

Abstract
Background
miRNAs are the most abundant class of small non-coding RNAs, and they are involved in post-transcriptional regulations, playing a crucial role in the refinement of genetic programming during plant development. Here we present a comprehensive picture of miRNA regulation in Vitis vinifera L. plant during its complete life cycle. Furthering our knowledge about the post-transcriptional regulation of plant development is fundamental to understand the biology of such an important crop.
Results
We analyzed 70 small RNA libraries, prepared from berries, inflorescences, tendrils, buds, carpels, stamens and other samples at different developmental stages. One-hundred and ten known and 175 novel miRNAs have been identified and a wide grapevine expression atlas has been described. The distribution of miRNA abundance reveals that 22 novel miRNAs are specific to stamen, and two of them are, interestingly, involved in ethylene biosynthesis, while only few miRNAs are highly specific to other organs. Thirty-eight miRNAs are present in all our samples, suggesting a role in key regulatory circuit. On the basis of miRNAs abundance and distribution across samples and on the estimated correlation, we suggest that miRNA expression define organ identity. We performed target prediction analysis and focused on miRNA expression analysis in berries and inflorescence during their development, providing an initial functional description of the identified miRNAs.
Conclusions
Our findings represent a very extensive miRNA expression atlas in grapevine, allowing the definition of how the spatio-temporal distribution of miRNAs defines organ identity. We describe miRNAs abundance in specific tissues not previously described in grapevine and contribute to future targeted functional analyses. Finally, we present a deep characterization of miRNA involvement in berry and inflorescence development, suggesting a role for miRNA-driven hormonal regulation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1610-5) contains supplementary material, which is available to authorized users.
Keywords: Grapevine, microRNAs, Deep Sequencing, RT-qPCR, Berries, Inflorescences, Plant development, Ethylene biosynthesis
Background
The completion of the grapevine (Vitis vinifera L.) genome sequence [1] and subsequent research to elucidate gene function and molecular mechanisms involved in plant development, yield and quality represent a milestone in the genomics of fruit tree species. Transcriptomic and functional analyses have greatly contributed to the description of the gene regulatory network underlying grapevine development [2–6] with a particular focus on berry ripening, due to its relevance in wine production and fresh fruit consumption. Vast collections of grapevine cultivars exist, making grapevine a model fruit tree species that offers the scientific community the opportunity to study genetic diversity.
Many studies have shed light on the regulatory layer mediated by small RNAs, [7–11] revealing their crucial role in the genetic programming and fine tuning of plant biology. MicroRNAs (miRNAs) are a class of small RNAs (21–24 nucleotide long) coded by specific endogenous genes, which act as negative gene regulators. miRNAs guide mRNA cleavage, translational repression and chromatin modification of corresponding target genes [12] and are involved in a variety of developmental processes, which include organ morphogenesis [7,13–16], transition from vegetative to reproductive plant growth [17,18] and responses to stress [19–21]. Other classes of small RNAs are involved in regulating genome methylation levels, and genome stability, thus keeping transposon and repetitive elements silent [22,23].
Considering the role that miRNAs and other small regulatory RNAs play in plant biology, any comprehensive genomic description should include a detailed structural and functional characterization of these regulatory molecules. It is necessary to detail the regulatory layer mediated by these molecules in grapevine, as well, not only because of its great economic importance, but also because grapevine has a complex life cycle, thus providing the opportunity to explore different developmental stages spanning a two-season period. Next generation sequencing technologies have strongly favoured the analyses of small RNA regulatory networks, detecting rare molecules and small RNA variants with unprecedented precision and sensitivity. There is a relatively large number of grapevine miRNAs deposited in miRBase (Release 20 www.mirbase.org) and/or reported in the literature [15, 24–31]. Their expression profile has been analyzed in a few critical tissues/organs, predominantly in developing berries and flowers.
Here we describe a genome-wide transcription atlas of miRNAs in grapevine, analyzing the spatio-temporal distribution of known and newly discovered miRNAs, in the widest range of grapevine samples considered thus far. The identification of targets mRNA by in silico prediction methods complements our characterization of the miRNA transcriptome and deepens our understanding of the biological functions of miRNAs in grapevine development.
Our aim is to establish a reference for the future development of targeted functional studies of miRNA regulation.
Results
Sequencing statistics
We constructed and sequenced 70 bar-coded libraries from Vitis vinifera L. cv. Corvina and the V. vinifera clone PN40024, the reference clone used for genome sequencing [1] (Table 1).
Table 1.
Samples list of Vitis vinifera cv Corvina and PN40024 clone, used for small RNA libraries
| Sample | Developmental stages | Replicates | Library codes | Modified E-L system stages |
|---|---|---|---|---|
| BUD | Latent bud | 2 | Bud_L | E-L 23 |
| Winter Bud | 2 | Bud_W | E-L 1 | |
| Bud burst (green tip) | 2 | Bud_B | E-L 4 | |
| Bud after burst (Rosette of leaf tips visible) | 2 | Bud_AB | E-L 5 | |
| LEAF | Young | 2 | Leaf_Y | EL-14 |
| Mature | 2 | Leaf_M | E-L 29 | |
| Leaf-Senescence | 2 | Leaf_S | E-L 43 | |
| STEM | Green stem | 2 | Stem_G | E-L 30 |
| Woody stem | 2 | Stem_W | E-L 43 | |
| ROOT | In vitro cultivation | 2 | Root | |
| TENDRIL | Young (Pool of tendrils from shoot bearing 7 separated leaves) | 2 | Tendril_Y | E-L 14 |
| Well-developed (Pool of tendrils from shoot bearing 12 separated leaves) | 2 | Tendril_WD | E-L 17 | |
| INFLORESCENCE | Young inflorescence | 2 | Inflorescence_Y | E-L 14 |
| Well-developed inflorescence | 2 | Inflorescence_WD | E-L 17 | |
| FLOWER | Beginning of flowering (10% caps off) | 2 | Flower_FB | E-L 20 |
| Flowering (50% caps off) | 2 | Flower_F | E-L 23 | |
| STAMEN | Pool of stamens from flowers at 10% and 50% caps off | 2 | Stamen | E-L 20/E-L 23 |
| POLLEN | Pollens from flowers at 80% caps off | 2 | Pollen | E-L 25 |
| CARPEL | Pool of carpels from flowers at 10% and 50% caps off | 2 | Carpel | E-L 20/E-L 23 |
| BERRY | Fruit set | 2 | Berry_FS | E-L 29 |
| Post fruit set | 2 | Berry_PFS | E-L 31 | |
| Pre-Veraison | 2 | Berry_PV | E-L 34 | |
| Veraison | 2 | Berry_V | E-L 35 | |
| Pre-ripening | 2 | Berry_MR | E-L 36 | |
| Ripening | 2 | Berry_R | E-L 38 | |
| Post-harvest withering (1st month) | 2 | Berry_PHWI | ||
| Post-harvest withering (2nd month) | 2 | Berry_PHWII | ||
| Post-harvest withering (3rd month) | 2 | Berry_PHWIII | ||
| SEED | Collected from green berries (Pool of berries from FS, PFS, PV) | 1 | Seed_G | |
| Collected from Mature berries (Pool of berries from V, MR, R) | 1 | Seed_M | ||
| RACHIS | Fruit set | 2 | Rachis_FS | E-L 29 |
| Post -fruit set | 2 | Rachis_PFS | E-L 31 | |
| Veraison | 2 | Rachis_V | E-L 35 | |
| Pre-ripening | 2 | Rachis_MR | E-L 36 | |
| Ripening | 2 | Rachis_R | E-L 38 | |
| Total Corvina | 35 | 68 | ||
| ROOT | Pool of in vitro roots of PN40024 clone | 1 | Root_PN | |
| LEAF | Pool of in vitro leaves of PN40024 clone | 1 | Leaf_PN | |
| Total PN40024 | 2 | 2 |
One database was established using the grapevine genome, and made available on the website (https://mpss.udel.edu/dbs/index.php?SITE=grape_sRNA_atlas), in order to store and assist the visualization of all the sequenced libraries.
Approximately 1.26 billion small RNA raw reads were produced. Adapter sequences were recognized and trimmed from 739,396,104 raw reads which correspond to 61,883,599 unique sequences, ranging from 18 to 34 nt (Additional file 1). A total of 377,328,013 reads represented by 27,318,558 unique sequences were perfectly mapped to the V. vinifera PN40024 reference genome [1], excluding reads mapping to rRNA, tRNA, snRNA, and snoRNA sequences.
In most samples, mapped reads show a similar pattern of size distribution, with distinct peaks at 21 and 24 nt (Fig. 1), in agreement with known products of Dicer-Like (DCL) protein cleavage. In particular, the 24 nt-long molecules were the most abundant class in latent buds, flowers (Flower_FB and Flower_F) and floral organs, such as carpels and stamens. The predominance in floral tissues of the 24 nt molecules, already described in grapevine by Wang et al. [30], may reflect the shift of the small RNA population towards siRNAs during plant development, shift linked to transposons and repeated sequences. The remaining samples had the highest proportion of 21 nt-long sequences, typically represented by miRNAs. The 21 nt class showed the highest degree of redundancy, suggesting that they derive from precursors which produce well-defined short sequences, whereas the 24-nt class showed a lower redundancy, indicating their origin in loci producing populations of heterogeneous small RNA molecules. Size profiling indicated an insufficient quality for further quantitative analyses of a few libraries (Fig. 1), namely those derived from roots, leaves in senescence, young leaves, woody stems, pollen, post-harvest berries and seeds, which were used only to identify known vvi-miRNAs and to predict novel miRNA candidates, but not for the expression analysis. These libraries did not present the typical 2-peaks profile expected in angiosperms [32] for small RNA sequencing data and/or were not sufficiently abundant in terms of distinct genome matched reads (Additional file 1).
Fig. 1.
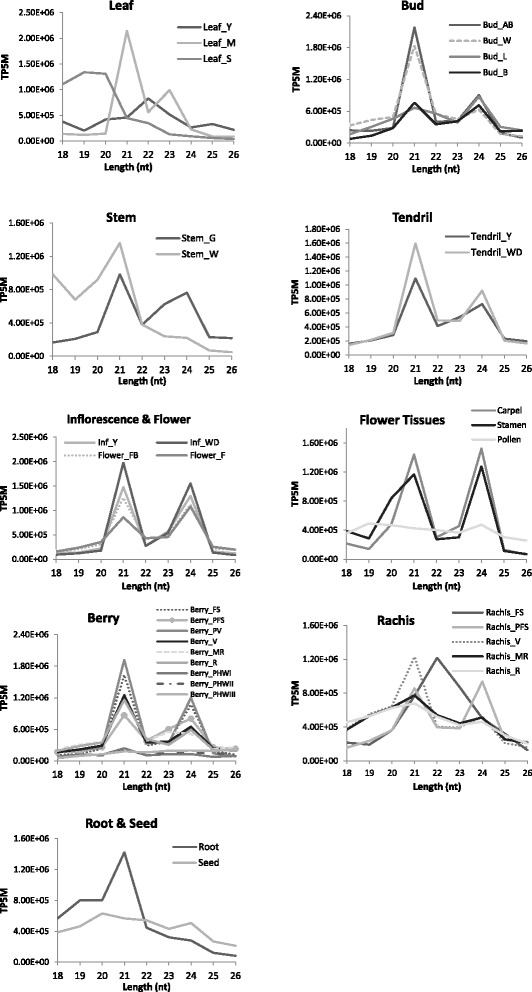
Size distribution of small RNAs from all samples of grapevine. X-axis: length in nucleotides (nt); Y-axis: summed normalized abundances for each size class as an average of two biological replicates. Library codes are indicated in Table 1
Identification of annotated and novel grapevine miRNAs
We applied a powerful miRNA identification pipeline, developed by Jeong, Zhai and collaborators [33,34], successfully applied to several species [35–40], to our set of 70 small RNA libraries in order to identify already annotated miRNAs (known) and to discover novel candidates.
We fed the pipeline with 32,214,912 and 3,393,929 distinct small RNA sequences from the 68 libraries of cultivar Corvina and the two libraries of clone PN40024, respectively.
