muscle fibres are hypernuclear, with double the number of myonuclei per unit volume so that the adult mdx mouse, as a whole, contains more than twice as many myonuclei as the WT mouse. This small myonuclear domain features in the hypotrophy in growing pre-myopathic mdx and diminishes further after the onset of myonecrosis and regeneration but is more than counter-balanced by the large numbers of myonuclei per fibre. Thus, the myogenic mechanisms in the mdx mouse are fully competent to maintain muscle mass, even in the context of massive persistent myofibre degeneration, and must entail extensive hyperplasia of the myogenic precursor pool. In accord with the finding of intact satellite cell number and function in old mdx mice [40], we have observed occasional BrdU labelling of mdx muscle up to 18 months (unpublished data). Reports of diminished satellite function with age in the mdx mouse (for example [17]) may be attributable to differences between in vitro and in vivo functional criteria [41,42]. Such exuberant myogenic activity in mdx muscle brings into question the utility of boosting myogenesis in this model [43,44]; it would be superfluous to the pathological and physiological demands on the muscle, which are accommodated adequately by the existing mechanisms.
Satellite cell numbers are maintained in mdx muscle
Beyond the first 4 weeks, satellite cell numbers on WT myofibres remain stable, while, in mdx muscle, the number per fibre rises steeply, as has been noted previously [17]. However, if normalized to myonuclear number, the satellite cell population remains stable at the approximately 2% level reported for sectioned mouse hindlimb muscles [45] and indistinguishable between mdx and WT muscles beyond the 3-week postnatal growth period. Regulation of satellite cell number has been viewed largely in terms of intrinsic, cell lineage-defined, satellite cell qualities that bias the proportion of symmetric versus asymmetric divisions, as influenced by spatial relationships to the basement membrane [46-50]. Our finding that satellite cell number is strongly related to myonuclear number in both WT and mdx muscles, even in the context of the abnormal numbers and spatial distributions of myonuclei seen in adult mdx muscle, suggests that feedback from the myonuclear population may also impinge on this important issue.
In DMD, the picture is clouded by uncertainty. Attempts to extract and culture myogenic cells have generally concluded that DMD myoblasts are fewer in number and less myogenic than those from healthy human muscle [51,52] and poor proliferation in vitro is widely interpreted as evidence of their failure as a causal feature of the pathogenesis [53-59]. At the same time, counts of satellite cells in situ have indicated that they are more frequent in DMD than in normal muscle [60-62]. But our ability to investigate the undoubted in vivo regeneration deficit in DMD by comparison with normal human muscle is limited by available technology. The discrepancy between the effective regeneration in the mdx mouse and its progressive failure in DMD has prompted the breeding of a telomerase-null mutation onto the mdx background, aiming to limit the supply of stem cells [63]. But the more general disruptive influence of telomere abnormalities on gene expression, in addition to their effects on cell proliferation, complicates interpretation of data from this animal.
Role of fibre branching in mdx hypertrophy
Since mean fibre diameter does not differ significantly between mdx and WT [16,21,64,65], hypertrophy is commonly assumed to reflect an increase in fibre numbers, although in histological sections, it is difficult to distinguish fibre number from fibre branching [18,19,66] and branching itself has recently been shown to be the main component of mdx muscle hypertrophy [19]. Thus, the hypertrophy in the mdx mouse involves two separate elements: a large increase in the number of myonuclei per fibre and extensive fibre branching that appears to arise from aberrant patterns of myoblast fusion [18,19,67,68].
Interestingly, branching has also been linked to susceptibility of fibres to work-induced damage [69-71], raising the interesting and unresolved question of why the most active degeneration occurs at a time before branches are detected while older muscles, with extensive fibre branching, show relatively little active pathology [16,21].
Central nucleation equates with hypernucleation
We were surprised to find that the strings of closely adjacent, non-juxta-sarcolemmal, ‘central nuclei’ effectively accounted for all of the hypernucleation in mdx muscle fibres. Although the number of peripheral nuclei per fibre remains constant, the excess being centrally located, BrdU marking shows that myonuclei in both positions are renewed during regeneration. The status of central nuclei, whether as an accommodation to the high rates of protein turnover in dystrophic muscle [15,26] or a fundamental defect in control of myonuclear number and placement, remains to be determined. Are they contributors to the myopathic process or a result of it [72]? In any event, it has implications for gene and exon-skipping therapies that are targeted at myonuclei. If these supernumerary myonuclei are transcriptionally active, they represent extra targets for such therapeutic agents; if not, they may act as counter-productive decoys. In non-rodent species, persistent central nucleation is not a conspicuous feature and hypernucleation has not been specifically investigated in muscular dystrophies.
