

ABSTRACT
Estrogens exert their activity through estrogen receptor alpha (ERalpha) to stimulate hypertrophy and hyperplasia in the uterus. A uterine epithelial ERalpha conditional knockout mouse model (Wnt7aCre+;Esr1f/f or cKO) demonstrated that ERalpha in the epithelial cells was dispensable for an initial uterine proliferative response to 17beta-estradiol (E2) but required for subsequent uterine biological responses. This study aimed to characterize the differential gene expression patterns induced by E2 in the presence or absence of epithelial ERalpha. RNA microarray analysis revealed that approximately 20% of the genes differentially expressed at 2 h were epithelial ERalpha independent, as they were preserved in the cKO uteri. This indicates that early uterine transcripts mediated by stromal ERalpha are sufficient to promote initial proliferative responses. However, more than 90% of the differentially expressed transcripts at 24 h were not regulated in the cKO, indicating that the majority of later transcriptional regulation required epithelial ERalpha, especially those involved in mitosis. This shows that loss of regulation of these later transcripts results in blunted subsequent uterine growth after 3 days of E2 treatment. Additionally, progesterone's ability to inhibit E2-induced epithelial cell proliferation was impaired, consistent with a uterine receptivity defect that contributes to cKO infertility. These transcriptional profiles correlate with our previously observed biological responses, in which the initial proliferative response is independent of epithelial ERalpha and thus dependent on stromal ERalpha, yet epithelial ERalpha is essential for subsequent tissue responsiveness.
Keywords: epithelium, estradiol/estradiol receptor, proliferation, stroma, uterus
INTRODUCTION
Mammalian uterine tissues undergo tissue remodeling during ovarian cycles and pregnancy [1]. The ovarian steroid hormone estrogen acts through estrogen receptors (ERs) to stimulate cell proliferation, differentiation, and growth in both reproductive and nonreproductive tissues [2]. In rodents, ER alpha (ERα) is expressed primarily in female reproductive tissues, including theca cells in the ovary, oviduct, uterus, and vagina as well as the mammary gland, whereas ER beta (ERβ) is expressed primarily in the granulosa cells in the ovary [3]. ERα is expressed in all uterine cell layers, including the epithelial cell layer and the mesenchymal (stromal and myometrial) cell layer [4].
In rodent uteri, 17β-estradiol (E2) treatment rapidly increases nuclear occupancy and ERα recruitment to targeted gene promoter regions [5]. Subsequently, early-phase gene products involved in fluid uptake, hyperemia, and immune cell infiltrations are synthesized [6, 7]. Later-phase physiological responses include waves of DNA synthesis and mitosis, changes in epithelial cell morphology from single cuboidal into columnar secretory epithelial cells, and increased immune cell infiltration [8]. The subsequent increase in cell number (hyperplasia) and cell size (hypertrophy) leads to an increase in total uterine wet weight. It has been shown that E2 stimulates cell proliferation strictly in the epithelial cells of ovariectomized adult mouse uteri [9]. We have previously shown that acute dosing of E2 induces two distinct genomic profiles in the uterus, including early and late phases [10]. Female mice with a global deletion of ERα (ERαKO) lack these E2-induced early and late genomic responses, whereas ERβKO mice show similar uterine responses to wild type (WT) [10]. Our previous findings indicate that ERα, not ERβ, regulates the genomic responses that are responsible for biological actions of E2 in the rodent uterus.
Epithelial-mesenchymal interaction is known to be important not only for embryonic development but also for cellular growth and differentiation of adult mammary gland, uterus, and prostate tissues [11]. Several studies used a tissue recombination strategy to demonstrate that E2 stimulated a rapid production of paracrine mitogenic signals, such as insulin-like growth factor 1 (Igf1) or other growth factors, through ERα in the stromal cells that subsequently induced uterine epithelial cell proliferation [12–15]. Recently, we generated a conditional deletion of epithelial ERα in the mouse uterus by crossing Wnt7aCre+ mice with floxed Esr1f/f mice (referred to as conditional knockout or cKO) to observe the physiological responsiveness in uterine tissue in vivo. Our findings demonstrated that the epithelial ERα is not necessary for the epithelial cell proliferative response of the uterus after 24 h of E2 treatment [16], which is consistent with the findings from developmental tissue recombination studies [17] but also illustrates that the same developmental paracrine signaling mechanism is retained in adult tissue. Normal levels of CCAAT enhancer binding protein beta (Cebpb, a transcription factor important for uterine proliferation [18]) and Igf1 are preserved in the absence of ERα in the epithelial cells [16]. However, lacking uterine epithelial ERα leads to an aberrant apoptotic event in epithelial cells after a prolonged (three consecutive days) E2 treatment [16]. We found that baculoviral inhibitors of apoptosis repeat-containing 1 (Birc1a) is absent in cKO uteri after prolonged E2 treatment, which may contribute to an increase in epithelial cell apoptosis. Additionally, female mice lacking epithelial ERα are infertile, in part due to an implantation defect [16]. These findings suggest that ERα, specifically in the uterine epithelial cells, is essential for complete uterine biological responses and establishment of successful pregnancy.
We hypothesized that loss of epithelial ERα in the uterine epithelial cells may not play a critical role in regulating expression of early uterine transcripts (2 h) that are responsible for initial E2-mediated events (24 h) in the uterus, including mitogenesis, but may disrupt late-phase transcriptional profiles (24 h) that lead to aberrant subsequent uterine responses (72 h). To test the hypothesis, we used our epithelial ERα cKO mice and treated with vehicle or E2 for 2 and 24 h to stimulate the early and late transcripts in the uteri. To understand the molecular mechanisms of downstream signaling of epithelial ERα during early and late time points, we analyzed our microarray data set from cKO and compared it with WT uterine samples using Ingenuity Pathway Analysis. Herein, we show that epithelial ERα was not involved in E2-induced early transcription events that contribute to epithelial proliferation. However, more than 90% of gene products induced by E2 at 24 h were missing in the cKO uteri, and these genes were found to be involved in cell cycle regulation, especially mitosis. Lacking these cell cycle-regulated genes may contribute to a blunted epithelial cell proliferation during the subsequent uterine growth observed in the cKO uteri.
MATERIALS AND METHODS
Animals and Experimental Procedures
A uterine epithelial cell-specific ERα deletion cKO mouse model was generated by crossing Wnt7aCre+ mice with Esr1f/f mice [16, 19]. Females with Wnt7aCre+, Esr1f/f, and Esr1+/+ genotypes were considered as WT control littermates. Adult females (8–10 wk old) were ovariectomized and housed for 10 days to eliminate endogenous ovarian steroids prior to the study. Animals were randomly assigned into groups and treated with sesame oil (vehicle) or 0.25 μg E2 in 100 μl sesame oil and euthanized for tissue collection at 2-, 24-, and 72-h time points as previously described [16]. For the 72-h time point, mice were injected with E2 every 24 h for three consecutive days. In some experiments, the ovariectomized females were treated with a series of E2 and progesterone (P4) injection with a nidatory dose of E2 prior to sacrifice (called “E+Pe”) to mimic the hormonal profile during the implantation window, as shown previously [20]. Briefly, animals were subcutaneously injected with E2 (100 ng/mouse in 100 μl sesame oil) for two consecutive days (D1 and D2). Mice were allowed to rest on D3–D5. On D6–D8, P4 at a dose of 1 mg/mouse in 100 μl sesame oil was injected for three consecutive days. At D9, a combination of P4 (1 mg/mouse) together with E2 (50 ng/mouse) was administered 18 h prior to tissue collection on D10. One uterine horn from each mouse in all treatment groups was snap frozen and stored at −80°C for RNA extraction. The contralateral uterine horn from the 24- and 72-h and E+Pe time points was also collected in 10% buffered formalin for hematoxylin and eosin (H&E) and immunohistochemical (IHC) analysis. Animals were handled according to National Institute of Environmental Health Sciences (NIEHS) Animal Care and Use Committee guidelines and in compliance with NIEHS-approved animal protocol.