First, we searched for known vvi-miRNAs in the Corvina data set and identified 98 unique sequences, representing 157 out of 186 known mature vvi-miRNAs. We manually investigated all the 157 known miRNA loci identified in the Corvina libraries, checking for the existence of the correct precursor and whether the annotated miRNAs corresponded to the most abundant one. We retrieved the highest abundant sequence and, when possible, also the complementary 3p or 5p sequence. For twenty-three miRNAs the most abundant sequence appeared as a different variant (isomiR) of the annotated miRNA, showing different length or a shift of the map position in the precursor (Additional file 2). For instance, the annotated sequence of vvi-miR171b showed an abundance of only 6 TP5M, whereas its variant, which starts 3 bp downstream of the annotated one, had an abundance of 4243 TP5M. The 3 bp shift of vvi-miR171b was already observed and deposited in miRBase. A difference in length was noted for 16 vvi-miRNAs from ten families (Additional file 2). In line with Jeong et al. [34], Devers et al. [41] and Marco et al. [42], we observed cases where the complementary sequence of the annotated vvi-miRNA was more abundant than or as abundant as the annotated sequence, suggesting that complementary sequences might have biological functions. This was true for five members of the vvi-miRNA169 family, for vvi-miR398a, vvi-miR479, and vvi-miR482. For three other loci, vvi-miR159c, vvi-miR169w and vvi-miRNA3639, we found two distinct duplexes produced from the same precursor, which we designated as vvi-miR159c.2, vvi-miR169w.2 and vvi-miRNA3639.1 (Additional file 3). The sequencing reads originating from vvi-miRNA159c and vvi-miRNA169w were arranged in phase, either in tandem (vvi-miR169w) or separated by a gap of 21 nucleotides (vvi-miR159c). In contrast, the two duplexes produced by vvi-miRNA3639 had a 13 bp overlapping region, and the most sequenced reads corresponded to the not annotated reads, as observed for the vvi-miRNA169w locus.
In conclusion, we confidently identified 88 unique, known sequences, corresponding to 110 known mature vvi-miRNAs from which, 80 also had the complementary (3p or 5p) sequence identified (Additional file 2). These identified vvi-miRNAs belong to 41 conserved plant miRNA families. Seven grapevine miRNA known families (vvi-miR828, vvi-miR845, vvi-miR3626, vvi-miR3630, vvi-miR3631, vvi-miR3636 and vvi-miR3638), were not identified in our libraries.
For the identification of novel miRNAs, the previous pipeline was used with Corvina and PN40024 libraries as input. In total, 905 unique miRNA candidates generated from 1216 precursors were kept. These candidates were compared to those in miRBase identifying 45 sequences with >90% similarity to known plant miRNAs, 32 were new members of 13 known Vitis families, and 13 candidates were members of 10 new Vitis families (Additional files 4, 5, 6 and 7). After subtracting these 45 sequences, 1171 completely novel grapevine miRNA candidates were retained.
Interestingly, we found two new members of the miRNA172 family, vvi-miRC172e and vvi-miRC172f, in antisense to vvi-miR172b and vvi-miR172a respectively. The 3’ sequences produced from both antisense duplexes were predicted to target Apetala2 (AP2) gene and other AP2-related genes, which are already well known targets of the miRNA172 family (Additional file 8). As for the 5’sequences of the duplexes, both had the DNA-directed RNA polymerase III subunit C3 as a predicted target (Additional file 8).
For the identification of completely novel miRNAs candidates, in order to prevent the selection of siRNA-like miRNAs, we selected from the list of candidates passing the pipeline’s filters, only 20, 21 and 22 bp long candidates. Their precursor secondary structures were then inspected manually (UEA sRNA toolkit, plant version [43]). Our analyses indicated that 132 novel vvi-miRNA candidates (109 from Corvina and 23 from PN40024) (Additional files 4, 5, 6 and 7) matched the criteria for miRNA annotation [44]. For ten sequences the complementary sequence (3p or 5p) was also retrieved with a high abundance, and is, hence, reported in Additional file 3. As observed in conserved miRNAs, around 45% (60 out of 132) of novel candidates began with a 5' uridine, which is a characteristic feature of miRNAs (Additional files 4 and 5).
Finally, we compared our novel miRNA candidates with those reported previously in grapevine, and not deposited in miRBase [27, 29–31] cross checking sequences, allowing some nucleotide shift, and, when available, the genomic coordinates. Only 31 miRNAs were previously identified. The mature sequences of 14 out of these candidates, were commonly retrieved in [27, 29, 30], while no sequences/precursors were in common with [31].
In silico identification of target genes
We predicted the targets for all the miRNA set here identified, comprising a total of 396 miRNA sequences (considering known, and novel miRNAs and their complementary sequences when is the case, from both Corvina and PN40024 data). A total of 2068 predicted targets were retrieved using TargetFinder [45]. Only targets with a score of 3.0 were considered and further investigated (Table 2, Additional files 8 and 9). On average, for each miRNA sequence, two targets were found, but we observed several cases in which one miRNA targets up to ten distinct coding genes. Since in silico prediction of miRNA targets is prone to false positive targets [46], to optimize both accuracy and sensitivity we checked our results with psRNATarget, a second in silico pipeline [47]. Seventy-one per cent of the predicted targets shown in Additional file 8 were confirmed (data not shown).
Table 2.
List of miRNA targets, discussed in the paper. List of miRNA and their respective targets as identified by TARGET FINDER, and divided per plant organ. GRAPE_IGGP_v1 12X assembly - v2 annotation was used. Only the miRNA/targets pair discussed in the paper are shown. All these miRNA targets are also confirmed by psRNATarget, with the exception of VIT_210s0003g00870, VIT_216s0022g02450, VIT_216s0100g00030
| Tissues | vvi-miRNA | Target | Predicted function |
|---|---|---|---|
| Inflorescence/Flowers | miR156a; miR156b-5p; miR156c-5p; miR156d-5p; miR156e; miR156f; miR156g-5p; miR156i | VIT_208s0007g06270 | Squamosa promoter-binding-like protein 9-like |
| VIT_211s0065g00170 | Squamosa promoter-binding-like protein 12-like | ||
| VIT_201s0010g03710 | Squamosa promoter-binding-like protein 2 | ||
| VIT_201s0011g00130 | Squamosa promoter-binding-like protein 6-like | ||
| VIT_201s0010g03910 | Squamosa promoter-binding-like protein 13-like | ||
| VIT_215s0021g02290 | Squamosa promoter-binding-like protein 7-like | ||
| VIT_217s0000g01260 | Squamosa promoter-binding-like protein 13 | ||
| VIT_217s0000g05020 | Squamosa promoter-binding-like protein 6-like | ||
| miR156a; miR156b-5p; miR156c-5p; miR156d-5p; miR156e | VIT_214s0068g01780 | Squamosa promoter-binding-like protein 16-like | |
| miR156b-5p; miR156c-5p; miR156d-5p; miR156e | VIT_219s0090g01180 | Uncharacterized protein | |
| miR156b-5p; miR156c-5p; miR156d-5p | VIT_215s0046g01020 | Uncharacterized protein loc100241322 | |
| miR156e | VIT_204s0008g02640 | Plastid-lipid-associated protein 8 | |
| miR172c-3p | VIT_213s0019g03550 | AP2 domain-containing transcription factor | |
| miR172c-3p; miR172d | VIT_207s0031g00220 | Transcription factor Apetala2 | |
| VIT_206s0004g03590 | Transcription factor Apetala2 | ||
| VIT_208s0040g03180 | AP2 domain-containing transcription factor | ||
| miR172d | VIT_200s0184g00102 | AP2 domain-containing transcription factor | |
| miR390-5p | VIT_210s0003g01890 | LRR receptor-like serine threonine-protein kinase RFK1 | |
| VIT_216s0098g01090 | Leucine-rich repeat receptor-like protein kinase PEPR1 | ||
| miR393a-5p; miR393b | VIT_214s0068g01330 | LRR receptor-like serine threonine-protein kinase RFK1 | |
| VIT_207s0104g01320 | Transport inhibitor response 1 | ||
| VIT_214s0030g01240 | Transport inhibitor response 1 | ||
| grape-m9269 | VIT_208s0007g05240 | Phosphatidylinositol-4-phosphate 5-kinase | |
| Stamen | grape-m5380; grape-m7774 | VIT_205s0049g00340 | 1-aminocyclopropane-1-carboxylate oxidase homolog 1 |
| VIT_200s1313g00010 | 1-aminocyclopropane-1-carboxylate oxidase homolog 1 | ||
| VIT_205s0049g00330 | 1-aminocyclopropane-1-carboxylate oxidase homolog 1 | ||
| VIT_205s0049g00370 | 1-aminocyclopropane-1-carboxylate oxidase homolog 1 | ||
| VIT_216s0022g02420 | Protein | ||
| VIT_216s0100g00030 | Cucumisin-like | ||
| VIT_216s0022g02450 | Protein | ||
| VIT_205s0049g00350 | 1-aminocyclopropane-1-carboxylate oxidase homolog 1 | ||
| Berry development | miR319b-3p; miR319c-3p; miR319f-3p; miR319g | VIT_213s0067g01630 | Transcription factor gamyb-like |
| VIT_210s0003g00870 | Transcription factor TCP4-like | ||
| miR319c-5p | VIT_214s0081g00480 | Protein | |
| miR395a; miR395c-3p; miR395d; miR395e-3p; miR395f-3p; miR395g-3p; miR395h-3p; miR395i-3p; miR395j-3p; miR395k-3p; miR395l-3p | VIT_207s0031g00940 | Sulphate bicarbonate oxalate exchanger and transporter SAT-1 | |
| miR395h-5p; miR395k-5p | VIT_214s0066g00910 | Protein | |
| miR395n | VIT_205s0020g04210 | ATP sulphurylase | |
| miR396a-3p | VIT_207s0031g02320 | Protein | |
| VIT_200s0208g00090 | Protein trichome birefringence-like 16 | ||
| miR396a-5p | VIT_210s0597g00040 | Pentatricopeptide repeat-containing protein | |
| VIT_202s0025g04910 | Growth-regulating factor | ||
| VIT_215s0048g01740 | Growth-regulating factor | ||
| VIT_200s0494g00010 | Transcription activator | ||
| VIT_218s0001g08650 | Uncharacterized protein loc100260890 | ||
| miR396a-5p; miR396c-5p; miR396d-5p | VIT_216s0039g01450 | Growth-regulating factor 1 | |
| VIT_209s0002g01350 | Growth-regulating factor 5 | ||
| VIT_211s0016g01250 | Growth-regulating factor 5 | ||
| VIT_208s0007g03760 | Growth-regulating factor 4 | ||
| VIT_216s0098g01080 | Protein | ||
| VIT_202s0025g02680 | Uncharacterized protein loc100258227 | ||
| miR396a-5p; miR396b-5p | VIT_207s0191g00220 | Uncharacterized protein loc100251922 | |
| miR396b-3p | VIT_201s0011g01220 | Double top | |
| miR396c-3p | VIT_205s0077g02140 | Glyoxysomal fatty acid beta-oxidation multifunctional protein MFP-A | |
| miR396d-3p | VIT_215s0024g00350 | TATA box binding protein associated factor-like protein | |
| grape-m6905 | VIT_213s0064g00290 | bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like | |
| VIT_213s0047g00940 | bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like | ||
| VIT_203s0110g00350 | Cinnamoyl reductase-like protein | ||
| VIT_213s0047g00990 | bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like | ||
| VIT_213s0064g00340 | Phenylacetaldehyde reductase | ||
| VIT_213s0067g00530 | bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like | ||
| VIT_213s0067g00460 | bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like | ||
| VIT_213s0047g00540 | bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like |
For each predicted miRNA:target pair, we calculated the Pearson correlation between the level of expression of the miRNA (discussed in the next paragraphs) and the expression values of the target gene available from the cv. Corvina transcriptomic atlas [4]. Considering all the common sample shared between our atlas and [4], and a p-value cutoff of 0.05, we identified the miRNAs negatively correlated to their predicted targets (Additional file 10)
Expression analyses of Corvina libraries
To evaluate the abundance of each miRNA identified in our libraries, we used the normalized abundance (TP5M) of each sequence in each library, which represents the relative cloning frequency. We considered, for further analyses, a miRNA as expressed only when both biological replicates met the set threshold of 10 TP5M. Accordingly, we eliminated only a few 3p or 5p sequences of known miRNA and nearly 40 newly predicted miRNAs that were below the set threshold. To establish the miRNA expression atlas, we used sequencing data of our Corvina libraries, discarding some samples that showed an insufficient sequencing-quality (see Sequencing Statistics for details).
Figure 2 shows the distribution of miRNAs expressed in each of the 25 different samples considered. The number of expressed miRNAs is relatively high in the 25 samples, ranging from 85 in ripening rachis to 176 in whole developed tendrils. Forty four miRNAs were classified as organ-specific (Fig. 3), i.e. miRNAs expressed exclusively in one organ, in at least one of its developmental stages. Stamen and buds expressed the highest number of organ-specific miRNAs. Of the 22 stamen-specific miRNAs, two have targets coding for a protein involved in TMV resistance (Additional file 8), while two (grape-m5380 and grape-m7774) have, among their targets, five genes coding for Aminocyclopropane-1-carboxylate oxidase homolog (ACC OX genes). Interestingly, grape-m7774 maps on chromosome five in a region where four out of the five targets map (Fig. 4). Thirty-eight miRNAs were identified as common to all samples; a part from grape-m3245, 3p and 5p sequence, all the other common miRNAs belong to known miRNA families.