It has been noted that there is a general inverse relationship between myonuclear domain and force production [73] that is contravened in mdx muscle, where a halving of myonuclear domain compared with WT is associated with a low specific force [14] adding to the idea that central positioning may reflect a disturbance of normal nuclear/sarcoplasmic relationships. The question of whether DMD and GRMD muscles have high myonuclear densities and whether the persistent central nucleation seen in adult WT mouse muscle after regeneration [20] is clearly of interest in this regard [74,75].
Conclusions
Observations
We have analysed the growth, loss and regeneration of skeletal muscle in the mdx mouse by comparison with the WT mouse.
From birth up to the onset of the myopathy, mdx mouse muscle grows at the same rate as WT mouse muscle but lags behind it in absolute size, with fewer myonuclei and smaller myonuclear domains than WT and contain excess lysosomal structures.
Satellite cells are less adherent to muscle fibres of the young pre-myopathic mdx mouse than those of the age-matched WT.
After onset of the myopathy, mdx muscles become hypertrophic and hypernucleated but the myonuclear domain shrinks to half of the WT size.
During the myopathic phase, mdx muscle fibres become increasingly branched and accumulate strings of myonuclei that are centrally positioned within the fibres.
These central nuclei account entirely for supernumerary myonuclei in mdx; the number of peripheral nuclei per fibre remains constant.
The number of satellite cells per fibre increases progressively in the mdx mouse, but the number per myonucleus remains constant and does not differ from that of the WT.
We describe two BrdU labelling protocols to permit quantitative comparison of muscle turnover in the mdx mouse.
Interpretations
Our data show no compromise of regeneration in limb muscles of the mdx mouse. Neither satellite cell number nor proliferative capacity is a limiting factor in the maintenance of muscle size and structure in this animal. We suggest that attention to defects in regenerative mechanisms in the mdx dystrophy are, as in aging muscle [42], subject to too strong a focus. This distracts from those aspects of the mdx myopathy that do closely simulate DMD pathology, such as defects in myofibre stability, where translation of findings from mouse to man is more likely to be of immediate practical value. A more detailed comparison of the cellular and molecular events that are common to DMD and mdx pathologies, or peculiar to one or the other, should also prove rewarding in this respect [7,32].
Acknowledgements
This work was supported by the Muscular Dystrophy Association USA grants MDA 200552 and MDA 115049. We also received support from the US Department of Defense (W81XWH-05-0334), National Institutes of Health (R01 NS029525, U54 HD053177, R24 HD050846 and P50AR060836) and Children’s National Medical Center Board of Visitors. KN is also supported by NIH K26OD011171, NIH P50AR060836, the MDA (translational grant), the US Department of Defense (W81XWH-05-1-0659, W81XWH-11-1-0782) and a pilot grant from Parent Project Muscular Dystrophy. TP was also supported by funding from the Foundation for the Eradication of Duchenne.
We thank Dr Jyoti Jaiswal for the advice on the measurement of phalloidin fluorescence.
Abbreviations
- BrdU
bromo-deoxy-uridine
- DMD
Duchenne muscular dystrophy
- EdU
5-ethynyl-2′-deoxyuridine
- GRMD
the golden retriever muscular dystrophy canine model of Duchenne muscular dystrophy
- mdx
a mouse on C57Bl/10ScSn background that bears a nonsense mutation in exon 23 of the dystrophin gene and thus a genetic homologue of Duchenne muscular dystrophy
- WT
the non-dystrophic C57Bl/10ScSn mouse strain
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WD was the major participant in the design performance and interpretation of data on fibre size and myonuclear number and in writing the manuscript. SD contributed to the experimental design and performance, interpretation of data and manuscript writing. HJ contributed to the experimental design and performance of much of the labelling work. TC performed part of the labelling work and helped with manuscript writing. AP contributed to the gathering and interpretation of data, BrdU labelling and interpretation of data. HG-D performed the statistical analysis of the data. KN contributed to the experimental design and manuscript writing. VG contributed to the gathering and interpretation of data and manuscript writing. SL contributed to the experimental design and performance, interpretation of data and manuscript writing. TP (corresponding author) was responsible for the design of experiments, interpretation of data and manuscript preparation. All authors read and approved the final manuscript.
