

ABSTRACT
Di-n-butyl phthalate (DBP) is present in many consumer products, such as infant, beauty, and medical products. Several studies have shown that DBP causes reproductive toxicity in rodents, but no studies have evaluated its effects on ovarian follicles. Therefore, we used a follicle culture system to evaluate the effects of DBP on antral follicle growth, cell cycle and apoptosis gene expression, cell cycle staging, atresia, and 17β-estradiol (E2) production. Antral follicles were isolated from adult CD-1 mice and exposed to DBP at 1, 10, 100, and 1000 μg/ml for 24 or 168 h. Follicles treated with vehicle or DBP at 1–100 μg/ml grew over time, but DBP at 1000 μg/ml significantly suppressed follicle growth. Regardless of effect on follicle growth, DBP-treated follicles had decreased mRNA for cyclins D2, E1, A2, and B1 and increased p21. Levels of the proapoptotic genes Bax, Bad, and Bok were not altered by DBP treatment, but DBP 1000 μg/ml increased levels of Bid and decreased levels of the antiapoptotic gene Bcl2. DBP-treated follicles contained significantly more cells in G1 phase, significantly less cells in S, and exhibited a trend for fewer cells in G2. Although DBP did not affect E2 production and atresia at 24 h, follicles treated with DBP had reduced levels of E2 at 96 h and underwent atresia at 168 h. These data suggest that DBP targets antral follicles and alters the expression of cell cycle and apoptosis factors, causes cell cycle arrest, decreases E2, and triggers atresia, depending on dose.
Keywords: 17β-estradiol, antral follicle, apoptosis, atresia, cell cycle, ovary, phthalate, toxicology
INTRODUCTION
Phthalates are industrial chemicals commonly used as plasticizers or solvents in many consumer products, such as infant care products and toys [1], beauty products (nail polish, perfume [2]), medical devices (blood bags, disposable gloves [3]), and the plastic coating of some oral medications [4]. Because of their ubiquitous presence and excessive worldwide production, exposure to phthalates can occur via ingestion, inhalation, and intravenous or dermal contact. Epidemiological studies have provided evidence supporting continuous daily exposure in humans by showing that more than 75% of human spot urine samples contained detectable levels of phthalates [2]. Interestingly, women were reported to have higher urinary phthalate levels than men [2]. Other epidemiological studies have shown associations between phthalate exposure and increased miscarriage rates [5] and urinary phthalate levels and increased pregnancy complications [6].
Dibutyl phthalate (DBP) is commonly found in cellulose acetate plastics, personal care products (e.g., nail polish, cosmetics), lacquers, varnishes, and the coating of some pharmaceuticals [7]. Human exposure has been estimated at 1–233 μg/kg/day in individuals taking medications coated with DBP [4] and 0.1–76 μg/kg/day in occupationally exposed groups [8]. Levels of monobutyl phthalate, a biomarker of DBP exposure, in serum have been estimated to range between 0.6 and 139 ng/ml (geometric mean = 13.54 ng/ml) in subjects included in the 1999–2000 National Health and Nutrition Examination Survey [9] and between 0 and 0.98 ng/ml in healthy Chinese children [10]. In urine, creatinine-adjusted MBP levels have been reported to range between 1.6 and 2760 μg/g [11].
In mice, prenatal exposure (70–2200 mg/kg, throughout gestation) to di-2-ethylhexyl phthalate (DEHP) and DBP leads to increased absorption of fetuses and increased incidence of external abnormalities in the offspring of dosed mice [12]. In rats, exposure to DEHP (2000 mg/kg/day; 1–12 days) causes prolonged estrous cycles, decreases circulating levels of 17β-estradiol, and alters ovulation rates [13]. Davis et al. [13] also identified the granulosa cell compartment as a target for phthalates by showing that DEHP-exposed ovaries contained antral follicles with smaller granulosa cell area compared to ovaries from vehicle-treated animals. More recently, DEHP (1–100 μg/ml × 96 h) and its metabolite, mono-2-ethyhexyl phthalate (MEHP; 0.1–10 μg/ml × 96 h), were shown to inhibit the growth of mouse antral follicles in vitro [14–16]. However, to our knowledge, no studies have evaluated the effects of DBP exposure on the adult ovary or antral follicle growth, survival, and steroidogenesis.
Ovarian follicles are the functional units of the ovary and exist in different stages of development: primordial, primary, secondary, and antral. The present study focused on antral follicles (200–350 μm in mice) because these follicles contain the oocyte for ovulation and are the major source of 17β-estradiol (E2) in cycling animals [17, 18]. Damage to the antral follicle population may result in altered levels of E2 and therefore altered ovarian function, which in turn may lead to infertility [19]. Further, decreased E2 levels may increase a woman's risk for disorders such as osteoporosis, cardiovascular disease, and depression [20–22].