Fig. 2.
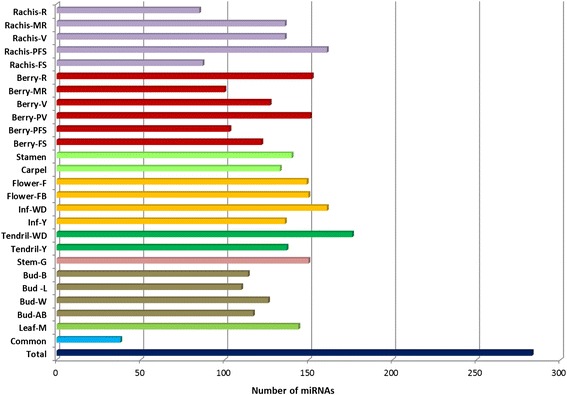
Number of miRNA expressed in 25 different grapevine samples used to define the atlas. X-axis: number of miRNAs expressed in individual samples; Y-axis: different samples. Common refers to the number of miRNA expressed in all the samples. Total is the whole set of miRNAs expressed above the set threshold of 10 TPM in both replicates
Fig. 3.
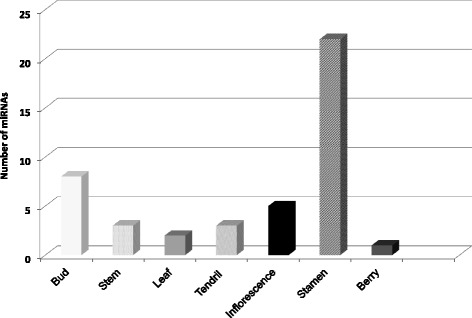
Number of organ specific miRNAs identified in Corvina derived libraries. X-axis: number of miRNAs; Y-axis: different organs
Fig. 4.
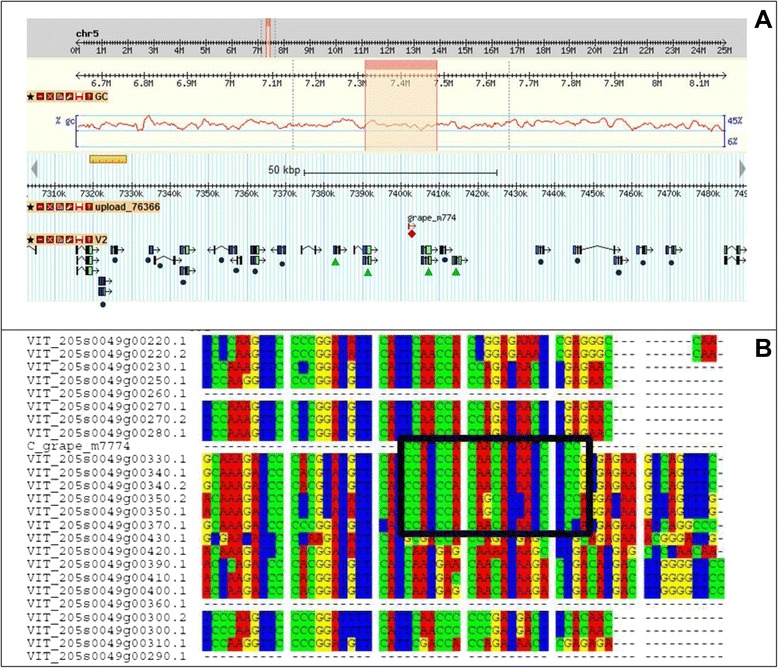
Genomic analyses of miRNA grape-m774 and its targets. A: The genomic region of chromosome five hosting grape-m774 and 19 genes coding for ACC oxidase, using Gbrowse (http://genomes.cribi.unipd.it). Closed diamond: miRNA; Closed dot: ACC oxidase homolog not target; Closed triangle: ACC oxidase homolog target of the miRNA. B: Alignment of the 19 ACC oxidase transcripts. When more than one transcript is present for a given gene, it is reported and indicated. The reverse complement of the miRNA is shown. Black box highlights the region of sequence similarity between miRNA and targets
To deepen the analysis on miRNAs distribution during plant development, we investigated each plant structure separately (i.e. berries, inflorescences/flowers, tendrils, rachis and buds) considering the distinct time points sampled. miRNAs expressed in all developmental phases of a given organ range from a minimum of 30.6% (54/176) in rachis, to a maximum of 68.3% in tendrils (Fig. 5). In the six stages of berry development 74 miRNAs (40%) are in common. The number of miRNAs present in only one developmental time point is very low, when compared to the total number of miRNAs expressed in the sample.
Fig. 5.
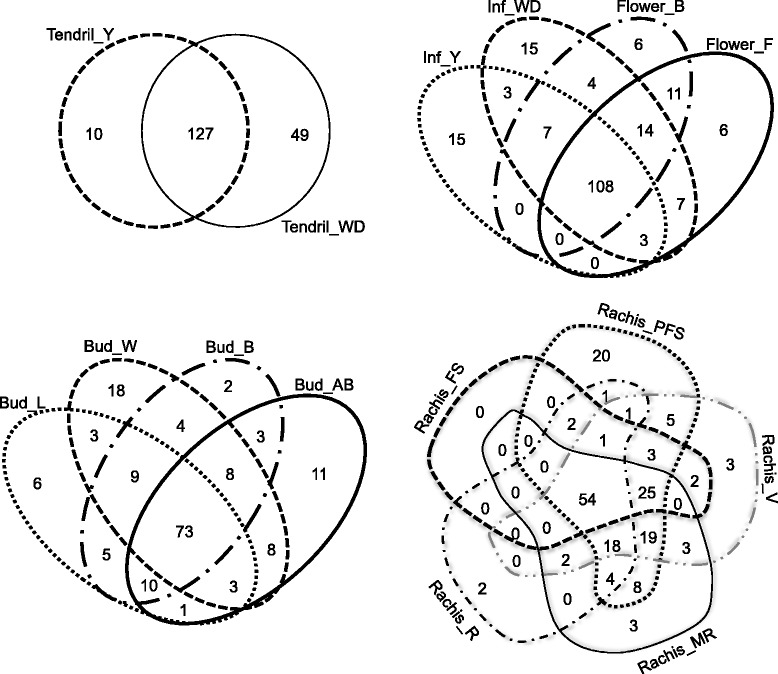
Shared miRNAs among the different developmental stage of each organ. The description of library codes is in Table 1
We analyzed the top 20 expressed miRNAs in each sample (Additional file 11) to check whether the most abundant miRNAs were shared among samples. Our analysis showed that in fact highly abundant (more than 1000 TP5M) miRNAs are similar in almost all the samples, but stamen and carpels, with vvi-miR166 family being by far the most abundant (above 30,000 TP5M and reaching 1 million TP5M), followed by vvi-miR3634-3p (a grapevine specific miRNA with unknown target), by vvi-miR159c.1-3p and by the novel candidate grape-m3245. vvi-miR159c.1-3p and four different members of miR319 family (above 59,000 or 70,000 TP5M), whose 3p sequence is predicted to target TCP4-like and a GAMYB-like transcription factors, are the most expressed in stamen and carpel. vvi-miR159c.1-3p is not predicted to target a MYB transcription factor, as is usual for miR159 molecules, but a gene encoding a Lactoylglutathione Lyase protein (Glyoxalase family), putatively involved in plant tolerance to stresses.
To analyze the overall expression profile of all the miRNAs in our atlas, we represented their expression values in a heat map, subjecting the normalized count of reads to hierarchical clustering (Fig. 6). Known miRNAs (Fig. 6A), and candidate miRNAs, putatively grapevine specific (Fig. 6B), were analyzed in two different heat maps, to reduce the complexity of the figure and allow the graphical representation of these two groups of miRNAs whose average expression level is very different: above 3000 TP5M for the group of known miRNAs and below 300 TP5M for the novel miRNAs. The expression profiles of the 109 novel candidates vvi-miRNA revealed that nine novel candidates are relatively highly expressed in most of the 25 samples (grape-m8278-3p, grape-m4553-3p, grape-m3245-3p, grape-m9163, grape-m3245-5p, grape-m7644, grape-m2399, grape-m9381, grape-m8993), while the remaining candidates showed a medium to low expression level, being enriched in few samples, such as grape-m4269 and grape-m 3469-5p expressed in flowers at beginning of flowering and at full bloom (Flower_FB and Flower_F), or as grape-m4250 and grape-m4277 expressed only in young inflorescences, and grape-m4553-5p and grape-m8278-5p, expressed in well developed tendrils, berry at veraison and berry at ripening (Fig. 6B). It is striking the presence of the 22 stamen-specific miRNAs, all belonging to our novel candidate group. Differently, the heat map representing the known miRNAs (Fig. 6A) reveals a reduced level of organ/tissue specificity and a high expression of numerous families, often nearly ubiquitous. Some miRNAs are specific to a few groups of samples, such as vvi-miR395-3p, which is strongly regulated during flower and berry development, or miRC477-3p mainly expressed in berries and rachis, and miR393 a,b -5p expressed in developing rachis and flowers.
Fig. 6.
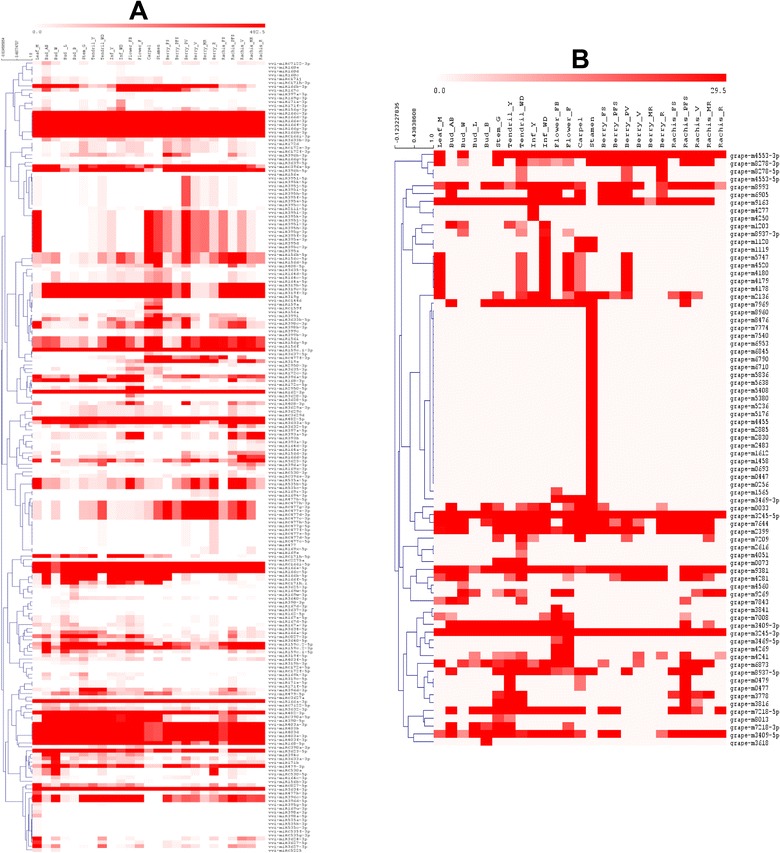
Heat map and hierarchical cluster analysis (HCL) of miRNAs identified in Corvina derived libraries. The HCL tree was generated with the average linkage clustering method. A: Heat map with known miRNAs including new members of known families. B: Heat map with novel miRNAs. miRNAs with an abundance below the set threshold of 10 TP5M in both replicates were set to zero. miRNAs that are zero in all the 25 samples were removed from these heat maps. Red and white represent high and low expression, respectively
miRNA profiles define organ identity
In order to define the relationships among the 25 different samples used in our atlas, a correlation matrix (Additional file 12), a dendrogram and a PCA (Fig. 7) were established. The distance was calculated using one minus Pearson correlation metric of normalized data, as explained in full details in Methods, to evaluate the level of similarity among the samples from the miRNA perspective. The correlation matrix identifies two main clusters (see Additional file 11): the first groups inflorescences, buds, tendrils and stem, the second includes all the developing berries and their respective rachis stages. The leaf is scarcely related to any of the samples, and far from the inflorescence/buds group. The carpel and stamen are part of a distinct group not correlated with any other tissue.
Fig. 7.