Uterine Epithelial Cell Height Measurement and Immunohistochemical Analyses
Formalin-fixed uterine samples collected at 24- and 72-h time points were sectioned and stained with H&E using standard histological protocol. Epithelial cell heights were measured in Image-Pro Plus Software, version 4.1.0.0 for Windows (Media Cybernetics, Inc.). Epithelial cell heights were measured using a “length measurement tool” by drawing lines from the basement membrane to the tip of the epithelial cells. Averages of nine points per uterine section were calculated. Fold increase of uterine epithelial cell heights from each group was calculated relative to WT vehicle-treated group. Uterine sections were also stained with ERα, Ki-67, and phosphohistone H3 and PR antibodies for IHC analyses as previously described [16, 21]. The ERβ antibody was purchased from Calbiochem (#PC168; EMD Millipore). The staining was performed on the Discovery XT Automated System (Ventana Medical Systems) with the OmniMap DAB anti-Rabbit Detection Kit according to the manufacturer's protocol (Ventana Medical Systems). The anti-ERβ was diluted at 1:500 and incubated for 1 h. HAND2 antibody (SC-9409; Santa Cruz) was diluted at 1:50 and incubated with uterine sections at 4°C overnight. All sections were dehydrated and coverslipped.
Microarray Analysis
For microarray analysis, adult WT and cKO females (8–10 wk old) were ovariectomized and treated with vehicle or 0.25 μg E2. The uterine samples were collected at 2 and 24 h after E2 administration and immediately frozen in liquid nitrogen. RNA was extracted using the RNeasy Mini Kit (Qiagen) and treated with DNaseI (Qiagen) according to the manufacturer's protocol. Gene expression analysis was conducted using Agilent Whole Mouse Genome 4 × 44 multiplex format oligo arrays (no. 014868; Agilent Technologies) as described previously [21]. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus [22] and are accessible through GEO Series accession number GSE23072 (WT samples) and GSE53812 (cKO samples). The gProcessedSignal data were obtained using Agilent Feature Extraction software (ver. 9.5), using the one-color defaults for all parameters. The Agilent Feature Extraction Software was used to perform error modeling, adjusting for additive and multiplicative noise. The resulting data were processed using Partek Genomics Suite (ver. 6.6Beta 6.11.1115). The data were log base 2-transformed and quantile normalized. ANOVA was used to detect differentially expressed genes between groups. Gene lists were generated using the following criteria: P < 0.01 and absolute value fold change ≥2. Hierarchical clustering was generated using the Partek default clustering method (shift genes to mean of zero and scale to standard deviation of one).
Verification of Microarray Results by Real-Time RT-PCR
RNA was extracted from the whole uteri using Trizol Reagent (Invitrogen, Life Technology) according to the manufacturer's protocol. Semiquantitative PCR and the analysis were carried out as previously described [16]. The expression values were calculated as fold change normalized to ribosomal protein L7 (Rpl7) expression, relative to WT vehicle. The primer sequences were designed using Primer Express (ver. 2.0; Applied Biosystems) and are listed in Supplemental Table S1 (all Supplemental Data are available online at www.biolreprod.org).
Image Analysis
Quantification of Ki-67 and phosphohistone H3 immunohistochemical analysis was determined using Definiens Tissue Studio version 3.6.1 (Definiens Inc.) and ImageJ version 1.47 (Wayne Rasband, National Institutes of Health), respectively. The amount of Ki67 signal in 1) uterine luminal epithelial cells and 2) uterine stromal cells (excluding the uterine glands) was determined by the algorithm in Definiens. This algorithm detected signals within designated regions of interest in the image and quantified the areas of stains within these regions of interest defined as marker areas. Using the accumulated data of immunohistochemical- and hematoxylin-stained areas from the regions of interest, the algorithm then calculated the percentage of marker present within the selected region(s) of interest. Phosphohistone H3-positive cells are not conducive to imaging by Definiens due to the punctate nature of positive nuclear regions. To quantify phosphohistone H3-positive cells, we used ImageJ software. Three images of positive cells from each sample were captured in the luminal and subepithelial stromal cell areas using Aperio ImageScope version 12.0.0.5039. The total number of nuclei within epithelial and stromal areas was counted and recorded as total cell count in each region using the Cell Counter Tool in ImageJ Plugins. Then the number of phosphohistone H3-positive cells was counted in the same region, and the percentage of phosphohistone H3-positive cells was calculated.
Statistical Analysis
Data were graphed and analyzed using GraphPad Prism version 6.0a for Mac OS X. All data are presented as mean ± SEM and were evaluated for statistically significant differences (P < 0.05) using a two-way ANOVA with the Tukey post hoc test, unless otherwise indicated.
RESULTS
Blunted Late Proliferative-Phase Response to E2 in cKO Uteri
To evaluate the function of ERα specifically in the epithelial cells during uterine proliferation, we generated mice lacking ERα in uterine epithelial cells using Wnt7aCre+ mice crossed with Esr1f/f mice as previously described [16]. IHC analysis of ERα demonstrated that ERα is expressed in all cell layers of WT uteri (Fig. 1A). Deletion of ERα was observed only in the luminal and glandular epithelial cells of cKO uteri (Fig. 1A) as previously reported [16]. Expression of ERα in the stromal cell layer was comparable between WT and cKO uteri (Fig. 1A). In the ovary, ERβ protein in the granulosa cells is expressed comparably in WT and cKO (Fig. 1B). Expression of ERβ was not detected in WT or cKO uterine samples (Fig. 1B), which suggests that loss of uterine epithelial ERα did not alter the expression level of ERβ in the cKO uteri.
FIG. 1.

Mice lacking ERα in uterine epithelial cells showed no alteration in uterine ERβ expression. ERα (A) and ERβ (B) immunohistochemistry (IHC) of WT control littermate (WT) and cKO animals. Representative images of uterine cross section (A) and ovarian and uterine cross section (B) are shown. Both WT and cKO ovarian sections, ERβ protein was detected in the granulosa cells. GE, glandular epithelium; LE, luminal epithelium; GC, granulosa cells. Bar = 100 μm.
E2 significantly increased the uterine epithelial cell height in both WT and cKO uteri after 24 and 72 h compared with the WT vehicle-treated group (Fig. 2, A and B). The increase of epithelial cell height was comparable between WT and cKO uteri 24 h after E2 treatment. However, the fold increase in cKO uterine epithelial cell height 72 h after E2 treatment was significantly lower than that of WT uteri (Fig. 2B).
FIG. 2.