Contributor Information
William Duddy, Email: william.duddy@upmc.fr.
Stephanie Duguez, Email: stephanie.duguez@upmc.fr.
Helen Johnston, Email: helenkjohnston@gmail.com.
Tatiana V Cohen, Email: CohenTa@kennedykrieger.org.
Aditi Phadke, Email: APhadke@childrensnational.org.
Heather Gordish-Dressman, Email: HGordish@childrensnational.org.
Kanneboyina Nagaraju, Email: KNagaraju@childrensnational.org.
Viola Gnocchi, Email: viola.gnocchi@me.com.
SiewHui Low, Email: low@ciwemb.edu.
Terence Partridge, Email: tpartridge@cnmcresearch.org.
References
- 1.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81:1189–92. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dangain J, Vrbova G. Muscle development in mdx mutant mice. Muscle Nerve. 1984;7:700–4. doi: 10.1002/mus.880070903. [DOI] [PubMed] [Google Scholar]
- 3.Tanabe Y, Esaki K, Nomura T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986;69:91–5. doi: 10.1007/BF00687043. [DOI] [PubMed] [Google Scholar]
- 4.Partridge TA. Animal models of muscular dystrophy – what can they teach us? Neuropathol Appl Neurobiol. 1991;17:353–63. doi: 10.1111/j.1365-2990.1991.tb00735.x. [DOI] [PubMed] [Google Scholar]
- 5.Harris JB, Slater CR. Animal models of muscular dystrophy. Br Med Bull. 1980;36:193–7. doi: 10.1093/oxfordjournals.bmb.a071637. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 7.Partridge TA. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013;280:4177–86. doi: 10.1111/febs.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao P, Hoffman EP. Embryonic myogenesis pathways in muscle regeneration. Dev Dyn. 2004;229:380–92. doi: 10.1002/dvdy.10457. [DOI] [PubMed] [Google Scholar]
- 9.Tseng BS, Zhao P, Pattison JS, Gordon SE, Granchelli JA, Madsen RW, et al. Regenerated mdx mouse skeletal muscle shows differential mRNA expression. J Appl Physiol (1985) 2002;93:537–45. doi: 10.1152/japplphysiol.00202.2002. [DOI] [PubMed] [Google Scholar]
- 10.Chen YW, Zhao P, Borup R, Hoffman EP. Expression profiling in the muscular dystrophies: identification of novel aspects of molecular pathophysiology. J Cell Biol. 2000;151:1321–36. doi: 10.1083/jcb.151.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duddy WJ, Cohen T, Duguez S, Partridge TA. The isolated muscle fibre as a model of disuse atrophy: characterization using PhAct, a method to quantify f-actin. Exp Cell Res. 2011;317:1979–93. doi: 10.1016/j.yexcr.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gnocchi VF, White RB, Ono Y, Ellis JA, Zammit PS. Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS One. 2009;4:e5205. doi: 10.1371/journal.pone.0005205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White RB, Bierinx AS, Gnocchi VF, Zammit PS. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol. 2010;10:21. doi: 10.1186/1471-213X-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coulton GR, Curtin NA, Morgan JE, Partridge TA. The mdx mouse skeletal muscle myopathy: II. Contractile properties. Neuropathol Appl Neurobiol. 1988;14:299–314. doi: 10.1111/j.1365-2990.1988.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 15.Duguez S, Duddy W, Johnston H, Laine J, Le Bihan MC, Brown KJ, et al. Dystrophin deficiency leads to disturbance of LAMP1-vesicle-associated protein secretion. Cell Mol Life Sci. 2013;70:2159–74. doi: 10.1007/s00018-012-1248-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coulton GR, Morgan JE, Partridge TA, Sloper JC. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol Appl Neurobiol. 1988;14:53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 17.Bockhold K, Rosenblatt J, Partridge T. Aging normal and dystrophic mouse muscle: analysis of myogenicity in cultures of living single fibres. Muscle Nerve. 1998;21:173–83. doi: 10.1002/(SICI)1097-4598(199802)21:2<173::AID-MUS4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Pichavant C, Pavlath GK. Incidence and severity of myofiber branching with regeneration and aging. Skelet Muscle. 2014;4:9. doi: 10.1186/2044-5040-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faber RM, Hall JK, Chamberlain JS, Banks GB. Myofiber branching rather than myofiber hyperplasia contributes to muscle hypertrophy in mdx mice. Skelet Muscle. 2014;4:10. doi: 10.1186/2044-5040-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pastoret C, Sebille A. Age-related differences in regeneration of dystrophic (MDX) and normal muscle in the mouse. Muscle Nerve. 1995;18:1147–54. doi: 10.1002/mus.880181011. [DOI] [PubMed] [Google Scholar]