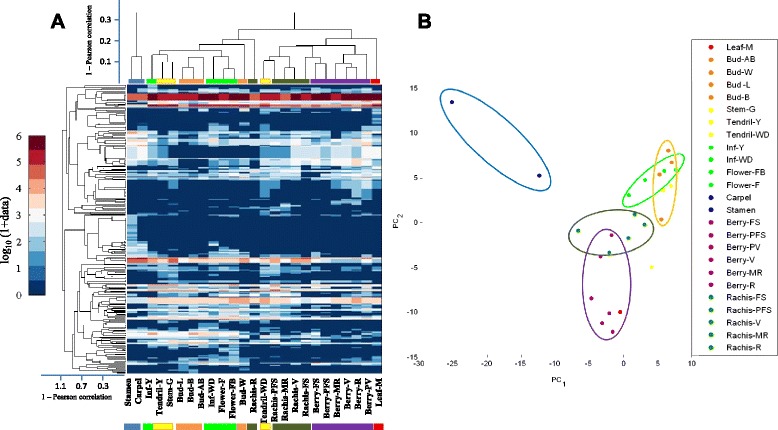
Clustergram and principal component analysis of the 25 different grapevine samples of the miRNA atlas. (A) The picture reports the hierarchical clustering analysis for both samples and miRNAs, the corresponding dendrogram and the data heat map. Data were normalized as described in Methods. The X-axis of each dendrogram is the 1 minus Pearson correlation distance metric. We used an optimal leaf order value which determines the leaf order that maximizes the similarity between neighbouring leaves. (B) A PCA analysis has been performed on the dataset. Data were preliminarily z-scored. X-axis represents the first components and the Y-axis the second component. The two components explain 34.5% of total variance. Each organ is associated to the same colour and ovals indicate clusters approximately related to organ identity. Blue for reproductive organs, purple for berries, red for leaf, orange for buds, yellow for stem and tendrils, light green for inflorescences and dark green for rachis. The same colours are used in the dendrogram. Each color represents a different cluster. One minus Pearson correlation was used as a metric distance
Dendrogram and PCA analyses (Figs. 7A and 7B) are consistent with correlation matrix, revealing the existence of the following functional groups: a) reproductive organs (stamen and carpel); b) leaves; both less correlated to the other following groups: c) green and maturing berries; d) rachis; e) inflorescences and opened flowers; f) buds and g) other green tissues. These analyses suggest that miRNAs reflect the functional specificity of different organs, as developmental stages of the same organ, and organ with a similar function are mainly grouped together, with few exceptions: well developed tendril that is closed to rachis, and not to other green tissues, the rachis at ripening stage and the woody bud that are less correlated to other rachis and buds samples, respectively.
Expression of miRNAs in berries and inflorescences
Considering the wide spectrum of sampling in our atlas, we focused on the spatio-temporal modulation of miRNA across inflorescences and berries development. We compared the abundance level of each miRNA (average of all replicates) among the four stages of inflorescences and, separately, among the six stages of developing berries. We investigated which miRNAs showed a fold change of at least five in at least one of all possible comparisons (Fig. 8). Additionally, we performed Real Time PCR (RT-qPCR) for eight of these miRNAs to confirm their abundance patterns (Fig. 9).
Fig. 8.
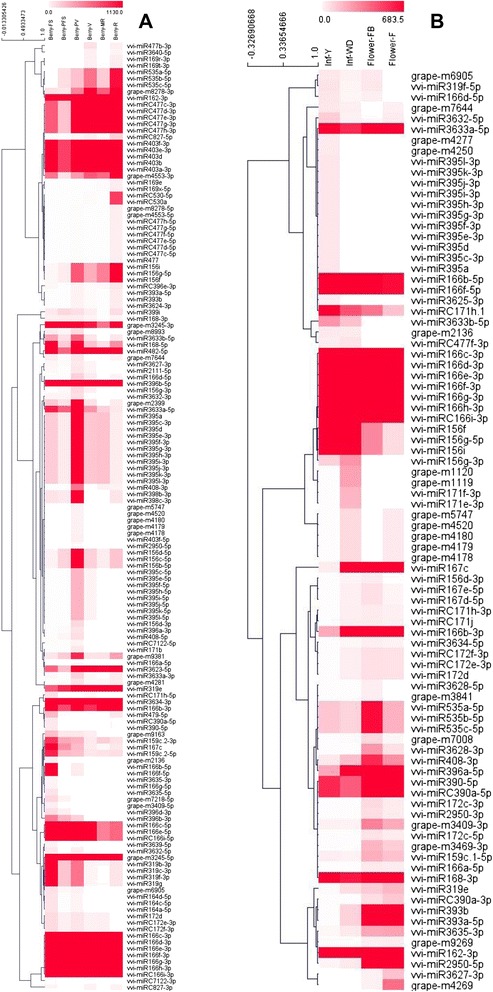
Heat map and hierarchical cluster analysis (HCL) of miRNAs with fold change greater than five. A: berries, B: flower tissues. The HCL tree was generated with the average linkage clustering method. Dark grey (red for online) and white represent high and low expression, respectively
Fig. 9.
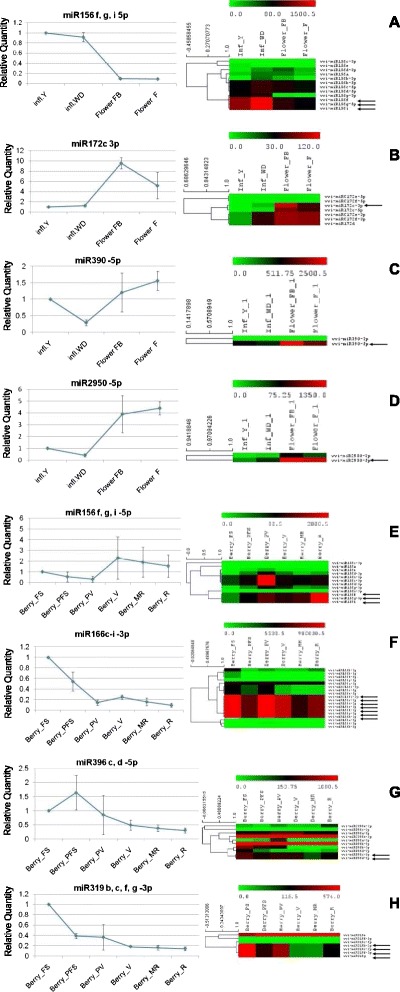
Real-Time PCR validation of deep sequencing data. Bars: standard error of the averages of each replicate. Heat maps on the right of each Real-Time graph refer to deep sequencing data. All the members of a given family are shown in heat maps, comprising 5p and 3p sequences. miRNAs tested by Real-Time PCR are indicated by an arrow. Panels A-D: miRNAs tested in the inflorescences/flowers samples; Panels E-H: miRNAs tested in the berries samples
In inflorescences, the well-known regulatory circuit involving miR156 and miR172 is confirmed by both sequencing and RT-qPCR data (Figs. 9A and 9B). As observed in other species [48–50], in grapevine an increase in miR156 levels corresponds to a low expression of miR172 and viceversa. From our data, miR156 family members target nine different SPL genes, six of which are negatively correlated to vvi-miR156 when considering their abundances (see Additional file 10).
Other two interesting miRNAs are vvi-miR390 and vvi-miR393, both involved in auxin signalling regulation, are up-regulated during inflorescence and flower development (Figs. 8B and 9C). miR390 regulates the Auxin Response Factor (ARF) through Trans-Acting siRNA locus 3 (TAS3), as experimentally validated in [28], while miR393 targets the auxin receptor TIR and an F-box protein [28] highlighting the importance of auxin regulation during flower development. Other conserved miRNAs, such as miR171 and miR395, show an interesting pattern of abundance in inflorescences (Fig. 8B), although their involvement in flower development is not clear. Our data show that miR171h decreases during inflorescence development, as already shown in Prunus [51]: in both Prunus and Vitis miR171 targets a GRAS family transcription factor, and from our data vvi-miR171b is negatively correlated to its predicted target VIT_202s0154g00400 (Additional file 10). miR395 family members are expressed in the young inflorescences (Infl_Y – 68.5 TP5M) and are then shut down in subsequent developmental stages. It is worth noting that while in open blooming flowers miR395 is below the set threshold of expression, its abundance is very strong in stamen (369.5 TP5M) and especially in carpel (658.5 TP5M), sampled from the same open flowers (Fig. 6A) This miRNA is also present in the berry at the fruit-set stage, peaking at pre-veraison (1571 TP5M), and decreasing from veraison through maturation, as already observed in our previous analyses [25] that suggests a role for this miRNA, regulating sulphate metabolism, at fruit setting and veraison.
Among the newly discovered miRNAs, showing a minimum fold change of five during flower development, we wish to highlight the role of grape-m6905, discussed later, and grape-m9269. This miRNA is below the set threshold of expression in the inflorescences (Infl-Y; Infl-WD) and is expressed at higher levels (around 50 TP5M) in flowers at the beginning of flowering and in open flowers (Flower_FB and Flower_F); it targets genes annotated as encoding aphosphatidylinositol-4-phosphate 5-kinase, known to mediate pollen tube growth in Arabidopsis and Nicotiana tabacum [52, 53].
Considering the six developmental stages of the berry and the relative heat map (Fig. 8A) we found some conserved miRNAs that reveal an interesting pattern of relative abundance. First of all, as observed in inflorescences, miR156 and miR172 are inversely correlated, and seem to be actively involved in fruit maturation: vvi-miR156 shows a gradual increase during ripening (as confirmed by RT-qPCR Fig. 9E), with a corresponding decrease of vvi-miR172. Furthermore, vvi-miR396, which targets Growth Regulating Factor genes (GRF) appears to be involved in berry development. Even though some discrepancies are visible between RT-qPCR and deep sequencing (Fig. 9G), both analyses indicate a clear reduction in abundance of this miRNA after veraison, when the berries enter the second growth phase, doubling their volume.
Vvi-miR166c/d/e/f/g/h/i-3p abundance profile is noteworthy: this molecule is the most abundant in our sequenced libraries. It shows a decrease after fruit set (Berry_FS), and peaks at veraison (Berry_V). Real Time PCR (Fig. 9F) does not show a strong increase at veraison, nor a peak at pre-veraison stage, raising some doubts on the huge abundance shown by sequencing (one third of the total sequences). Among newly discovered miRNAs, grape-m6905 is interesting in the context of both flower and berry development. It is switched off only in open flowers (Flower_F) and it accumulates (69.5 TP5M) in carpels, and, later in development, shows a peak (138.5 TP5M) in berries at the fruit-set (Berry_FS). It is then nearly switched off (below 25 TP5M) after veraison. This miRNA targets genes coding for enzymes (bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like DFR-like and cinnamoylreductase-like) involved in secondary metabolite biosynthesis, such as flavonoids and anthocyanin. The DFR gene transcript (VIT_213s0064g00290) is negatively correlated to its miRNA grape-m6905 (see Additional file 10), considering their relative abundances, thus reinforcing our in silico target prediction.
Discussion
To comprehensively define and describe the genetic programming of grapevine biology and development, it is important to extensively study genetic and epigenetic regulations driven by miRNAs, the most representative class of small non-coding RNAs. With the grapevine genome sequenced [1] and the extensive advances in next generation sequencing technologies, numerous miRNAs have been predicted in grapevine [24–31]. However, all these studies are based on few grapevine samples, mainly berries, inflorescences, leaves or tendrils at one/two developmental stages; therefore an expression atlas, covering a much wider spectrum of plant structures during grapevine life cycle, is required to provide a detailed overview of miRNA regulation and a solid base for functional studies.
miRNA populations in grapevine
The sequencing of 70 small RNA libraries, constructed from samples of Corvina cv. and the clone PN40024, produced a total of 1,219,741,533 and 39,268,213 reads respectively. Taking into consideration the enormous inventory of small RNAs in plants and their huge complexity, the identification of a relatively small portion of miRNAs from this vast stock posed a challenge [34]. By combining high-throughput data with a well-established and stringent bioinformatics pipeline and the in silico prediction of miRNA targets, we studied 110 conserved and 175 novel vvi-miRNA.
The conserved vvi-miRNAs identified represent 41 families out of the 48 annotated in grapevine. The absence, in our data, of 7 annotated miRNA families is ascribed to their low abundance in the analyzed samples (i.e. for miR828; miR3626; miR3631; miR3636 and miR3638), or to their nature of siRNA-like miRNAs, since most of them are not annotated with high confidence in miRBase (as explained in Kozomara and Griffiths-Jones, [54]). However we cannot rule out the possibility of differences between the Corvina cultivar and the reference genome sequence derived from the clone PN40024. For example, miR3630 is present in our libraries, but was discarded by the pipeline because of a sub-optimal pairing between the miRNA and its complementary sequence. miR845 was not found in any of our libraries, confirming previous papers [25] and miRBase findings that report a very faint expression of vvi-miR845a and vvi-miR845b.