Loss of uterine epithelial ERα led to a blunted late-phase uterine proliferative response after E2 treatment. A) H&E staining of uterine sections from WT and cKO mice treated with vehicle or E2 for 24 or 72 h. Adult females were ovariectomized and treated with E2 (0.25 μg/mouse) for 24 h or every day consecutively for 3 days (72 h). Uterine sections were stained with H&E, and epithelial cell heights were measured using ImagePro software as indicated in Materials and Methods. Yellow lines indicate examples of epithelial cell height measured. B) Uterine epithelial cell height after E2 treatment of WT and cKO animals. Fold increase of uterine epithelial cell heights from each group was calculated relative to WT vehicle-treated group (arbitrary unit). Bar graphs represent mean ± SEM, n = 4–7 mice/group. ** and ***P < 0.01 and 0.001, respectively; significant difference when compared to vehicle-treated group within genotype. ns, no significant difference when compared between WT E2 24 h and cKO E2 24 h. #P < 0.0001, significant difference when compared between WT E2 72 h and cKO E2 72 h.
Dampened E2-Induced Uterine Transcripts in the Absence of Epithelial ERα
Our previous findings demonstrated that ERα-dependent genomic responses in the uterus mirrored the biphasic physiological response to E2 treatment [10]. Because we observed comparable E2-induced uterine responses between WT and cKO uteri at 24 h but a significantly lower response in cKO uteri at 72 h, we hypothesized that the early transcriptional profile (2 h) would not be altered in the absence of epithelial ERα, whereas the late transcripts (24 h) would be disrupted and lead to aberrant subsequent uterine responses in cKO uteri. To test our hypothesis, a microarray analysis was performed. We used uterine samples from WT and cKO treated with vehicle or E2 for 2 h (early transcripts) or 24 h (late transcripts) to evaluate the functions of E2-responsive transcripts that might underlie physiological end points at 24 and 72 h, respectively. Unsupervised hierarchical clustering demonstrated that the transcriptional profiles of WT and cKO were similar when treated with E2 for 2 h, indicating similarities of transcripts regulated by E2 regardless of the presence of epithelial ERα (Fig. 3A). Venn diagrams demonstrate that approximately only 20% of the transcripts regulated by E2 at 2 h in WT were also E2 regulated in cKO uteri (overlap of WT and cKO; Fig. 3B), indicating epithelial ERα-independency. More than 95% of the gene expression at 24 h was regulated by E2 only in WT uteri, suggesting that the late phase was regulated primarily by epithelial ERα (Fig. 3C). These results indicate that uterine epithelial ERα plays crucial roles for E2-induced gene transcription with varying degrees for the early (2 h) and late (24 h) responses, but the contribution of the epithelial cells is more pronounced at 24 h, which is consistent with morphological changes observed in the early- and late-phase responses, respectively.
FIG. 3.

Microarray analysis of uterine transcripts from WT versus cKO treated with vehicle, E2 2 h, or E2 24 h. A) Unsupervised hierarchical clustering of all replicates from all genotypes and treatment groups using Partek Genomic Suites with the following criteria: raw intensity of at least one sample of each probe ≥ 100, gene is considered differentially expressed when P < 0.01, |fold change| ≥ 2, n = 3 animals/group. Green or red represent normalized expression that is less or greater than the mean of all conditions, respectively. B and C) Venn diagram of transcripts differentially expressed relative to vehicle in WT and cKO at (B) 2 h and (C) 24 h after E2 treatment using similar statistical criteria as A. Numbers in overlapping region are epithelial ERα-independent transcripts. Transcripts differentially expressed only in WT group are epithelial ERα-dependent transcripts.
Genes Involved in Proliferation of Epithelial Cells Expressed Independent of Epithelial ERα at 2 h
To characterize the molecular and cellular functions of ERα-independent genes in uteri 2 or 24 h after E2 treatment, microarray data sets were analyzed using Ingenuity Pathway Analysis. Indeed, at 2 h, common E2-regulated genes observed in both WT and cKO uteri were from signaling pathways for cellular growth and proliferation, gene expression, and cellular development (Table 1). A list of genes involved in proliferation of epithelial cells is presented in Supplemental Table S2. Selected genes from the proliferation of epithelial cells category were validated using real-time PCR analysis. Mitotic arrest deficient 2-like protein 1 (Mad2l1), vascular endothelial growth factor A (Vegfa), inhibin beta B (Inhbb), progesterone receptor (Pgr), and FBJ murine osteosarcoma viral oncogene homolog (Fos) were significantly up-regulated, whereas patch 1 (Ptch1) was significantly down-regulated in WT and cKO uteri after 2 h of E2 treatment (Fig. 4A). Microarray results also showed that cyclin-dependent kinase inhibitor 1a (Cdkn1a), Cebpb, and Igf1 were increased in both WT and cKO uteri after 2 h of E2 treatment (Supplemental Table S2), which is consistent with our previous findings [16]. This indicates that genes, most likely from the stroma, are involved in the proliferation of epithelial cells, remain E2 responsive in cKO, and are sufficient to stimulate epithelial cell proliferation in the absence of epithelial ERα.
TABLE 1.
Epithelial ERα-independent molecular and cellular functions that are commonly regulated in both WT and cKO uteri 2 or 24 h after E2 treatment.
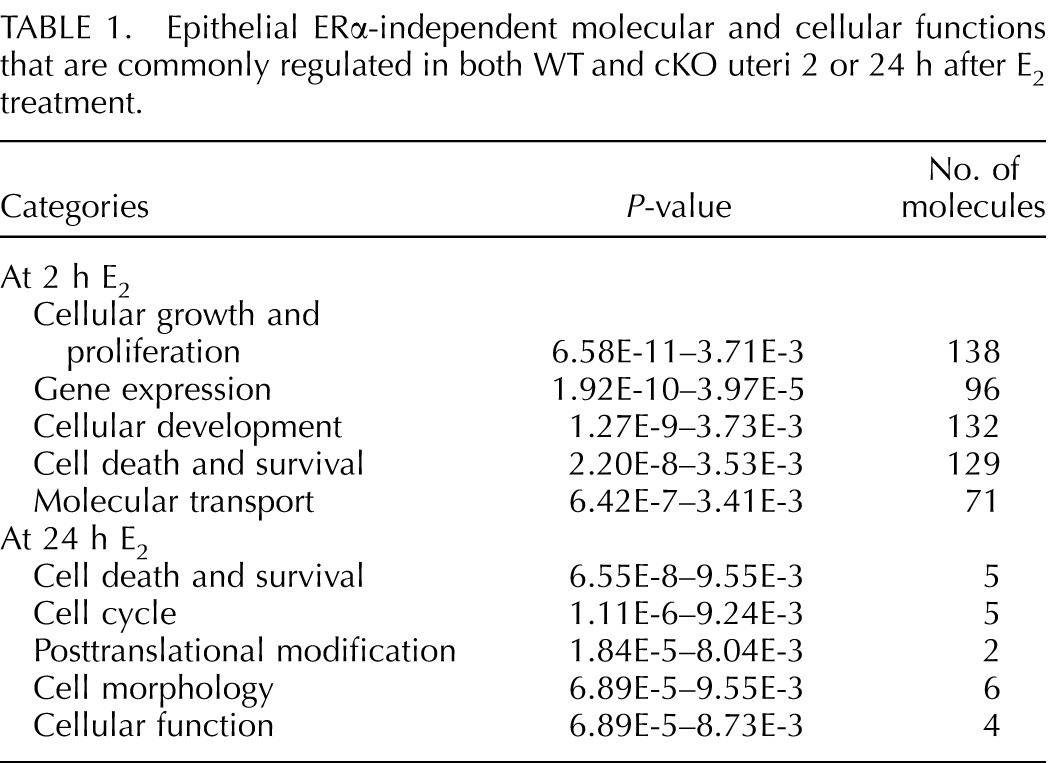
FIG. 4.