- 21.Carnwath JW, Shotton DM. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J Neurol Sci. 1987;80:39–54. doi: 10.1016/0022-510X(87)90219-X. [DOI] [PubMed] [Google Scholar]
- 22.Merrick D, Stadler LK, Larner D, Smith J. Muscular dystrophy begins early in embryonic development deriving from stem cell loss and disrupted skeletal muscle formation. Dis Model Mech. 2009;2:374–88. doi: 10.1242/dmm.001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brohl D, Vasyutina E, Czajkowski MT, Griger J, Rassek C, Rahn HP, et al. Colonization of the satellite cell niche by skeletal muscle progenitor cells depends on Notch signals. Dev Cell. 2012;23:469–81. doi: 10.1016/j.devcel.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 24.Sunada Y, Campbell KP. Dystrophin-glycoprotein complex: molecular organization and critical roles in skeletal muscle. Curr Opin Neurol. 1995;8:379–84. doi: 10.1097/00019052-199510000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Rayavarapu S, Coley W, Cakir E, Jahnke V, Takeda S, Aoki Y, et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol Cell Proteomics. 2013;12:1061–73. doi: 10.1074/mcp.M112.023127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radley-Crabb HG, Marini JC, Sosa HA, Castillo LI, Grounds MD, Fiorotto ML. Dystropathology increases energy expenditure and protein turnover in the mdx mouse model of Duchenne muscular dystrophy. PLoS One. 2014;9 doi: 10.1371/journal.pone.0089277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jahnke VE, Van Der Meulen JH, Johnston HK, Ghimbovschi S, Partridge T, Hoffman EP, et al. Metabolic remodeling agents show beneficial effects in the dystrophin-deficient mdx mouse model. Skelet Muscle. 2012;2:16. doi: 10.1186/2044-5040-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet. 2009;18:482–96. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neal A, Boldrin L, Morgan JE. The satellite cell in male and female, developing and adult mouse muscle: distinct stem cells for growth and regeneration. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheek DB. Human growth — methods of approach. Aust Paediatr J. 1977;13:77–87. doi: 10.1111/j.1440-1754.1977.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 31.Cheek DB, Petrucco OM, Gillespie A, Ness D, Green RC. Muscle cell growth and the distribution of water and electrolyte in human pregnancy. Early Hum Dev. 1985;11:293–5. doi: 10.1016/0378-3782(85)90083-0. [DOI] [PubMed] [Google Scholar]
- 32.Dadgar S, Wang Z, Johnston H, Kesari A, Nagaraju K, Chen Y-W, et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J Cell Biol. 2014;207:139–58. doi: 10.1083/jcb.201402079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornegay JN, Childers MK, Bogan DJ, Bogan JR, Nghiem P, Wang J, et al. The paradox of muscle hypertrophy in muscular dystrophy. Phys Med Rehabil Clin N Am. 2012;23:149–72. doi: 10.1016/j.pmr.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearce GW, Pearce JMS, Walton JN. The Duchenne type muscular dystrophy: histopathological studies of the carrier state. Brain. 1966;89:109–20. doi: 10.1093/brain/89.1.109. [DOI] [PubMed] [Google Scholar]
- 35.Kornegay JN, Cundiff DD, Bogan DJ, Bogan JR, Okamura CS. The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul Disord. 2003;13:493–500. doi: 10.1016/S0960-8966(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 36.Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Grange RW. Golden retriever muscular dystrophy (GRMD): developing and maintaining a colony and physiological functional measurements. Methods Mol Biol. 2011;709:105–23. doi: 10.1007/978-1-61737-982-6_7. [DOI] [PubMed] [Google Scholar]