The process of ovarian folliculogenesis is characterized by proliferation of the follicular cells that surround the oocyte and growth of the oocyte itself [18]. On activation, granulosa cells in primordial follicles enter the cell cycle, and their accelerated proliferation results in follicle growth to the antral and preovulatory stages [18]. To progress through the cell cycle, cells must complete four distinct phases known as gap 1 (G1), synthesis (S), gap 2 (G2), and mitosis (M). The processes of cell cycle progression and cell proliferation are controlled by three groups of regulatory proteins: cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors (CKIs; reviewed in Graña and Reddy [23]). Cyclins are expressed in an oscillatory fashion throughout the cell cycle and, by physically binding CDKs, facilitate phosphorylation of the complex and activation of its kinase activity. The complex formed by cyclin D and CDK4/6 is critical for entry into the cell cycle and progression through the G1 phase. Likewise, the complex formed by cyclin E and CDK2 is required for the G1/S transition, cyclin A-CDK1 for progression through S phase, and cyclin B-CDK1 for initiation of M phase. In contrast, CKIs, like p21, induce cell cycle arrest by inhibiting the activity of cyclin-CDK complexes. The importance of cyclin D2 in granulosa cell proliferation has been demonstrated by studies showing that cyclin D2-null mice have significantly smaller follicles, decreased granulosa cell proliferation, and impaired ovulation [24].
Although many follicles begin development during each estrous or menstrual cycle, only a few will be ovulated. In fact, the vast majority of follicles will undergo atresia, an apoptotic process [18, 25]. Activation of apoptosis involves a tight balance between the proapoptotic proteins BAX, BAD, BOK, BID, and BBC3 (formerly known as PUMA), among others, and antiapoptotic proteins commonly represented by BCL2 (reviewed in Hengartner [26]). Previous studies of follicle growth and atresia in various species have shown that proliferating granulosa cells appear to be more susceptible to apoptosis. Specifically, resting granulosa cells and those in early G1 have been shown to be relatively less susceptible to apoptosis than those at the G1/S transition [27].
Therefore, the main objective of this study was to use an isolated mouse antral follicle culture system to determine the direct effects of DBP on mouse antral follicles. We hypothesized that DBP exposure would result in impaired antral follicle growth, survival, and steroidogenesis. To test our hypothesis, we exposed isolated antral follicles to increasing concentrations of DBP and determined the effect of DBP on antral follicle growth, cell cycle and apoptosis gene expression, cell cycle staging, atresia, and E2 production. Because we used doses of DBP that are above those detected in humans, the present study should not be considered a direct risk assessment analysis. Instead, the present work was meant to help identify targets of DBP toxicity in the ovary, elucidate the mechanisms of DBP toxicity in antral follicles, and, by doing so, support future environmentally relevant risk assessment studies.
MATERIALS AND METHODS
Chemicals
DBP, dimethylsulfoxide (DMSO), ITS (insulin, transferrin, selenium), penicillin and streptomycin, bovine serum albumin (BSA), and DNAse I were obtained from Sigma-Aldrich (St. Louis, MO). Alpha-minimal essential media (α-MEM), H-199 media, collagenase type I, and propidium iodide were obtained from Life Technologies (Grand Island, NY). Human recombinant follicle-stimulating hormone (rFSH) was obtained from Dr. A.F. Parlow from the National Hormone and Peptide Program (Harbor-UCLA Medical Center, Torrance, CA), charcoal-stripped fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Lawrenceville, GA), and RNAse A was obtained from Worthington Biochemical Company (Lakewood, NJ).
Animals
Cycling female CD-1 mice (35–37 days old) were obtained from or bred in-house from animals purchased from Charles River Laboratories (Charles River, CA). Animals were housed four mice per cage at the University of Illinois College of Veterinary Medicine Central Animal Facility. Food and water were provided ad libitum. Temperature was maintained at 22 ± 1°C, and animals were subjected to 12L:12D cycles. Animals were euthanized at 35–37 days old by carbon dioxide (CO2) inhalation followed by cervical dislocation. The ovaries were removed and antral follicles isolated as described below. All experiments and methods involving animals were approved by the University of Illinois Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Experimental Animals [28].
Antral Follicle Culture
Mice were euthanized by CO2 inhalation followed by cervical dislocation. Ovaries were removed and antral follicles mechanically isolated based on relative size (200–350 μm) and placed in culture as previously described [29]. Follicles were isolated from three to four mice per culture with approximately 15–30 antral follicles obtained from each mouse. Following isolation, antral follicles were placed in individual wells of 96-well culture plates with 75 μl of unsupplemented α-MEM prior to treatment. Each follicle culture experiment consisted of 8–15 follicles per treatment.