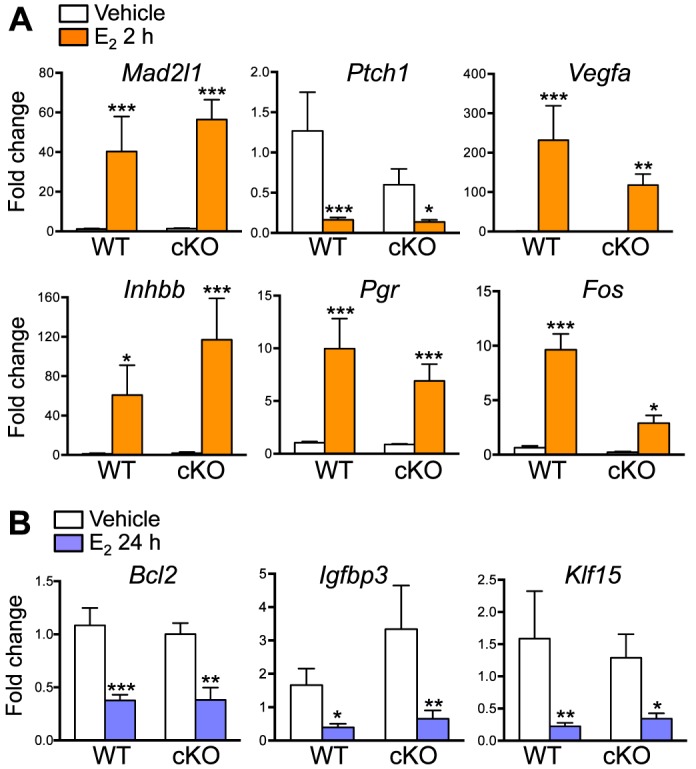
Uterine epithelial ERα-independent transcripts 2 (A) and 24 (B) h after E2 treatment. Validated gene expression using real-time PCR analysis of uterine samples from WT and cKO treated with E2 and collected 2 (A) or 24 (B) h after the treatment, n = 4–7 animals/group. Bar graphs represent mean ± SEM. *, **, and ***P < 0.05, 0.01, and 0.001, respectively; significant difference when compared to the vehicle-treated group within genotype.
At 24 h, there were fewer genes that were commonly regulated by E2 in both WT and cKO uteri (Fig. 3C) compared to those at 2 h. Ingenuity Pathway Analysis demonstrated that limited numbers of overlapping E2-regulated molecules were detected in both WT and cKO at 24 h (Table 1). Transcripts represented in these molecular and cellular functions were involved in cell death and survival, cell cycle, posttranslational modification, cell morphology, and cellular function (Table 1). Genes that were involved in cell death and survival network (Supplemental Table S3) were validated, including B-cell CLL/lymphoma 2 (Bcl2) and insulin-like growth factor binding protein 3 (Igfbp3), which were significantly down-regulated in both WT and cKO uteri (Fig. 4B). In addition, we also found that Krüppel-like factor 15 (Klf15), a progesterone (P4)-regulated gene important for uterine epithelial cell proliferation [23], was also significantly suppressed in both WT and cKO uteri 24 h after E2 treatment (Fig. 4B).
Lack of E2 Regulation of Cell Cycle Genes After 24 h in the Absence of Epithelial ERα
Genes involved in cellular development, gene expression, cell morphology, cell signaling, and energy production were preferentially regulated in WT 2 h after E2 treatment (Table 2). Interestingly, Wingless MMTV integration site (WNT)/β-catenin signaling and regulation of the epithelial-mesenchymal transition pathway were categorized as epithelial ERα-dependent canonical pathways, as these genes were significantly regulated only in E2-treated WT samples (Supplemental Table S4). Of note, there are some genes in cKO uteri that showed more than absolute twofold change; however, the P-values of those genes were greater than 0.01, which did not meet our statistical criteria. Real-time PCR analysis showed that dickkopf 2 homolog (Dkk2) was significantly down-regulated and that wingless-type MMTV integration site family, member 7B (Wnt7b), was up-regulated only in WT uteri at 2 h after E2 treatment (Fig. 5A). Additionally, we found that kallikrein 1-related peptidase b5 (Klk1b5), an E2-regulated gene [24, 25], was significantly up-regulated only in WT uteri. Expression levels of Dkk2, Wnt7b, and Klk1b5 were unchanged in cKO uteri when treated with E2 compared to vehicle control (Fig. 5A).
TABLE 2.
Molecular and cellular functions that are observed only in WT uteri 2 or 24 h after E2 treatment.
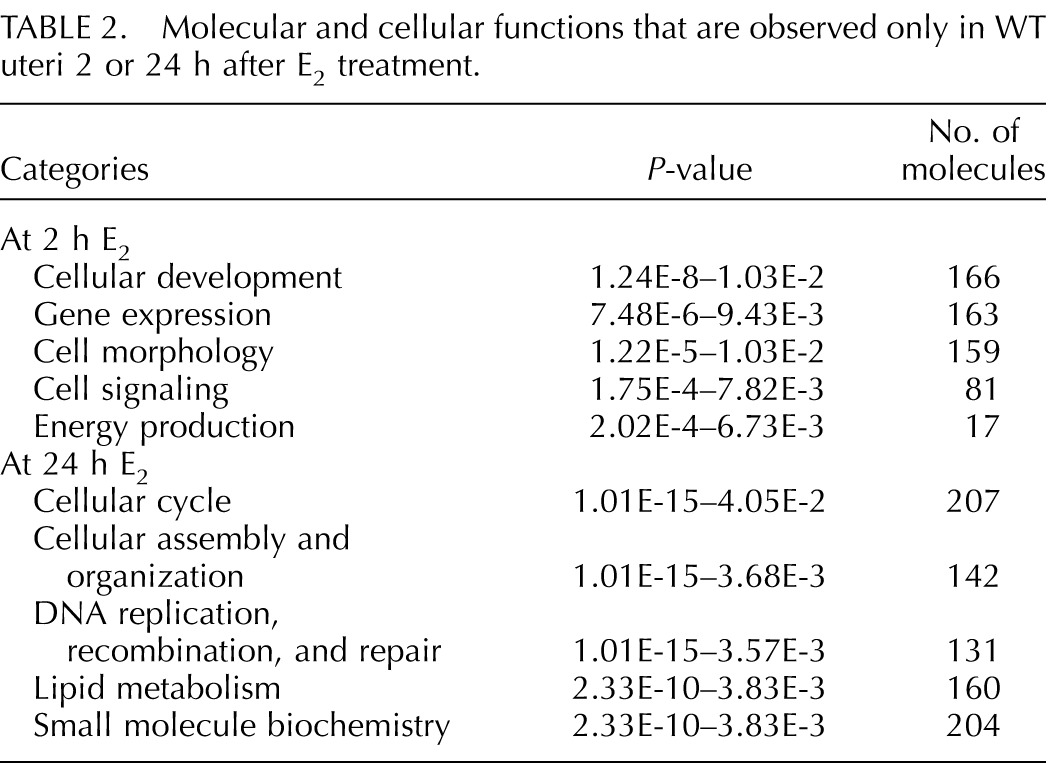
FIG. 5.