Of the 175 novel grapevine miRNA candidates unravelled and described in our analysis, 45 showed similarity with other plant miRNAs deposited in miRBase. Interestingly, two of those were generated in antisense to two members of the vvi-miRNA172 family: vvi-miRC172e and vvi-miRC172f were reverse complementary (RC) to the known vvi-miR172b and vvi-miR172a, respectively. Shao et al. [55] reported these molecules in Vitis as members of a novel category of miRNAs, named RC-miRNAs. They have a small regulatory activity by cleaving target mRNAs, and might possess siRNA-like activity by mediating DNA methylation [56]. Very few papers have reported this class of miRNAs, first identified in animals [57–59]. Comparing novel miRNAs identified in the present paper (either completely novel or similar to other plant miRNAs) with those reported from the hybrids of V. vinifera and V. labrusca [29, 30] and from other V. vinifera cultivars [31] we found only 31 shared sequences. Similarly, comparing our newly identified Corvina and PN40024 miRNAs, only two miRNAs are in common. These findings strongly suggest that, apart from a group of well-defined miRNAs, each Vitis variety and species has its own specific set of miRNAs, or exhibit a different spatio-temporal pattern expression because of a different genetic background or an effect of genome plasticity, as already suggested by Alabi and colleagues [31].
Moreover, our data reinforce recent findings that the population of miRNAs is highly heterogeneous across species and varieties, since the processing of miRNA precursors is not always precise and equal under every condition. We found different sequence variants (isomiR) for each miRNA and we also showed that in some loci, the sequence on the opposite arm of the annotated miRNA (usually known as miRNA*) had a higher read number than their respective annotated sequence. Although miRNAs* was conventionally been thought to be degraded and non-functional [7, 60, 61], it is now clear that they accumulate in the cells, and are able to down-regulate mRNA targets [40–42, 62]. It was also shown that the protein complex guiding target inhibition (named RISC) may be loaded with one or the other sequence of the miRNA duplex [63, 64].
We found two conserved loci (vvi-miR159c, vvi-miR169w) and one Vitis-specific locus (vvi-miRNA3639) producing additional small RNA duplexes besides the annotated ones (Additional file 12). All the three alternative duplexes identified show a two-nucleotide 3’-end overhang, typical of RNase III products and the vvi-miR159c and vvi-miR169w are arranged in phase [40]. The observation that multiple unique small-RNA reads could be generated from the same miRNA precursor has already been investigated by other authors [34, 55, 65, 66]. Much evidence suggests that these molecules, different from the canonical mature miRNAs, widely exist in plants and some are actually authentic miRNAs, with predicted targets.
Fluctuations in miRNA abundance define organ identity
We established a miRNA expression atlas in the grapevine cultivar Corvina, including 25 different samples, and 354 miRNA sequences. This atlas, because of its dimension, represents a crucial step in the development of future targeted functional studies.
We analyzed the distribution of known and novel miRNAs and their relative abundance, in several plant structures such as berries, inflorescences, tendrils, buds, carpels, stamens, leaf, stem and rachis.
Considering the normalized miRNA abundances, we observed that known and conserved miRNAs are, on average, more abundant (average expression 15,000 TP5M) than novel grapevine-specific miRNAs (average expression around 150 TP5M), confirming that recently evolved molecules are less expressed and generally more tissue-specific (Fig. 6) [30, 34, 67–69]. Moreover, the analysis of the top-most expressed miRNAs suggests that miRNAs expression trend is characterized by the presence of few abundant miRNAs (more than 1000 TP5M) (Additional file 11), while the majority of the miRNAs accumulates at a lower level in the different samples.
We then compared the distribution of the miRNAs across our samples: the set of miRNAs expressed appears highly variable: only 38 miRNAs are expressed in all our samples and few miRNAs are highly organ specific miRNAs (for example only three miRNAs are specific to the stem), with the exception of stamen, where 22 miRNAs appear to be specific. Wang and collaborators [30], similarly, identified in grapevine a small number of tissue-specific miRNAs, but a large number of miRNAs expressed in all their six samples. This result is not in contrast with our findings, if we consider the higher number of samples used in the present work. Indeed, if we consider only the various developmental stages of a given organ, we identify a high proportion (more than 40%) of expressed miRNAs as shared among samples. This suggests that these miRNAs are needed for organ-identity and functionality, and that less miRNAs are involved in a specific developmental phase. Rachis has a different behaviour, since only 30% of miRNAs are shared among all the five stages considered, while many molecules are stage-specific (i.e. 20 miRNAs expressed only in Rachis at post fruit set, when compared to all other rachis samples) or shared by three or four stages (Fig. 5). This would seem to indicate that different developing stages of rachis are characterized by a heterogeneous miRNA population that drives a well-defined gene regulatory network.
Hierarchical clustering analyses, together with PCA and correlation matrix, sustain the evidences that miRNAs influence organ identity, highlighting the similarities among grapevine samples derived from the same plant structure. The correlation matrix (Additional file 12) together with the dendrogram (Fig. 7A) and the PCA (Fig. 7B) show that stamen and carpel are distant from all other samples, and moderately related to each other. Data from all our analyses suggest the presence of two major clusters: one including all the berries and their rachis, and the second comprising inflorescences, buds and the green vegetative tissues. Leaf is always distant from the rest of the considered samples, closer to berries than to other green tissues.
These analyses suggest that fluctuations in the levels of the miRNAs in different samples, without high organ specificity, may have a crucial functional role, in defining organ identity and developmental programming.
This observation differs from what described in Fasoli et al. [4], who performed a transcriptomics analysis on the same samples we used, showing that grapevine tissues undergo a transcriptional reprogramming during maturation. In their work, grapevine tissues were mainly clustered based on the maturity stage rather than organ identity, with green berries more similar to young leaves and tendrils than to ripe berries. This means that miRNAs may act differently from other protein coding genes, establishing a regulatory layer that strictly influences organ identity.
miRNA involved in berry and inflorescence development
The regulation of grapevine flowering is still poorly characterized [70], and our results highlight some key regulatory steps driven by miRNAs. Similarly, our studies on berry development enrich present knowledge on miRNA involvement in fruit (berry) maturation, only partially disclosed in tomato [71, 72].
In inflorescence, a well recognized role is played by miR156-miR172 regulation: miR156 decreases during inflorescence development while miR172 increases (Figs. 9A and 9B). This is because SPL, the target of miR156, is an elicitor of miR172 promoter region which, in turn, targets APETALA2. This regulatory unit is fundamental for vegetative to reproductive changes and for the subsequent flowering onset and flower organ identity [49, 73, 74]. Our data also suggest a role of miRNAs in the hormonal regulation of floral development: in particular for auxin and ethylene. Auxin-regulating miRNAs are clearly modulated in our data: miR390, targeting ARF via ta-siRNA production, and miR393, targeting auxin receptors are both up-regulated (Fig. 8B) at late inflorescence development (Flower_FB and Flower_F), with open flowers, thus suggesting a role for this hormone in the first stages of inflorescence development. A previous work in the hybrid Summer Black (V. vinifera x V. labrusca) showed a down-regulation of miR390 and miR393 in open flowers, compared to young inflorescences [27]. This difference could be an indication of a diverse mechanism governing auxin signalling during inflorescence development in the two Vitis species. V. labrusca and V. vinifera were already shown to have a different auxin accumulation pattern, in berry development as well [75]. Among the stamen specific miRNAs, two miRNAs (grape-m5380 and grape-m7774) were predicted to target five ACC oxidases, key enzymes in ethylene biosynthesis, suggesting a role for these miRNAs in hormone regulation during anther and pollen development. In grapevine, ethylene production peaks at anthesis before berry fruit set (phenological stage EL 23 – EL 26), inducing fruitlet abscission [76]. Subsequently, its level rises until pre-veraison stage, and declines at maturity [77]. Ethylene treatment of berries at pre-veraison induces an over expression of numerous grapevine-specific miRNAs [30]. Although these miRNAs were not detected in our libraries, this evidence supports the existence of ethylene responsive miRNAs. The role of ethylene in anther development has not been extensively studied [78, 79]. Recently it was shown that ACC oxidase gene is more expressed in female flowers of wild Vitis than in hermaphrodite or male flowers, corroborating our hypothesis of an active role of ethylene in female structure development [80]. Interestingly, grape-m7774 maps in a chromosomal region where four out of its targets also map. A total of 19 genes coding for ACC oxidase map in this region (Fig. 4) suggesting that grape-m7774 could have arisen from a tandem duplication of a target gene, followed by genetic drift, leaving only a short region of complementarity for the gene target, a mechanism suggested for miRNA evolution [81]. Among the newly discovered miRNAs, we found that grape-m9269 might be potentially involved in pollen growth, as it was predicted to target a gene coding for a Phosphatidylinositol-4-phosphate 5-kinase [52, 53], responsible for the synthesis of the minor membrane phospholipid phosphatidyl inositol - 4,5-bisphosphate. This enzyme is involved in the modification of the plasma membrane during pollen tube elongation. In agreement with this putative role, grape-m9269 is expressed in open flowers and flowers at the beginning of blooming (Flower_F and Flower_FB), but is absent in stamens and carpels. This observation suggests a role in confining the target gene expression in the organs where the pollen grains grow and develop the pollen tubes.
Considering berry development, our atlas provides interesting hints on the maturation of a non-climacteric fruit, in which the regulatory role played by miRNAs has been scarcely investigated. Some of the most conserved miRNAs reveal intriguing profiles: among others, miR156 and miR164 seem to be involved in berry maturation, in an ethylene independent manner. SPL genes, targeted by vvi-miR156, decrease during berry ripening in grapevine [82] and, accordingly, we showed that vvi-miR156 is more expressed in the last stage of berry maturation. In tomato, an epimutation in an SPL gene is responsible for Cnr mutants, which result in colorless fruits with loss of cell-to-cell adhesion [82]. SPL genes are upstream of ethylene synthesis in tomato, activating maturation through alternative programs [83]. Furthermore, in Arabidopsis, an over expression of miR156 determines a reduced SPL genes activity and, by a subsequent regulation of several genes, lead to an accumulation of anthocyanins [84], likely connected to DFR gene activation [85], a flavonoid biosynthetic gene. These data reinforce the idea vvi-miR156 may be responsible for inducing maturation programs in grapevine berries, in an ethylene independent manner, through SPL genes regulatory networks and anthocyanins accumulation, secondary metabolites accumulating in mature berries of red grapevine varieties.
vvi-miR164 is also modulated during berry development (125.5 TP5M in berry at fruit set, then declines below the threshold of expression), confirming previous observations [24, 86] that show its gradual decrease during ripening. vvi-miR164 target gene is VvNAC33 [28, 86], reported to increase during ripening [87]. Interestingly, its tomato homolog LeNOR exerts a direct role in berry ripening progression, being upstream of ethylene regulatory signals such as CNR [88, 89]
Considering the particular interest of secondary metabolites during berry maturation, and the large variety of phenylpropanoid compounds present in grapevine, two miRNAs caught our attention because their abundance patter may indicate a connection of miRNAs function to secondary metabolism. Vvi-miR395, whose abundance decreases from pre-veraison to maturity, as already reported in [25], down-regulates a class 2-sulphate transporter and it has been usually involved in sulphur uptake and assimilation, or in nutrient starvation response [90–92]. Class 3 sulphate transporters were shown to be down-regulated during late berry maturation, reinforcing the role of sulphate metabolism in berry ripening [93]. The link between sulphur depletion and flavonoid and stilbene biosynthetic pathways was studied in grapevine by Tavares et al. [94] in both photosynthetic and non photosynthetic tissues. In this respect, it is plausible that miR395, by regulating sulphate transporter, may be implicated in the activation of phenylpropanoid pathways.
The grapevine-specific miRNA grape-m6905, whose expression peaks in carpels and in berries at fruit set, being down-regulated at the onset of ripening, was predicted, instead, to target seven genes (Additional file 8) whose amino acid sequences share an identity ≥70% and are annotated as bifunctional dihydroflavonol 4-reductase flavanone 4-reductase-like (DFR-like) and cinnamoylCoA reductase-like proteins (CCR). DFR is a structural gene of the flavonoid biosynthesis pathway, specifically involved in anthocyanin and proanthocyanins biosynthesis [95], while CCR (GO:0006694; GO:0008207) might be involved in steroid and phenylpropanoid biosynthesis. Brassinosteroids (BR) are important regulators of ripening onset in grapevine [75], and induce the expression of the anthocyanin biosynthesis genes, such as DFR, when exogenously applied to berry skins [96]. BR endogenous level during berry ripening in grapevine accumulates in flower and early berry development (2 weeks after flowering, coincident with our berry at fruit set), and at late maturation stage [75]. Accordingly, grapevine genes involved in BR synthesis are up-regulated during berry development [75]. A reduction of grape- m6905 abundance right at the onset of ripening could coincide with an activation of BR synthesis and consecutively with berry maturation programs.