- 37.Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;4548:1–9. doi: 10.1016/0022-510x(94)00276-t. [DOI] [PubMed] [Google Scholar]
- 38.Anderson JE, Bressler BH, Ovalle WK. Functional regeneration in the hindlimb skeletal muscle of the mdx mouse. J Muscle Res Cell Motil. 1988;9:499–515. doi: 10.1007/BF01738755. [DOI] [PubMed] [Google Scholar]
- 39.Anderson JE, Ovalle WK, Bressler BH. Electron microscopic and autoradiographic characterization of hindlimb muscle regeneration in the mdx mouse. Anat Rec. 1987;219:243–57. doi: 10.1002/ar.1092190305. [DOI] [PubMed] [Google Scholar]
- 40.Boldrin L, Zammit PS, Morgan JE. Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 2014;14:20–9. doi: 10.1016/j.scr.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007;25:885–94. doi: 10.1634/stemcells.2006-0372. [DOI] [PubMed] [Google Scholar]
- 42.Grounds MD. Therapies for sarcopenia and regeneration of old skeletal muscles: more a case of old tissue architecture than old stem cells. Bioarchitecture. 2014;4:81–7. doi: 10.4161/bioa.29668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang YX, Bentzinger CF, Rudnicki MA. Treating muscular dystrophy by stimulating intrinsic repair. Regen Med. 2013;8:237–40. doi: 10.2217/rme.13.27. [DOI] [PubMed] [Google Scholar]
- 44.Mozzetta C, Consalvi S, Saccone V, Tierney M, Diamantini A, Mitchell KJ, et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old mdx mice. EMBO Mol Med. 2013;5:626–39. doi: 10.1002/emmm.201202096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson MC, Schultz E. Age related differences in absolute numbers of skeletal muscle satellite cells. Muscle Nerve. 1983;6:574–80. doi: 10.1002/mus.880060807. [DOI] [PubMed] [Google Scholar]
- 46.Le Grand F, Jones AE, Seale V, Scime A, Rudnicki MA. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009;4:535–47. doi: 10.1016/j.stem.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuang S, Gillespie MA, Rudnicki MA. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell. 2008;2:22–31. doi: 10.1016/j.stem.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 48.Shinin V, Gayraud-Morel B, Gomes D, Tajbakhsh S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol. 2006;8:677–87. doi: 10.1038/ncb1425. [DOI] [PubMed] [Google Scholar]
- 49.Conboy MJ, Karasov AO, Rando TA. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biol. 2007;5 doi: 10.1371/journal.pbio.0050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rando TA. The immortal strand hypothesis: segregation and reconstruction. Cell. 2007;129:1239–42. doi: 10.1016/j.cell.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 51.Webster C, Blau HM. Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: implications for cell and gene therapy. Somat Cel Mol Genetics. 1990;16:557–65. doi: 10.1007/BF01233096. [DOI] [PubMed] [Google Scholar]
- 52.Webster C, Filippi G, Rinaldi A, Mastropaolo C, Tondi M, Siniscalco M, et al. The myoblast defect identified in Duchenne muscular dystrophy is not a primary expression of the DMD mutation. Clonal analysis of myoblasts from five double heterozygotes for two X-linked loci: DMD and G6PD. Hum Genet. 1986;74:74–80. doi: 10.1007/BF00278789. [DOI] [PubMed] [Google Scholar]
- 53.Lipton BH. Skeletal muscle regeneration in muscular dystrophy. In: Mauro A, editor. Muscle regeneration. New York: Raven; 1979. pp. 31–40. [Google Scholar]
- 54.Grounds MD, Yablonka-Reuveni Z. Molecular and cell biology of skeletal muscle regeneration. Mol Cell Biol Hum Dis Ser. 1993;3:210–56. doi: 10.1007/978-94-011-1528-5_9. [DOI] [PubMed] [Google Scholar]
- 55.Decary S, Hamida CB, Mouly V, Barbet JP, Hentati F, Butler-Browne GS. Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children. Neuromuscul Disord. 2000;10:113–20. doi: 10.1016/S0960-8966(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 56.Renault V, Thornell LE, Eriksson PO, Butler-Browne G, Mouly V, Thorne LE. Regenerative potential of human skeletal muscle during aging. Aging Cell. 2002;1:132–9. doi: 10.1046/j.1474-9728.2002.00017.x. [DOI] [PubMed] [Google Scholar]