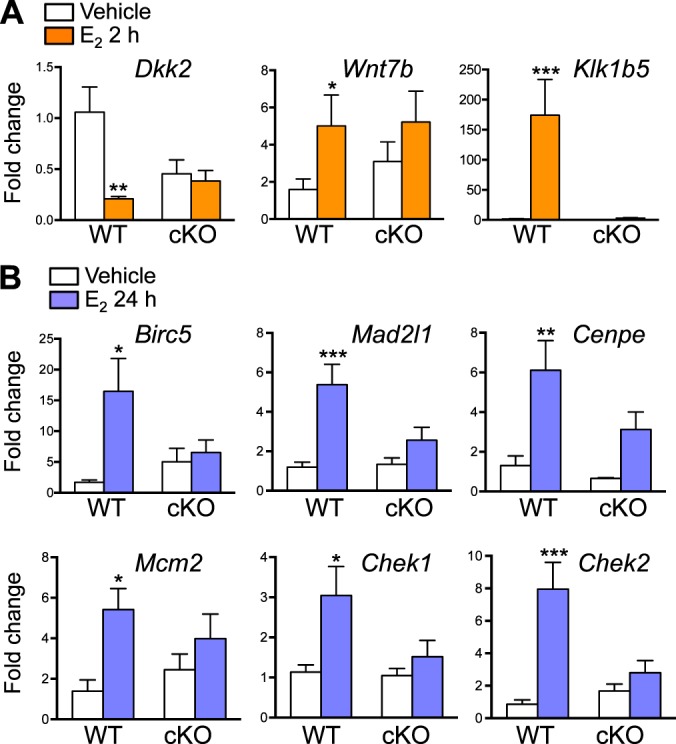
Uterine epithelial ERα-dependent transcripts 2 (A) and 24 (B) h after E2 treatment. Validated gene expression using real-time PCR analysis of uterine samples from WT and cKO treated with E2 and collected 2 (A) or 24 (B) h after the treatment, n = 4–7 animals/group. Bar graphs represent mean ± SEM. *, **, and ***P < 0.05, 0.01, and 0.001, respectively; significant difference when compared to the vehicle-treated group within genotype.
Genes that were not regulated after 24 h of E2 treatment in the absence of epithelial ERα are listed by various molecular and cellular functions in Table 2. Mitosis was one of the top altered categories in the cellular cycle (Supplemental Table S5). We found that a number of genes in the mitosis category were unchanged in cKO after E2 treatment, whereas all the genes in this category were up-regulated in WT uteri. Genes involved in mitosis include baculoviral IAP repeat containing 5 (Birc5), Mad2l1, centromere protein E (Cenpe), checkpoint kinase 1 (Chek1), and Chek2 (Fig. 5B). In addition, we found that minichromosome maintenance deficient 2 mitotin (Mcm2), a molecule required for DNA synthesis [23], was up-regulated by E2 treatment in WT but not in cKO uteri (Fig. 5B). Together, our findings suggest that a blunted uterine proliferative response occurs in the absence of epithelial ERα, which may be due to a lack E2 regulation of genes selectivity in epithelial cells required for mitosis.
Disrupted P4 Inhibition of E2-Induced Epithelial Cell Proliferation in cKO Uteri
Because a number of mitotic genes were unchanged in the cKO uteri 24 h after E2 treatment, we reasoned that lack of these gene products would lead to a proliferative defect in uterine stromal cells. It is known that the inhibition of epithelial cell growth and the induction of stromal cell proliferation are crucial for embryo implantation and decidual response. We previously determined that a lack of epithelial ERα causes female infertility partly due to defective implantation [16]. To mimic the hormonal profile during implantation, we administered E2 followed by P4 and then P4 with a nidatory dose of E2 18 h prior to tissue collection (called “E+Pe”) to WT or cKO females. In the vehicle-treated group, both WT and cKO uteri showed minimal uterine epithelial proliferation due to a lack of hormones as reported previously [9, 26]. As expected, E+Pe significantly increased uterine weight in WT females (Fig. 6A). Evaluation of the presence of Ki-67 (proliferative marker) or phosphohistone H3 (mitotic marker) demonstrated that, in WT uteri, E+Pe treatment induced the proliferation of stromal cells, whereas basal proliferation in the epithelial cells remained unchanged (Fig. 6, B–E). In cKO uteri, treatment with E+Pe showed a dampened uterine weight increase when compared to the WT-treated group (Fig. 6A). Although there was a slight decrease of the proliferation index (Ki-67 and phosphohistone H3) in the stromal cells of cKO uteri compared with WT uteri (Fig. 6, B–E), there were no significant differences in the Ki-67 and phosphohistone H3-positive cells between E+Pe-treated WT and cKO stroma. However, the Ki-67- and phosphohistone H3-positive cells in the epithelia were significantly higher in cKO compared to WT uteri (Fig. 6, C and E). This suggests that lack of uterine epithelial ERα does not affect stromal cell mitosis and proliferation but leads to an inability to appropriately arrest epithelial cell proliferation. Additionally, leukemia inhibitory factor (Lif) [27] and Indian hedgehog (Ihh) [28, 29], both required for uterine receptivity and induced in WT uteri, were not induced in the cKO (Fig. 7A). In the vehicle-treated WT and cKO uteri, PR is expressed exclusively in the luminal and glandular epithelial cells (Fig. 7B). Treatment with E+Pe induced the stromal PR and decreased epithelial PR expression in both WT and cKO uteri when compared to vehicle treatment (Fig. 7B). Moreover, expression of HAND2, a PR-regulated transcription factor expressed in uterine stromal cells during implantation [30], was comparably expressed in cKO and WT uteri (Fig. 7C). This suggests that the expression of PR and its downstream effector (HAND2) in the stromal cells were not disrupted by a lack of epithelial ERα. In summary, loss of epithelial ERα disrupted progesterone's ability to inhibit E2-induced epithelial cell proliferation but did not affect uterine stromal cell proliferation.
FIG. 6.

Epithelial ERα mediates P4 inhibition of E2-induced epithelial cell proliferation. A) Uterine weight increased in WT and cKO ovariectomized animals after a series of E2 and P4 with nidatory E2 treatments (called “E+Pe”), as described in Materials and Methods, compared to vehicle control, n = 5–8 animals/group. Bar graphs represent mean ± SEM. ***P < 0.001; significant difference when compared to vehicle control within genotype. #P < 0.0001, significant difference when compared between WT E+Pe and cKO E+Pe. B–D) Immunohistochemical analyses and quantification of Ki-67 (B and C) and phosphohistone H3 (D and E) in WT and cKO uterine cross sections. Representative images are shown (n = 5). Bar = 100 μm. C and E) Cells with positive staining of Ki-67 and phosphohistone H3 in the E+Pe-treated groups were counted (detail described in Materials and Methods) and presented as percentage of total cell count (luminal epithelial vs. stroma). Ki-67-positive cells were quantified from the whole uterine cross section. Phosphohistone H3-positive cells were quantified from the luminal and subepithelial stromal cells area. * and ***P < 0.05 and 0.001, significant difference between designated groups. ns, no significant difference.
FIG. 7.

Epithelial ERα mediates A) Lif and Ihh induction in WT and cKO ovariectomized animals after E+Pe treatments. Bar graphs represent mean ± SEM, n = 5–8 animals/group. * and ***P < 0.05 and 0.001, respectively; significant difference when compared to vehicle control within genotype. Loss of epithelial ERα does not alter expression pattern of PR (B) and HAND2 (C) when treated with E+Pe. Representative images are shown (n = 5).