Our results on miRNA involvement in berry and inflorescence development highlight the importance of studying species-specific and conserved miRNAs in the context of specific metabolic pathways, such as berry maturation and secondary metabolism, and in specific organs, such as stamen and carpel, to uncover new roles for miRNAs, not limited to the conventional involvement in plant growth and development regulation.
Conclusions
In the present study, we applied next-generation sequencing technology to study miRNA involvement in grapevine development. Our small RNA libraries, covering a large set of plant structures at different developing stages, allowed us to identify not only110 known miRNAs but also 175 novel miRNA candidates, considerably extending the number of miRNAs described in grapevine.
Our work assesses the spatio-temporal distribution of miRNAs, defining the role of miRNAs in sculpturing organ identity and fine tuning developmental programs. Combining target prediction and expression profile, we shed light on the functional role of some miRNAs adding insight on inflorescence and fruit development, reinforcing, among others, the role of the miR156/miR172 regulatory circuit in both inflorescence and berry development and suggesting a role of miRNAs in regulating hormonal-driven fruit maturation and inflorescence development. For example, stamen specific miRNAs are putatively involved in regulating ethylene production, stimulating stamen development.
Our atlas aims at becoming the reference for the future development of targeted functional studies, a first indispensable step towards the definition of miRNA involvement in grapevine development.
Methods
Plant material
All samples of Vitis vinifera L. cv Corvina were collected from a 7-year-old vineyard in Montorio, during 2008/2009 growing seasons.
Thirty five grapevine samples (listed in Table 1) from 13 different plant structures at different developmental stages were collected, sampling two biological replicates, except for seeds. All samples used in this study are those used for the transcriptomics atlas [4], from which RNA was isolated. Developmental stages were recorded according to the modified Eichorn and Lorenz system [97, 98] (for a detailed description of each sample see Additional file 13 and Fasoli et al. [4].
Leaves and roots at juvenile stage were collected from Vitis vinifera clone PN40024 grown in vitro on half strength Murashige and Skoog [99] MS medium with 15 g/l sucrose, at 25 °C with a 16 h light cycle.
RNA extraction and library preparation
The small RNA fraction was isolated by two different methods, depending on tissue types. Specifically, small RNAs from leaf, stem, flower, tendril, anther, and carpel, were extracted with miR Premier™ MicroRNA Isolation Kit (Sigma-Aldrich®), following the manufacturer’s protocol. Small RNAs from bud, berry, rachis were first extracted using Plant RNA Isolation Reagent (PRIR – Life Technologies™) and followed by the miR Premier™ MicroRNA Isolation Kit (Sigma-Aldrich®). A total of 100 mg of ground tissue was used as input material. Small RNA quality and quantity were evaluated with a Nanodrop 1000 spectrometer (Thermoscientific). RNA integrity was determined on an Agilent 2100 Bioanalyzer using a small RNA chip (Agilent Technologies) according to the manufacturer’s instructions.
Small RNA libraries were constructed as previously described [25] following the TruSeq Small RNA Sample Preparation guide (Illumina Inc.). Seventy small RNA libraries were constructed starting from 50 ng of small RNA. Bar-coded libraries were amplified with 15 cycles PCR, prior to 6% PAGE gel purification. Corvina derived libraries were grouped in pools containing 6/7 libraries each and sequenced on an Illumina Hiseq2000 (Illumina Inc.). Small RNA libraries from PN40024 were constructed following the Small RNA Sample Prep Kit (Illumina Inc.), starting from 2 μg of small RNAs, and sequenced independently on a GAIIx Illumina Sequencer (Illumina Inc.). Library sequencing was performed by IGA Technology Services (Udine, Italy).
The sequencing data was submitted to GEO – NCBI under the accession number GSE59802.
Computational analysis of sequencing data
Raw reads were trimmed by removing adaptor sequences and selecting reads between 18 and 34 nt long, which were then mapped to the reference Vitis vinifera L. genomic sequence (PN40024 [1] using Bowtie [100]. Perfect matches were retained, excluding those matching rRNAs, tRNAs, snRNAs and snoRNAs.
The abundance of each sequence was normalized to overcome sequencing bias. Data were normalized using the linear count scaling method [101] calculated as:
Where n_base is 5,000,000 and TP5M is Transcript Per 5 Million.
Identification of annotated and novel vvi-miRNA
Annotated vvi-miRNAs (known vvi-miRNAs) were characterized in the 68 small RNA Corvina derived libraries, whereas novel vvi-miRNAs were identified using the whole set of Corvina and PN40024 libraries, by independently applying the miRNA identification pipeline. The analysis was performed as described by Jeong and colleagues [33] and Zhai and co-authors [34].
Finally, annotated vvi-miRNAs identified by the pipeline were passed through a careful manual inspection, evaluating their precursors and retrieving the most abundant isoform within the reads mapping on each precursor.
To discover novel vvi-miRNA candidates, we retained only those for which the most abundant sequence was 20, 21 or 22 nt long. All novel vvi-miRNA candidates were compared against miRBase version 20 to identify high-similarity homologs. Novel candidates were manually evaluated for precursor secondary structures, using the plant version of the UEA sRNA hairpin folding and annotation tool, with default settings [43]. For both annotated and novel miRNAs, complementary 3p/5p were retrieved when identified by the pipeline or when it was possible to recognize them according to the miRBase annotation.
miRNA accumulation analysis
We defined a miRNA as “expressed” only when the values of both biological replicates were greater than or equal to the threshold set at 10TP5M. Any miRNA below this threshold was considered as “not expressed”. We defined a miRNA as “organ-specific” when it was present only in a given organ, in at least one of the developmental stages of that given organ and its abundance exceeded the threshold of 10TP5M. The final miRNA expression counts were obtained by calculating the average of the two normalized biological replicates.
Target prediction and analysis
miRNA targets were identified using TargetFinder, release 1.6 (www.carringtonlab.org [44]). miRNAs were matched against the grape mRNA and long non-coding RNA sequences version 2.1 (V2.1_mrna.fa downloaded from http://genomes.cribi.unipd.it/grape/). Analyses were conducted setting a score cut off of four but results show targets with a score ≤ 3, to reduce false positives. psRNATarget [47] was used, with a score cut off 4.0, UPE:50.9 to check previous predictions.
We computed Pearson correlation values among all considered miRNA:target pairs, using the normalization procedure indicated in the following paragraph of the "Methods" section. To obtain a proper comparison, we also log10 transformed the values of the available expression data coming from [4], considering all the 24 common samples shared between the two datasets. A p-value cutoff was set to 0.05 and we selected all those pairs showing a negative correlation.
Correlation and PCA analyses
Normalized reads (NR) of all miRNAs detected and predicted in this study were normalized for correlation analysis using the log10 (1 + NR) of expression value (normalized reads) profiles. The log10 (1 + NR) normalization was chosen first because of the very large range of expression values which produced a log-unimodal distribution and significant biases in the un-normalized correlation analysis. Second, we added a unity to the expression value due to the presence of zero entries. With this last choice, a value of zero still corresponds to zero of the log10 (1 + NR) function, thus making consistent the comparisons between profiles. As a distance metric we used the usual “one minus Pearson correlation” to compare the values of miRNA in 68 samples by taking the average of the log-normalized NR reads. The log-normalized values were also used to evaluate the reproducibility of our replicates (Additional file 14). A Principal Component Analysis (PCA) has been performed on the same dataset.
Real-Time PCR on mature miRNAs
Total RNA was extracted from two biological replicates of inflorescence and berries samples, using the Spectrum™ Plant Total RNA Kit (Sigma-Aldrich®), and subsequently DNase treated using DNA-Free™ Kit (Life Technologies™). RNA quality was checked using a spectrophotometer (DU640 Beckman) and its integrity was determined on an Agilent 2100 Bioanalyzer using a RNA 6000 Nano Kit (Agilent Technologies). Treated total RNA (200 ng) was subjected to pulsed reverse transcription following Varkonyi-Gasic [102], with minor modifications and using Superscript® III (Life Technologies™) and stem-loop specific primers. Absence of genomic DNA was verified by performing a control PCR on the Actin gene (VIT_204s0044g00580), whose primers span an intron, checking amplicon length.
RT-qPCR was performed using the SYBR Green PCR Master Mix (Life Technologies™), with reaction volumes of 25 μl, three technical replicates and running each plate on the 7300 Real-Time PCR System (Life Technologies™). Eight different miRNAs were tested using a poly-ubiquitin transcript (VIT_219s0177g00040, [103] and a snRNA U6 transcript (u6wg_chr6_15577690_15577792_+) as internal standards. Primers are listed in Additional file 15. The relative quantification of each miRNA tested was calculated from Ct value, using the 2-ΔΔCt method.
Availability of supporting data
The sequence data supporting the results of this article are available in the GEO – NCBI data libraries under accession number GSE59802. Additionally, a publicly available database was set up at the following website https://mpss.udel.edu/dbs/index.php?SITE=grape_sRNA_atlas, hosting our small RNA libraries.
Acknowledgements
This work was supported by the Doctoral School in Life Sciences of Scuola Superiore Sant’Anna and by Italian Ministry of Agricultural, Food and Forestry Policies (MIPAF): Vigneto and RINGO projects: [grant numbers: D.M. 11275/7303/2011; D.M.27724/7303/11]. Work in the Meyers lab is supported by funding from the US National Science Foundation. We wish to thank Mayumi Nakano for assistance with the websites of small RNA databases.
Abbreviations
- ACC ox
Aminocyclopropane-1-carboxylate oxidase homolog
- AP2
APETALA 2 transcription factor
- ARF
Auxin Response Factor
- BR
Brassinosteroids
- CCR
Cinnamoyl CoA reductase-like proteins
- CNR
Colorless non-ripening mutation
- DCL
Dicer-Like protein
- DFR
Bifunctional dihydroflavonol 4-reductase flavanone 4-reductase
- grape-mXXXX
Novel grapevine microRNA candidate
- GRAS
Transcription factor of the family Gibberellin-Insensitive, Repressor of ga1-3, Scarecrow
- GRF
Growth Regulating Factor
- isomiR
Sequences that have variations with respect to the reference miRNA sequence
- LeNOR
Lycopersicon esculentum non-ripening
- miRNA*
microRNA star, the complementary strands of functional mature miRNA
- MS
Murashige and Skoog medium
- MYB
Transcription factor containing a Myb DNA binding domain
- NR
Normalized reads
- PCA
Principal Component analysis
- PRIR
Plant RNA Isolation Reagent
- RC-miRNAs
Reverse complementary microRNAs
- RISC
RNA-induced silencing complex
- RT-qPCR
Quantitative real-time polymerase chain reaction
- snoRNA
small nucleolar RNA
- snRNA
small nuclear RNA
- SPL
Squamosa Promoter binding Like protein
- TAS3
Trans-Acting siRNA locus 3
- TCP
Transcription factor of the family Teosinte branched 1 – Cycloidea- PCF
- TMV
Tobacco Mosaic Virus
- TP5M
Tags per 5 million
- vvi-miRC
Vitis vinifera miRNA candidate
- vvi-miRNAs
Vitis vinifera microRNAs
- VvNAC
Vitis vinifera NAM ATAF1/2 and CUC transcription factor
Additional files
Small RNA Libraries Sequencing Statistics. A pool of 8 barcoded libraries were sequenced in a single channel, except for libraries constructed with tissues of PN40024 clone which were sequenced in a separate and individual channel. The description of library codes is indicated in Table 1.
Known vvi-miRNAs identified in Corvina-derived libraries. List of reliable vvi-miRNAs from known miRNA precursors identified in Corvina-derived libraries.
Stem-loop structure of two miRNA loci generating more than one duplex. Dot-bracket notation representing RNA stem-loop structure of some examples of miRNA loci, generating more than one distinct duplex from the same precursor. For each precursor MIR locus and mapping position (chromosome, strand, coordinates) are presented. Below the dot-bracket notation, distinct miRNA duplexes are aligned and the sum of the abundance (TP5M) of the reads is shown in parenthesis. The most abundant duplex is highlighted.
Novel vvi-miRNAs identified in Corvina-derived libraries. List of novel Vitis vinifera miRNAs identified in small RNA libraries of Corvina-derived tissues. A temporary name has been given to each miRNA sequence either using a sequential numbering associated with the abbreviation mirC (miRNA Candidate) for those similar to other known plant miRNAs or using “grape-m” with a random numbering for those completely new candidates.