- 57.Blau HM, Webster C, Pavlath GK, Chiu CP. Evidence for defective myoblasts in Duchenne muscular dystrophy. Adv Exp Med Biol. 1985;182:85–110. doi: 10.1007/978-1-4684-4907-5_7. [DOI] [PubMed] [Google Scholar]
- 58.Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 1983;80:4856–60. doi: 10.1073/pnas.80.15.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang NC, Rudnicki MA. Satellite cells: the architects of skeletal muscle. Curr Top Dev Biol. 2014;107:161–81. doi: 10.1016/B978-0-12-416022-4.00006-8. [DOI] [PubMed] [Google Scholar]
- 60.Kottlors M, Kirschner J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010;340:541–8. doi: 10.1007/s00441-010-0976-6. [DOI] [PubMed] [Google Scholar]
- 61.Watkins SC, Cullen MJ. A quantitative study of myonuclear and satellite cell nuclear size in Duchenne’s muscular dystrophy, polymyositis and normal human skeletal muscle. Anat Rec. 1988;222:6–11. doi: 10.1002/ar.1092220103. [DOI] [PubMed] [Google Scholar]
- 62.Maier F, Bornemann A. Comparison of the muscle fiber diameter and satellite cell frequency in human muscle biopsies. Muscle Nerve. 1999;22:578–83. doi: 10.1002/(SICI)1097-4598(199905)22:5<578::AID-MUS5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 63.Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell. 2010;142:1059–71. doi: 10.1016/j.cell.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pagel CN, Partridge TA. Covert persistence of mdx mouse myopathy is revealed by acute and chronic effects of irradiation. J Neurol Sci. 1999;164:103–16. doi: 10.1016/S0022-510X(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 65.Wakeford S, Watt DJ, Partridge TA. X-irradiation improves mdx mouse muscle as a model of myofiber loss in DMD. Muscle Nerve. 1991;14:42–50. doi: 10.1002/mus.880140108. [DOI] [PubMed] [Google Scholar]
- 66.Wernig A, Irintchev A, Weisshaupt P. Muscle injury, cross-sectional area and fibre type distribution in mouse soleus after intermittent wheel-running. J Physiol. 1990;428:639–52. doi: 10.1113/jphysiol.1990.sp018232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Partridge TA, Morgan JE. Multiple insights from myogenic cell transplants. Hum Gene Ther. 2014;25:404–5. doi: 10.1089/hum.2014.035. [DOI] [PubMed] [Google Scholar]
- 68.Blaveri K, Heslop L, Yu DS, Rosenblatt JD, Gross JG, Partridge TA, et al. Patterns of repair of dystrophic mouse muscle: studies on isolated fibers. Dev Dyn. 1999;216:244–56. doi: 10.1002/(SICI)1097-0177(199911)216:3<244::AID-DVDY3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 69.Chan S, Head SI. The role of branched fibres in the pathogenesis of Duchenne muscular dystrophy. Exp Physiol. 2011;96:564–71. doi: 10.1113/expphysiol.2010.056713. [DOI] [PubMed] [Google Scholar]
- 70.Head SI. Branched fibres in old dystrophic mdx muscle are associated with mechanical weakening of the sarcolemma, abnormal Ca2+ transients and a breakdown of Ca2+ homeostasis during fatigue. Exp Physiol. 2010;95:641–56. doi: 10.1113/expphysiol.2009.052019. [DOI] [PubMed] [Google Scholar]
- 71.Chan S, Head SI, Morley JW. Branched fibers in dystrophic mdx muscle are associated with a loss of force following lengthening contractions. Am J Physiol Cell Physiol. 2007;293:C985–92. doi: 10.1152/ajpcell.00128.2007. [DOI] [PubMed] [Google Scholar]
- 72.Metzger T, Gache V, Xu M, Cadot B, Folker ES, Richardson BE, et al. MAP and kinesin-dependent nuclear positioning is required for skeletal muscle function. Nature. 2012;484:120–4. doi: 10.1038/nature10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qaisar R, Larsson L. What determines myonuclear domain size? Indian J Physiol Pharmacol. 2014;58:1–12. [PubMed] [Google Scholar]
- 74.Hernandez-Caballero E, Herrera-Gonzalez NE, Salamanca-Gomez F, Arenas-Aranda DJ. Role of telomere length in subtelomeric gene expression and its possible relation to cellular senescence. BMB Rep. 2009;42:747–51. doi: 10.5483/BMBRep.2009.42.11.747. [DOI] [PubMed] [Google Scholar]
- 75.Lackner DH, Hayashi MT, Cesare AJ, Karlseder J. A genomics approach identifies senescence-specific gene expression regulation. Aging Cell. 2014;13:946–50. doi: 10.1111/acel.12234. [DOI] [PMC free article] [PubMed] [Google Scholar]