DISCUSSION
Our findings, together with our previous studies, demonstrate that stromal and not epithelial ERα is crucial for regulating gene expression in the acute or early phase of estrogenic responses in the uterus that lead to initial epithelial cell proliferation. Early transcriptional responses (2 h), which underlying initial biological response at 24 h, are preserved in the absence of epithelial ERα (Fig. 8A). However, the deletion of epithelial ERα disrupts the expression of E2-regulated late transcripts involved in cell cycle progression and thereby impairs subsequent epithelial cell growth and proliferation. These findings indicate that late transcriptional responses (24 h), underlying subsequent or continuous growth (72 h), and depend on epithelial ERα (Fig. 8B). During the receptivity window, uterine epithelial cells ceases proliferation in order for the embryo to attach, while stromal cells undergo proliferation supporting embryo implantation. Our model demonstrated that the epithelial ERα is indispensable for P4 inhibition of epithelial cell proliferation (Fig. 8C). We also showed that epithelial ERα is crucial for implantation markers expressed in the epithelial cells (Lif and Ihh). However, stromal PR and HAND2 expression were induced in the absence of epithelial ERα, indicating that epithelial ERα does not account for expression of PR and its downstream signaling molecule (HAND2) in the stromal cells. This model suggests that epithelial ERα also regulates other molecule(s) responsible for inhibition of epithelial proliferation regardless of HAND2 expression.
FIG. 8.

Working model illustrating epithelial ERα function during uterine proliferation in response to E2. A) Epithelial ERα-independent, early transcriptional phase (2 h) of E2-induced initial epithelial cell proliferation (24 h). E2 activates stromal ERα to produce and secrete mitogenic signals (such as growth factors and potentially other molecules shown in Supplemental Table S1) and stimulate uterine epithelial cell proliferation. This initial proliferative event is not regulated by epithelial ERα. B) Epithelial ERα-dependent, late transcriptional phase (24 h) of E2-induced subsequent epithelial cell growth and proliferation (72 h). Both epithelial and stromal ERα are pivotal for a maximal uterine biological response regulated by E2. At this stage, epithelial ERα may induce transcripts needed to generate autocrine signals or provide positive feedback to the stromal cells to induce gene products involved in mitosis and cell cycle progression as well as inhibition of apoptosis that led to an ultimate physiological end point induced by E2. C) Inhibition of epithelial cell proliferation and induction of implantation markers during the uterine receptivity window require the expression of epithelial ERα. Stromal cell proliferation during this window appears to be independent from epithelial ERα expression.
Epithelial-stromal (or epithelial-mesenchymal) interactions are crucial for tissue development, growth, and differentiation [11]. In mouse uterine tissues, genetic fate mapping has shown that progenitor stromal cells provide the replacement of epithelial cells during tissue remodeling after parturition [19]. In the oviduct, tissue recombination studies demonstrate that the origin of stromal cells underlying epithelium determines the epithelial cell development, characteristics, and ultimately the epithelial cell functions [31]. These studies indicate that stromal cells in the mesenchyme origin determine cell fate and functionality and stimulate the proliferative phase of the epithelial cell responses in female reproductive tracts. In our model, we analyzed the transcriptional profiles in whole uterine tissue in response to E2 in the absence of epithelial ERα. The findings from this study indicate that early-phase E2-induced transcriptional changes in the uteri are independent of epithelial ERα. We demonstrated that gene expression induced by E2 was not due to compensation via ERβ increased expression in the uterus due to the loss of ERα but more likely through the remaining stromal ERα. To date, ours is the first finding demonstrating in vivo that signals regulated by ERα in the epithelial cells were indispensable for optimal uterine transcript expression and physiological functions induced by E2. Moreover, we also showed that epithelial ERα is required for P4-mediated inhibition of epithelial cell proliferative responses, a process crucial for embryo implantation.
In the initial phase of E2-induced uterine responses, we observed a comparable increase of epithelial cell height in this study and DNA synthesis and cell proliferation in our previous findings [16] in both WT and cKO animals 24 h after E2 treatment. Herein, we explored in more detail the uterine transcripts during the early phase (2 h) and found that approximately 20% of differentially regulated transcripts were shared between WT and cKO uteri (ERα-independent transcripts). Molecules involved in proliferation of epithelial cells and transcription of RNA, such as Mad2l1, Vegfa, Inhbb, Pgr, and Fos, as well as other transcripts shown in Supplemental Table S2, were regulated similarly in WT and cKO uteri. Additionally, our previous findings showed that IGF-1 treatment stimulates uterine epithelial proliferation in the cKO as similar to WT uteri [16]. Basic transcription element binding protein 1 (Bteb1, also called Klf9), a growth factor-regulated gene [32], was expressed comparably in cKO and WT uteri that were treated with IGF1 (our unpublished data), indicating epithelial ERα-independent activation of growth factor-mediated signals. Together, these findings suggest that epithelial ERα is not responsible for E2-induced early gene response (2 h) and that signals produced from genomic action of E2 in cKO uteri at 2 h is dependent on stromal ERα and sufficient to induce epithelial cell proliferation.
WNT signaling regulates left-right patterning and cell development and differentiation through cell-cell communication [33]. In mouse uteri, E2 increases expression of Wnt4, Wnt5a, and Fzd2 (a WNT molecule receptor) but decreases secreted frizzled-related protein-2 (SFRP-2; negative regulator of WNT signaling) expression [34]. Overexpression of SFRP-2 inhibited E2-induced uterine proliferation [34], indicating that the activation of the WNT/β-catenin canonical pathway is involved in E2-induced uterine epithelial cell proliferation. However, Hou et al. [34] showed global deletion of ERα did not affect E2-regulated expression of WNT signaling molecules in the uteri. In contrast, we found that WNT/β-catenin signaling was one of the top canonical pathways regulated in an epithelial ERα-dependent manner. We have validated that E2-mediated regulation of Dkk2 and Wnt7b (signaling molecules in WNT/β-catenin pathway) was lost in the absence of epithelial ERα. However, lacking E2 regulation of these molecules in WNT/β-catenin pathway in cKO uteri did not alter the initial (24 h) proliferative response. Recently, a study using uterine stromal cell-specific knockout of β-catenin demonstrated that loss of stromal β-catenin does not affect E2-induced mitosis in the uterine epithelial cells [35]. Together with our results, this suggests that epithelial ERα regulates expression of WNT/β-catenin signaling molecules, but these molecules were not the only E2-induced signals needed for epithelial cell proliferation. It is possible that the ERα-independent regulation of uterine WNT/β-catenin molecules observed in the Hou study was due to the residual splice variant of ERα in the original ERα KO model [36, 37].