Novel vvi-miRNAs identified in PN40024-derived libraries. List of novel Vitis vinifera miRNAs identified in small RNA libraries from tissues of the PN40024 clone. A temporary name has been given to each miRNA sequence either using a sequential numbering associated with the abbreviation miRC (miRNA Candidate) for those similar to other known plant miRNAs or using “grape-m” with a random numbering for those completely new candidates.
Secondary structure of novel miRNAs identified in Corvina-derived libraries. List of all RNA secondary structures of novel miRNA precursors, predicted using the RNA folding tool of the UEA sRNA toolkit – Plant version [43]. The sequence of the mature miRNA is highlighted in red and the complementary sequence (miRNA*) is highlighted in pink when present.
Secondary structure of novel miRNAs identified in PN40024-derived libraries. List of all RNA secondary structures of novel miRNA precursors, predicted using the RNA folding tool of the UEA sRNA toolkit – Plant version [43]. The sequence of the mature miRNA is highlighted in red and the complementary sequence (miRNA*) is highlighted in pink when present.
Putative targets of known and novel vvi-miRNAs. List of putative targets as identified by TARGET FINDER. Only targets with score ≥ 3 are listed. GRAPE_IGGP_v1 12X assembly of Vitis vinifera genome and the v2 annotation were used.
Number of putative miRNA targets. Number of putative miRNA target within the (A) PN40024 and (B) Corvina data sets, identified using the TargetFinder (release 1.6). Novel and known refer to the different classes of miRNAs.
miRNA - target correlations. From a list of 933 predicted miRNA - target pairs we could find 815 pairs for which the target gene expression values are available from a public dataset [4]. The table reports the Pearson correlations of the pairs for which the P value is less than 0.05. The P value is computed for testing the hypothesis of no correlation against the alternative that there is a non-zero correlation. The annotations reported are the same as in Additional file 8.
Top 20 expressed miRNAs per tissue/developmental stage. Top 20 expressed miRNAs in each tissue/developmental stage studied. Reads refers to the average of TP5M of both replicates.
Correlation matrix of the 25 grapevine samples used to define the miRNA atlas. One minus Pearson correlation was used as a metric distance. Main clusters identified are highlighted with a red box.
Additional plant material details.
Pearson correlation between replicates. Values of Pearson correlation between replicates using log2 transformed data.
Primers used for Real Time PCR (RT-qPCR).
Footnotes
Jayakumar Belli Kullan and Daniela Lopes Paim Pinto contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MP and MEP conceived the study, MEP and EM participated in its design and coordination. JBK , EM performed all the experiments. DLPP EB performed in silico analysis for miRNA and target prediction; BCM assisted in silico analysis set up; LF performed correlation, PCA and clustering analyses; MF SZ GBT collected all the materials; EM, DLPP and JBK wrote the paper. MEP, MF, SZ, EB, GBT, MP, BCM, LF critically revised the manuscript and all authors have read and given approval of the version to be published.
Contributor Information
Jayakumar Belli Kullan, Email: kullan@sun.ac.za.
Daniela Lopes Paim Pinto, Email: d.lopespaimpinto@sssup.it.
Edoardo Bertolini, Email: e.bertolini@sssup.it.
Marianna Fasoli, Email: marianna.fasoli@univr.it.
Sara Zenoni, Email: sara.zenoni@univr.it.
Giovanni Battista Tornielli, Email: giovannibattista.tornielli@univr.it.
Mario Pezzotti, Email: mario.pezzotti@univr.it.
Blake C. Meyers, Email: meyers@dbi.udel.edu
Lorenzo Farina, Email: farina@dis.uniroma1.it.
Mario Enrico Pè, Email: marioenrico.pe@sssup.it.
Erica Mica, Email: erica.mica@entecra.it.
References
- 1.Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–7. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 2.Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, et al. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010;152:1787–95. doi: 10.1104/pp.109.149716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamboni A, Di Carli M, Guzzo F, Stocchero M, Zenoni S, Ferrarini A, et al. Identification of putative stage-specific grapevine berry biomarkers and omics data integration into networks. Plant Physiol. 2010;154:1439–59. doi: 10.1104/pp.110.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fasoli M, Dal Santo S, Zenoni S, Tornielli GB, Farina L, Zamboni A, et al. The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. Plant Cell. 2012;3489–3505. [DOI] [PMC free article] [PubMed]
- 5.Tornielli G, Zamboni A, Zenoni S, Delledonne M, Pezzotti M: Transcriptomics and metabolomics for the analysis of grape berry development. In: The Biochemistry of the Grape Berry. Edited by Gerós H, Chaves MM, Delrot S. Sharjah United Arab Emirates: Bentham Science Publisher 2012, 218–240
- 6.Dal Santo S, Tornielli GB, Zenoni S, Fasoli M, Farina L, Anesi A, et al. The plasticity of the grapevine berry transcriptome. Genome Biol. 2013;14:r54. doi: 10.1186/gb-2013-14-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–85. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 8.Mallory AC, Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nat Genet. 2006;38(Suppl):S31–6. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- 9.Simon SA, Zhai J, Zeng J, Meyers BC. The cornucopia of small RNAs in plant genomes. Rice. 2008;1:52–62. doi: 10.1007/s12284-008-9008-5. [DOI] [Google Scholar]
- 10.Chen X. Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol. 2009;25:21–44. doi: 10.1146/annurev.cellbio.042308.113417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz-Ferrer V, Voinnet O. Roles of plant small RNAs in biotic stress responses. Annu Rev Plant Biol. 2009;60:485–510. doi: 10.1146/annurev.arplant.043008.092111. [DOI] [PubMed] [Google Scholar]
- 12.Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301:336–8. doi: 10.1126/science.1085242. [DOI] [PubMed] [Google Scholar]
- 13.Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004;303:2022–5. doi: 10.1126/science.1088060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aukerman MJ, Sakai H. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2 -like target genes. Plant Cell. 2003;15:2730–41. doi: 10.1105/tpc.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Han J, Liu C, Kibet K, Kayesh E, Shangguan L, et al. Identification of microRNAs from Amur grape (Vitis amurensis Rupr.) by deep sequencing and analysis of microRNA variations with bioinformatics. BMC Genomics. 2012;13:122. doi: 10.1186/1471-2164-13-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Floyd SK, Bowman JL. Gene regulation: ancient microRNA target sequences in plants. Nature. 2004;428:485–6. doi: 10.1038/428485a. [DOI] [PubMed] [Google Scholar]
- 17.Achard P, Herr A, Baulcombe DC, Harberd NP. Modulation of floral development by a gibberellin-regulated microRNA. Development. 2004;131:3357–65. doi: 10.1242/dev.01206. [DOI] [PubMed] [Google Scholar]
- 18.Lauter N, Kampani A, Carlson S, Goebel M, Moose SP. microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc Natl Acad Sci U S A. 2005;102:9412–7. doi: 10.1073/pnas.0503927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trindade I, Capitão C, Dalmay T, Fevereiro MP, Dos Santos DM. miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta. 2010;231:705–16. doi: 10.1007/s00425-009-1078-0. [DOI] [PubMed] [Google Scholar]
- 20.Khraiwesh B, Zhu J-K, Zhu J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta. 1819;2012:137–48. doi: 10.1016/j.bbagrm.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17:196–203. doi: 10.1016/j.tplants.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Van Wolfswinkel JC, Ketting RF. The role of small non-coding RNAs in genome stability and chromatin organization. J Cell Sci. 2010;123:1825–39. doi: 10.1242/jcs.061713. [DOI] [PubMed] [Google Scholar]
- 23.Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet. 2013;14:100–12. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carra A, Mica E, Gambino G, Pindo M, Moser C, Pè ME, et al. Cloning and characterization of small non-coding RNAs from grape. Plant J. 2009;59:750–63. doi: 10.1111/j.1365-313X.2009.03906.x. [DOI] [PubMed] [Google Scholar]
- 25.Mica E, Piccolo V, Delledonne M, Ferrarini A, Pezzotti M, Casati C, et al. Correction: High throughput approaches reveal splicing of primary microRNA transcripts and tissue specific expression of mature microRNAs in Vitis vinifera. BMC Genomics. 2010;11:109. doi: 10.1186/1471-2164-11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, et al. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62:960–76. doi: 10.1111/j.0960-7412.2010.04208.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Wang X, Kibet NK, Song C, Zhang C, Li X, et al. Deep sequencing of grapevine flower and berry short RNA library for discovery of novel microRNAs and validation of precise sequences of grapevine microRNAs deposited in miRBase. Physiol Plant. 2011;143:64–81. doi: 10.1111/j.1399-3054.2011.01481.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang C, Han J, Korir NK, Wang X, Liu H, Li X, et al. Characterization of target mRNAs for grapevine microRNAs with an integrated strategy of modified RLM-RACE, newly developed PPM-RACE and qPCRs. J Plant Physiol. 2013;170:943–57. doi: 10.1016/j.jplph.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Han J, Fang J, Wang C, Yin Y, Sun X, Leng X, et al. Grapevine microRNAs responsive to exogenous gibberellin. BMC Genomics. 2014;15:111. doi: 10.1186/1471-2164-15-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Leng X, Zhang Y, Kayesh E, Zhang Y, Sun X, et al. Transcriptome-wide analysis of dynamic variations in regulation modes of grapevine microRNAs on their target genes during grapevine development. Plant Mol Biol. 2014;84:269–85. doi: 10.1007/s11103-013-0132-2. [DOI] [PubMed] [Google Scholar]
- 31.Alabi OJ, Zheng Y, Jagadeeswaran G, Sunkar R, Naidu RA. High-throughput sequence analysis of small RNAs in grapevine (Vitis vinifera L.) affected by grapevine leafroll disease. Mol Plant Pathol. 2012;13:1060–76. doi: 10.1111/j.1364-3703.2012.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montes RAC, de Fátima R-CF, De Paoli E, Accerbi M, Rymarquis LA, Mahalingam G, et al. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat Commun. 2014;5:3722. doi: 10.1038/ncomms4722. [DOI] [PubMed] [Google Scholar]
- 33.Zhai J, Jeong DH, de Paoli E, Park S, Rosen BD, Li Y, et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011;25:2540–53. doi: 10.1101/gad.177527.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeong D-H, Park S, Zhai J, Gurazada SGR, De Paoli E, Meyers BC, et al. Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell. 2011;23:4185–207. doi: 10.1105/tpc.111.089045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu F, Liu Q, Chen L, Kuang J, Walk T, Wang J, et al. Genome-wide identification of soybean microRNAs and their targets reveals their organ-specificity and responses to phosphate starvation. BMC Genomics. 2013;14:66. doi: 10.1186/1471-2164-14-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeong DH, Schmidt S, Rymarquis L, Park S, Ganssmann M, German M, et al. Parallel analysis of RNA ends enhances global investigation of microRNAs and target RNAs of Brachypodium distachyon. Genome Biol. 2013;14:R145. doi: 10.1186/gb-2013-14-12-r145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arikit S, Xia R, Kakrana A, Kun Huang K, Zhai J, Zhe Z, et al. An atlas of soybean small RNAs identifies phased siRNAs from hundreds of coding genes. Plant Cell. 2014;26:4584–601. doi: 10.1105/tpc.114.131847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandey R, Joshi G, Bhardwaj AR, Agarwal M, Katiyar-Agarwal SA. Comprehensive genome-wide study on tissue-specific and abiotic stress-specific miRNAs in triticum aestivum. PLoS One. 2014;9:e95800. doi: 10.1371/journal.pone.0095800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu H, Yu D, Liu H. Bioinformatics analysis of small RNAs in pima (gossypium barbadense L.) PLoS One. 2015;10:e0116826. doi: 10.1371/journal.pone.0116826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong D-H, Thatcher SR, Brown RSH, Zhai J, Park S, Rymarquis LA, et al. Comprehensive investigation of microRNAs enhanced by analysis of sequence variants, expression patterns, ARGONAUTE loading, and target cleavage. Plant Physiol. 2013;162:1225–45. doi: 10.1104/pp.113.219873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devers EA, Branscheid A, May P, Krajinski F. Stars and symbiosis: MicroRNA- and MicroRNA*-mediated transcript cleavage involved in arbuscular mycorrhizal symbiosis. Plant Physiol. 2011;156:1990–2010. doi: 10.1104/pp.111.172627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marco A, MacPherson JI, Ronshaugen M, Griffiths-Jones S. MicroRNAs from the same precursor have different targeting properties. Silence. 2012;3:8. doi: 10.1186/1758-907X-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moxon S, Schwach F, Dalmay T, MacLean D, Studholme DJ, Moulton V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008;24:2252–3. doi: 10.1093/bioinformatics/btn428. [DOI] [PubMed] [Google Scholar]