Ingenuity pathway analyses of our microarray data sets demonstrated that the top cellular and molecular function at 24 h regulated by epithelial ERα was cell cycle progression. A total of 207 molecules in the cell cycle category that were differentially expressed in the WT were not significantly regulated in cKO uteri. Other E2-regulated cell cycle mediators during mitosis that were selectively regulated in WT uteri were cyclin, cell division cycle, centromere protein, E2F transcription factor, and kinesin (Kif) family members. Previous studies in our laboratory demonstrated that expression of Mad2l1, a component of mitotic spindle assembly checkpoint, peaks 2 h after E2 treatment and declines but remains elevated 24 h after E2 treatment [10]. In the present study, we found that Mad2l1 was induced in both WT and cKO uteri 2 h after E2 treatment. However, loss of epithelial ERα led to a return of Mad2l1 to basal level 24 h after E2 treatment. It is possible that epithelial ERα may provide a positive feedback to stromal cells to generate a maximal paracrine mitogenic signal to stimulate epithelial cell proliferation (Fig. 8B). Alternatively, the Mad2l1 pattern may also suggest that the initial rise in Mad2l1 is due to stromal ERα and that epithelial ERα is required for the subsequent late-phase expression involved in the proliferative response. Overall, these results suggest that proper communication between epithelial and stromal cells is required to properly maintain the optimal proliferative response in the uterus.
We observed unique transcripts expressed only in cKO uteri, which may reflect differences resulting from tissue development in the presence or absence of epithelial ERα. However, we did not observe any overt morphological defect in cKO uteri. In addition, we also reported that a lack of epithelial ERα cause an aberrant epithelial cell apoptosis subsequent to proliferation in part due to a lack of Birc1a expression [16]. In this study, we also showed that inhibitor of apoptosis, Birc5, was not significantly up-regulated in cKO uteri after 24 h of E2 treatment (Supplemental Table S5). However, ingenuity pathway analysis did not indicate a significant change in the apoptotic signaling pathway in cKO uteri at either 2 or 24 h of E2 treatment compared to WT uteri. It is possible that E2-regulated transcripts involved in apoptosis may be significantly altered later than the 24-h E2 treatment in cKO uteri or the changes in apoptosis gene(s) occurred only in the epithelial cells and were diluted by the whole uteri extract, which obscured the difference. Future analyses using laser capture microdissection to distinguish the alteration in expression profile exclusively in epithelial versus stromal cells in the absence of epithelial ERα would allow detection of such subtle differences.
Our previous findings showed that there are no implantation sites in female mice lacking epithelial ERα and that these females are sterile [16]. To understand the role of epithelial ERα and identify signaling molecules downstream of epithelial ERα during implantation, we administered ovarian steroids (E+Pe) to mimic the hormonal profile during the uterine receptive window. In the ovariectomized vehicle-treated mice, the labeling index is extremely low or at a minimally detectable level of cell activity in the absence of ovarian hormones [9, 26]. We have observed the similarly low Ki-67 and phosphohistone H3-positive staining in both WT and cKO uteri. In this experiment of P4 inhibition of epithelial cell proliferation, we compared the uterine proliferative responses in epithelial versus stromal cells only in the animals that received the E+Pe treatment in the presence or absence of epithelial ERα. In the absence of uterine epithelial ERα, we observed an incomplete inhibition of epithelial cell proliferation compared to WT uteri. This finding indicates that the epithelial ERα is required for P4 inhibition of E2-induced proliferation. Due to the limitation of the software we used and the enrichment of the areas we analyzed as described in Materials and Methods, the direct comparison between the numbers of Ki-67 and phosphohistone H3-positive cells in epithelial versus stromal cells cannot be made from our analyses. The inhibitory effect on epithelial cells is needed to provide a receptive uterine environment for embryo attachment. Studies using uterine epithelial cell specific knockout of PR demonstrated aberrant P4 inhibition of E2-induced epithelial cell proliferation [38]. However, we have shown here that deletion of epithelial ERα has no effect on mRNA level or protein expression of PR [16]. Therefore, a lack of inhibitory effect on epithelial cell proliferation was not due to loss of PR expression.
We found that Lif, an E2-regulated secretory molecule in uterine glandular epithelial cells [27, 39], was not induced during the uterine receptive window in the absence of epithelial ERα. In addition, global deletion of Ihh, a P4-regulated uterine gene, caused an implantation defect and a lack of stromal cell proliferation during the preimplantation period [28]. However, recent studies from our laboratory and others reported that E2 also induces Ihh expression in mouse uteri [29, 40]; however, the induction was minimal when compared to the response induced by P4. We found that the uterine Ihh transcript was not increased in the cKO mice compared to a strong induction in the WT uteri. We also found that expression of Ptch1, the Ihh receptor, was comparable between WT and cKO uteri. This may be due to the fact that expression of Ptch1 is restricted in the uterine stromal cells [28] and would thus be regulated independently from epithelial ERα. Together, these findings suggest that E2 induces the luminal and glandular epithelial ERα regulation of Lif and Ihh during implantation. Lack of Lif and Ihh alters the uterine receptivity and decreases uterine proliferation during implantation in the cKO uteri. This study provides a more complete understanding of an ERα cell-specific functionality and gene-selective expression regarding the interaction of epithelium and stroma in eliciting uterine E2 responses.
ACKNOWLEDGMENT
We thank Drs. Grant D. Orvis and Richard R. Behringer for providing Wnt7aCre+ animals; Drs. Kevin Gerrish and Liwen Li for initial microarray analysis; the NIEHS histology and immunohistology core for tissue processing and H&E and ERβ staining; Julie Foley, Norris Flager, and Elizabeth Ney for image analysis; Brianna Pockette for Ki67 staining; Drs. Shannon Whirledge and Wendy Jefferson for critical review of this manuscript; and Comparative Medicine Branch for animal care and surgery.
Footnotes
Supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences to K.S.K. (Z01ES70065).
REFERENCES
- Tabibzadeh S. The signals and molecular pathways involved in human menstruation, a unique process of tissue destruction and remodelling. Mol Hum Reprod. 1996;2:77–92. doi: 10.1093/molehr/2.2.77. [DOI] [PubMed] [Google Scholar]
- Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology. 1997;138:4613–4621. doi: 10.1210/endo.138.11.5496. [DOI] [PubMed] [Google Scholar]
- Wang H, Eriksson H, Sahlin L. Estrogen receptors alpha and beta in the female reproductive tract of the rat during the estrous cycle. Biol Reprod. 2000;63:1331–1340. doi: 10.1095/biolreprod63.5.1331. [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Li L, Grimm SA, Chen Y, Liu L, Li Y, Bushel PR, Fargo D, Korach KS. Research resource: whole-genome estrogen receptor alpha binding in mouse uterine tissue revealed by ChIP-seq. Mol Endocrinol. 2012;26:887–898. doi: 10.1210/me.2011-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JS, Jensen SM, Lunceford JK, Kahn MW, Zheng Y, Falase EA, Lyttle CR, Teuscher C. Evidence for the genetic control of estradiol-regulated responses. Implications for variation in normal and pathological hormone-dependent phenotypes. Am J Pathol. 1997;150:2223–2230. [PMC free article] [PubMed] [Google Scholar]
- Perez MC, Furth EE, Matzumura PD, Lyttle CR. Role of eosinophils in uterine responses to estrogen. Biol Reprod. 1996;54:249–254. doi: 10.1095/biolreprod54.1.249. [DOI] [PubMed] [Google Scholar]
- Pollard JW, Pacey J, Cheng SV, Jordan EG. Estrogens and cell death in murine uterine luminal epithelium. Cell Tissue Res. 1987;249:533–540. doi: 10.1007/BF00217324. [DOI] [PubMed] [Google Scholar]
- Quarmby VE, Korach KS. The influence of 17 beta-estradiol on patterns of cell division in the uterus. Endocrinology. 1984;114:694–702. doi: 10.1210/endo-114-3-694. [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, Korach KS. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol Endocrinol. 2003;17:2070–2083. doi: 10.1210/me.2003-0146. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Chung LW, Shannon JM, Reese BA. Stromal-epithelial interactions in sex differentiation. Biol Reprod. 1980;22:19–42. doi: 10.1095/biolreprod22.1.19. [DOI] [PubMed] [Google Scholar]
- DiAugustine RP, Petrusz P, Bell GI, Brown CF, Korach KS, McLachlan JA, Teng CT. Influence of estrogens on mouse uterine epidermal growth factor precursor protein and messenger ribonucleic acid. Endocrinology. 1988;122:2355–2363. doi: 10.1210/endo-122-6-2355. [DOI] [PubMed] [Google Scholar]
- Zhu L, Pollard JW. Estradiol-17beta regulates mouse uterine epithelial cell proliferation through insulin-like growth factor 1 signaling. Proc Natl Acad Sci U S A. 2007;104:15847–15851. doi: 10.1073/pnas.0705749104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom YK, Young P, Wiesen JF, Miettinen PJ, Derynck R, Werb Z, Cunha GR. Uterine and vaginal organ growth requires epidermal growth factor receptor signaling from stroma. Endocrinology. 1998;139:913–921. doi: 10.1210/endo.139.3.5817. [DOI] [PubMed] [Google Scholar]
- Nelson KG, Takahashi T, Lee DC, Luetteke NC, Bossert NL, Ross K, Eitzman BE, McLachlan JA. Transforming growth factor-alpha is a potential mediator of estrogen action in the mouse uterus. Endocrinology. 1992;131:1657–1664. doi: 10.1210/endo.131.4.1396310. [DOI] [PubMed] [Google Scholar]
- Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Uterine epithelial estrogen receptor α is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci U S A. 2010;107:19272–19277. doi: 10.1073/pnas.1013226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke PS, Buchanan DL, Young P, Setiawan T, Brody J, Korach KS, Taylor J, Lubahn DB, Cunha GR. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc Natl Acad Sci U S A. 1997;94:6535–6540. doi: 10.1073/pnas.94.12.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantena SR, Kannan A, Cheon YP, Li Q, Johnson PF, Bagchi IC, Bagchi MK. C/EBPbeta is a critical mediator of steroid hormone-regulated cell proliferation and differentiation in the uterine epithelium and stroma. Proc Natl Acad Sci U S A. 2006;103:1870–1875. doi: 10.1073/pnas.0507261103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Orvis GD, Wang Y, Behringer RR. Stromal-to-epithelial transition during postpartum endometrial regeneration. PLoS One. 2012;7:e44285. doi: 10.1371/journal.pone.0044285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Zhu L, Deng Y, Pollard JW. Microarray analysis of uterine epithelial gene expression during the implantation window in the mouse. Endocrinology. 2006;147:4904–4916. doi: 10.1210/en.2006-0140. [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Korach KS. Estrogenic activity of bisphenol A and 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE) demonstrated in mouse uterine gene profiles. Environ Health Perspect. 2011;119:63–70. doi: 10.1289/ehp.1002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Pollard JW. KLF15 negatively regulates estrogen-induced epithelial cell proliferation by inhibition of DNA replication licensing. Proc Natl Acad Sci U S A. 2012;109:E1334–1343. doi: 10.1073/pnas.1118515109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes G, Figueroa CD, Corthorn J. Temporospatial changes of kallikrein-like enzymes during the estrous cycle and pregnancy in the rat uterus. Biol Reprod. 1996;55:236–245. doi: 10.1095/biolreprod55.2.236. [DOI] [PubMed] [Google Scholar]
- Rajapakse S, Yamano N, Ogiwara K, Hirata K, Takahashi S, Takahashi T. Estrogen-dependent expression of the tissue kallikrein gene (Klk1) in the mouse uterus and its implications for endometrial tissue growth. Mol Reprod Dev. 2007;74:1053–1063. doi: 10.1002/mrd.20567. [DOI] [PubMed] [Google Scholar]
- Martin L, Finn CA, Trinder G. Hypertrophy and hyperplasia in the mouse uterus after oestrogen treatment: an autoradiographic study. J Endocrinol. 1973;56:133–144. doi: 10.1677/joe.0.0560133. [DOI] [PubMed] [Google Scholar]
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Kontgen F, Abbondanzo SJ. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature. 1992;359:76–79. doi: 10.1038/359076a0. [DOI] [PubMed] [Google Scholar]
- Lee K, Jeong J, Kwak I, Yu CT, Lanske B, Soegiarto DW, Toftgard R, Tsai MJ, Tsai S, Lydon JP, DeMayo FJ. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat Genet. 2006;38:1204–1209. doi: 10.1038/ng1874. [DOI] [PubMed] [Google Scholar]
- Wakitani S, Hondo E, Phichitraslip T, Stewart CL, Kiso Y. Upregulation of Indian hedgehog gene in the uterine epithelium by leukemia inhibitory factor during mouse implantation. J Reprod Dev. 2008;54:113–116. doi: 10.1262/jrd.19120. [DOI] [PubMed] [Google Scholar]
- Li Q, Kannan A, DeMayo FJ, Lydon JP, Cooke PS, Yamagishi H, Srivastava D, Bagchi MK, Bagchi IC. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science. 2011;331:912–916. doi: 10.1126/science.1197454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanouchi H, Umezu T, Tomooka Y. Reconstruction of oviduct and demonstration of epithelial fate determination in mice. Biol Reprod. 2010;82:528–533. doi: 10.1095/biolreprod.109.078329. [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Collins J, Grissom S, Deroo B, Korach KS. Global uterine genomics in vivo: microarray evaluation of the estrogen receptor alpha-growth factor cross-talk mechanism. Mol Endocrinol. 2005;19:657–668. doi: 10.1210/me.2004-0142. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Hou X, Tan Y, Li M, Dey SK, Das SK. Canonical Wnt signaling is critical to estrogen-mediated uterine growth. Mol Endocrinol. 2004;18:3035–3049. doi: 10.1210/me.2004-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Patterson AL, Teixeira JM, Pru JK. Endometrial stromal beta-catenin is required for steroid-dependent mesenchymal-epithelial cross talk and decidualization. Reprod Biol Endocrinol. 2012;10:75. doi: 10.1186/1477-7827-10-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, Smithies O, Korach KS. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol. 1995;9:1441–1454. doi: 10.1210/mend.9.11.8584021. [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J. 2010;24:4660–4667. doi: 10.1096/fj.10-163428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Rubel CA, Large MJ, Wetendorf M, Fernandez-Valdivia R, Jeong JW, Spencer TE, Behringer RR, Lydon JP, Demayo FJ. Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 2012;26:1218–1227. doi: 10.1096/fj.11-193334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filant J, Spencer TE. Cell-specific transcriptional profiling reveals candidate mechanisms regulating development and function of uterine epithelia in mice. Biol Reprod. 2013;89:86. doi: 10.1095/biolreprod.113.111971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Li L, Grimm SA, Winuthayanon W, Hamilton KJ, Pockette B, Rubel CA, Pedersen LC, Fargo D, Lanz RB, DeMayo FJ, Schutz G, et al. Novel DNA motif binding activity observed in vivo with an estrogen receptor alpha mutant mouse. Mol Endocrinol. 2014;28:899–911. doi: 10.1210/me.2014-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]