- 44.Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, et al. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20:3186–90. doi: 10.1105/tpc.108.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNAGenes. PLoS One. 2007;2:e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srivastava PK, Moturu TR, Pandey P, Baldwin IT, Pandey SP. A comparison of performance of plant miRNA target prediction tools and the characterization of features for genome-wide target prediction. BMC Genomics. 2014;15:348. doi: 10.1186/1471-2164-15-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155–9. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chuck G, Cigan AM, Saeteurn K, Hake S. The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat Genet. 2007;39:544–9. doi: 10.1038/ng2001. [DOI] [PubMed] [Google Scholar]
- 49.Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009;138:750–9. doi: 10.1016/j.cell.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu QH, Helliwell CA. Regulation of flowering time and floral patterning by miR172. J Exp Bot. 2011;62:487–95. doi: 10.1093/jxb/erq295. [DOI] [PubMed] [Google Scholar]
- 51.Wang T, Pan H, Wang J, Yang W, Cheng T, Zhang Q. Identification and profiling of novel and conserved microRNAs during the flower opening process in Prunus mume via deep sequencing. Mol Genet Genomics. 2014;289:169–83. doi: 10.1007/s00438-013-0800-6. [DOI] [PubMed] [Google Scholar]
- 52.Ischebeck T, Stenzel I, Heilmann I. Type B phosphatidylinositol-4-phosphate 5-kinases mediate Arabidopsis and Nicotiana tabacum pollen tube growth by regulating apical pectin secretion. Plant Cell. 2008;20:3312–30. doi: 10.1105/tpc.108.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stenzel I, Ischebeck T, Quint M, Heilmann I. Variable regions of PI4P 5-kinases direct PtdIns(4,5)P2 toward alternative regulatory functions in tobacco pollen tubes. Frontiers Plant Sci. 2011;2:114. doi: 10.3389/fpls.2011.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kozomara A, Griffiths-Jones S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shao C, Ma X, Xu X, Wang H, Meng Y. Genome-wide identification of reverse complementary microRNA genes in plants. PLoS One. 2012;7:e46991. doi: 10.1371/journal.pone.0046991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu J, Zhang H, Ding Y. Identification of conserved microRNAs and their targets in the model legume Lotus japonicus. J Biotechnol. 2013;164:520–4. doi: 10.1016/j.jbiotec.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 57.Bender W. MicroRNAs in the Drosophila bithorax complex. Genes Dev. 2008;22:14–9. doi: 10.1101/gad.1614208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stark A, Bushati N, Jan CH, Kheradpour P, Hodges E, Brennecke J, et al. A single Hox locus in Drosophila produces functional microRNAs from opposite DNA strands. Genes Dev. 2008;22:8–13. doi: 10.1101/gad.1613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tyler DM, Okamura K, Chung WJ, Hagen JW, Berezikov E, Hannon GJ, et al. Functionally distinct regulatory RNAs generated by bidirectional transcription and processing of microRNA loci. Genes Dev. 2008;22:26–36. doi: 10.1101/gad.1615208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 61.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Zhao H, Gao S, Wang WC, Katiyar-Agarwal S, Huang H, et al. Arabidopsis argonaute 2 regulates innate immunity via miRNA393*-mediated silencing of a Golgi-localized SNARE gene, MEMB12. Mol Cell. 2011;42:356–66. doi: 10.1016/j.molcel.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, et al. Mammalian microRNAs: Experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Griffiths-Jones S, Hui JHL, Marco A, Ronshaugen M. MicroRNA evolution by arm switching. EMBO Rep. 2011;12:172–7. doi: 10.1038/embor.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurihara Y, Watanabe Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci U S A. 2004;101:12753–8. doi: 10.1073/pnas.0403115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang W, Gao S, Zhou X, Xia J, Chellappan P, Zhou X, et al. Multiple distinct small RNAs originate from the same microRNA precursors. Genome Biol. 2010;11:R81. doi: 10.1186/gb-2010-11-8-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Voinnet O. Origin, biogenesis, and activity of plant MicroRNAs. Cell. 2009;136:669–87. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- 68.Jagadeeswaran G, Zheng Y, Sumathipala N, Jiang H, Arrese EL, Soulages JL, et al. Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics. 2010;11:52. doi: 10.1186/1471-2164-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim B, Yu H-J, Park S-G, Shin JY, Oh M, Kim N, et al. Identification and profiling of novel microRNAs in the Brassica rapa genome based on small RNA deep sequencing. BMC Plant Biol. 2012;12:218. doi: 10.1186/1471-2229-12-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carmona MJ, Chaïb J, Martínez-Zapater JM, Thomas MR. A molecular genetic perspective of reproductive development in grapevine. J Exp Bot. 2008;59:2579–96. doi: 10.1093/jxb/ern160. [DOI] [PubMed] [Google Scholar]
- 71.Karlova R, van Haarst JC, Maliepaard C, van de Geest H, Bovy AG, Lammers M, et al. Identification of microRNA targets in tomato fruit development using high-throughput sequencing and degradome analysis. J Exp Bot. 2013;64:1863–78. doi: 10.1093/jxb/ert049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao C, Ju Z, Cao D, Zhai B, Qin G, Zhu H, et al. MicroRNA profiling analysis throughout tomato fruit development and ripening reveals potential regulatory role of RIN on microRNAs accumulation. Plant Biotechnol J. 2015;13:370–82. doi: 10.1111/pbi.12297. [DOI] [PubMed] [Google Scholar]
- 73.Wang JW, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138:738–49. doi: 10.1016/j.cell.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 74.Yant L, Mathieu J, Dinh TT, Ott F, Lanz C, Wollmann H, et al. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell. 2010;22:2156–70. doi: 10.1105/tpc.110.075606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Symons GM, Davies C, Shavrukov Y, Dry IB, Reid JB, Thomas MR. Grapes on steroids. Brassinosteroids are involved in grape berry ripening. Plant Physiol. 2006;140:150–8. doi: 10.1104/pp.105.070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hilt C, Bessis R. Abscission of grapevine fruitlets in relation to ethylene biosynthesis. Vitis. 2003;42:1–3. [Google Scholar]
- 77.Chervin C, El-Kereamy A, Roustan J-P, Latché A, Lamon J, Bouzayen M. Ethylene seems required for the berry development and ripening in grape, a non-climacteric fruit. Plant Sci. 2004;167:1301–5. doi: 10.1016/j.plantsci.2004.06.026. [DOI] [Google Scholar]
- 78.Duan QH, Wang DH, Xu ZH, Bai SN. Stamen development in Arabidopsis is arrested by organ-specific overexpression of a cucumber ethylene synthesis gene CsACO2. Planta. 2008;228:537–43. doi: 10.1007/s00425-008-0756-7. [DOI] [PubMed] [Google Scholar]
- 79.Boualem A, Troadec C, Kovalski I, Sari MA, Perl-Treves R, Bendahmane A. A conserved ethylene biosynthesis enzyme leads to andromonoecy in two Cucumis species. PLoS One. 2009;4:e6144. doi: 10.1371/journal.pone.0006144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ramos MJN, Coito JL, Silva HG, Cunha J, Costa MMR, Rocheta M. Flower development and sex specification in wild grapevine. BMC Genomics. 2014;15:1095. doi: 10.1186/1471-2164-15-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allen E, Xie Z, Gustafson AM, Sung G-H, Spatafora JW, Carrington JC. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004;36:1282–90. doi: 10.1038/ng1478. [DOI] [PubMed] [Google Scholar]
- 82.Manning K, Tör M, Poole M, Hong Y, Thompson AJ, King GJ, et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet. 2006;38:948–52. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 83.Giovannoni JJ. Fruit ripening mutants yield insights into ripening control. Curr Opin Plant Biol. 2007;10:283–9. doi: 10.1016/j.pbi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 84.Gou J-Y, Felippes FF, Liu C-J, Weigel D, Wang J-W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell. 2011;23:1512–22. doi: 10.1105/tpc.111.084525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cui L-G, Shan J-X, Shi M, Gao J-P, Lin H-X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014;80:1108–17. doi: 10.1111/tpj.12712. [DOI] [PubMed] [Google Scholar]
- 86.Sun X, Korir NK, Han J, Shangguan LF, Kayesh E, Leng XP, et al. Characterization of grapevine microR164 and its target genes. Mol Biol Rep. 2012;39:9463–72. doi: 10.1007/s11033-012-1811-9. [DOI] [PubMed] [Google Scholar]
- 87.Deluc LG, Grimplet J, Wheatley MD, Tillett RL, Quilici DR, Osborne C, et al. Transcriptomic and metabolite analyses of Cabernet Sauvignon grape berry development. BMC Genomics. 2007;8:429. doi: 10.1186/1471-2164-8-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tigchelaar E, Tomes M, Kerr E, Barman R: A new fruit ripening mutant, non-ripening (nor). Rep Tomato Genet Coop 1973;23:33.
- 89.Vrebalov J, Ruezinsky D, Padmanabhan V, White R, Medrano D, Drake R, et al. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (rin) locus. Science. 2002;296:343–6. doi: 10.1126/science.1068181. [DOI] [PubMed] [Google Scholar]
- 90.Matthewman CA, Kawashima CG, Húska D, Csorba T, Dalmay T, Kopriva S. miR395 is a general component of the sulfate assimilation regulatory network in Arabidopsis. FEBS Lett. 2012;586:3242–8. doi: 10.1016/j.febslet.2012.06.044. [DOI] [PubMed] [Google Scholar]
- 91.Kawashima CG, Matthewman CA, Huang S, Lee B-R, Yoshimoto N, Koprivova A, et al. Interplay of SLIM1 and miR395 in the regulation of sulfate assimilation in Arabidopsis. Plant J. 2011;66:863–76. doi: 10.1111/j.1365-313X.2011.04547.x. [DOI] [PubMed] [Google Scholar]
- 92.Liang G, Yu D. Reciprocal regulation among miR395, APS and SULTR2;1 in Arabidopsis thaliana. Plant Signal Behav. 2010;5:1257–9. doi: 10.4161/psb.5.10.12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guillaumie S, Fouquet R, Kappel C, Camps C, Terrier N, Moncomble D, et al. Transcriptional analysis of late ripening stages of grapevine berry. BMC Plant Biol. 2011;11:165. doi: 10.1186/1471-2229-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tavares S, Vesentini D, Fernandes JC, Ferreira RB, Laureano O, Ricardo-Da-Silva JM, et al. Vitis vinifera secondary metabolism as affected by sulfate depletion: diagnosis through phenylpropanoid pathway genes and metabolites. Plant Physiol Biochem. 2013;66:118–26. doi: 10.1016/j.plaphy.2013.01.022. [DOI] [PubMed] [Google Scholar]
- 95.Honda C, Kotoda N, Wada M, Kondo S, Kobayashi S, Soejima J, et al. Anthocyanin biosynthetic genes are coordinately expressed during red coloration in apple skin. Plant Physiol Biochem. 2002;40:955–62. doi: 10.1016/S0981-9428(02)01454-7. [DOI] [Google Scholar]
- 96.Luan L, Zhang Z, Xi Z, Huo SS, Ma LN. Brassinosteroids regulate anthocyanin biosynthesis in the ripening of grape berries. South African J Enol Viticulture. 2013;34:196–203. [Google Scholar]
- 97.Eichhorn KW, Lorenz DH. Phänologische Entwicklungsstadien der Rebe. Nachrichtenbl Deut Pflanzenschutz. 1977;29:119–20. [Google Scholar]
- 98.Lorenz DH, Eichhorn KW, Blei Holder H, Klose R, Meier U, Weber E. Phänologische Entwicklungsstadien der Weinrebe (Vitis vinifera L.: ssp. vinifera) Vitic Enol Sci. 1994;49:66–70. [Google Scholar]
- 99.Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant. 1962;15:473–97. doi: 10.1111/j.1399-3054.1962.tb08052.x. [DOI] [Google Scholar]
- 100.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jeong D-H, German MA, Rymarquis LA, Thatcher SR, Green PJ. Abiotic stress-associated miRNAs: detection and functional analysis. Methods Mol Biol. 2010;592:203–30. doi: 10.1007/978-1-60327-005-2_14. [DOI] [PubMed] [Google Scholar]
- 102.Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods. 2007;3:12. doi: 10.1186/1746-4811-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marè C, Aprile A, Roncaglia E, Tocci E, Corino LG, De Bellis L, et al. Rootstock and soil induce transcriptome modulation of phenylpropanoid pathway in grape leaves. J Plant Interact. 2013;8:334–49. doi: 10.1080/17429145.2012.754958. [DOI] [Google Scholar]