A stock solution of DBP was prepared using DMSO as a solvent. Various concentrations (1333.33, 133.33, 13.33, and 1.33 mg/ml) were prepared to ensure that an equal volume of each chemical could be added to each well to control for solvent concentration. Final concentrations of DBP in culture were 1, 10, and 100, and 1000 μg/ml. Because no other studies have evaluated the effects of DBP on ovarian follicles in vitro, the present doses were selected based on studies that have reported toxicity of DBP in males and females. Bao et al. [30] reported that in vivo exposure to DBP produced significantly different protein expression profiles in male rat testes (0.1–10 mg/kg/day) and altered reproductive organ morphology (100–500 mg/kg/day). Further, in utero exposure to DBP at 850 mg/kg/day has been shown to cause anorectal malformations and megacolon in male rats [31]). Oral exposure to DBP (500 and 1000 mg/kg/day) from weaning through puberty, mating, and gestation resulted in increased midpregnancy abortions and decreased ex vivo ovarian hormone production in female rats [32].
Treatment groups included four doses of DBP (1, 10, 100, and 1000 μg/ml) and a vehicle control consisting of DMSO (0.075%). All dosing solutions were prepared individually in supplemented α-MEM with an equal volume of chemical added for each dose to control for the amount of vehicle in each preparation. Supplemented α-MEM contained 5% FBS, 1% ITS (10 ng/ml insulin, 5.5 ng/ml transferrin, 5.5 ng/ml selenium), 100 U/ml penicillin, 100 mg/ml streptomycin, and 5 IU/ml rFSH. For treatment, unsupplemented media was replaced with 150 μl supplemented α-MEM containing vehicle or DBP at the various doses. Then follicles were incubated for 24 or 168 h at 37°C in 5% CO2. In the 168-h cultures, media were removed after 96 h and replaced with fresh supplemented media containing treatment. Media samples from individual follicles (24 and 168 h) were stored at −80°C until further analysis.
Analysis of Follicular Growth
Follicle growth was assessed by measuring the diameter of each follicle as described previously [29]. Follicles with diameters of 200 μm or greater were considered antral [33] based on the histological appearance of these follicles. At least three separate culture experiments were performed for each chemical treatment. Follicle diameter measurements were converted to percent change relative to 0 h and averaged among treatment groups. The resulting data were compared between the chemical treatments over time.
Histological Evaluation of Antral Follicles and Atresia Rating
At the end of culture, media were removed, and each individual follicle was processed for analysis of atresia as previously described [34, 35]. Briefly, follicles were fixed with Dietrich fixative for at least 24 h. Following fixation, follicles were dehydrated and prepared for plastic embedding using a Technovit 7100 The Sliceable kit (Heraeus Kulzer, Wehrheim, Germany) according to the manufacturer's instructions. Once embedded, follicles were placed in a dissecator for at least 24 h prior to sectioning. Follicle sections (2 μm) were obtained using an ultramicrotome, mounted on glass slides, and stained with Lee Methylene Blue Basic:Fuchsin stain. Slides were coverslipped and allowed to dry for at least 24 h before evaluation. Each follicle section was examined for level of atresia as evidenced by the presence of apoptotic bodies and reported as the average of all ratings observed throughout the follicle [34–36]. Follicles were rated on a scale of 1–4 for the presence of apoptotic bodies: 1 for healthy, 2 for 1%–10%, 3 for 10%–30%, and 4 for more than 30% apoptotic bodies. All atresia ratings were assigned without knowledge of treatment group.
Measurement of E2 and Progesterone Levels
The medium from each individual follicle was collected at the end of culture for the 24-h cultures or at 96 h (during media replenishment) for the 168-h cultures and stored at −80°C. Media samples were randomly selected from all experiments and subjected to ELISA. Levels of E2 and progesterone in media were measured using kits from DRG International (Mountainside, NJ) according to the manufacturer's instructions. The minimum detection limits were 9.71 pg/ml for E2 and 0.045 ng/ml for progesterone, the intra-assay coefficients of variation (CV) were 4.7% for E2 and 6.42% for progesterone, and the interassay CVs were 7.8% for E2 and 6.63% for progesterone. Sex steroid hormone data consisted of values from 12–18 individual follicles obtained from three to six separate culture experiments. Individual follicle hormone data were normalized to follicle diameter at 24 or 96 h, as appropriate.
Quantitative RT-PCR
At the end of the culture period, follicles were immediately snap frozen and stored at −80°C for subsequent quantitative PCR (qPCR) analysis as described previously [29]. Total RNA was extracted from pooled follicles (10–16 follicles per treatment) using an RNeasy Micro Kit (Qiagen, Valencia, CA) and incubated with DNAse (Qiagen; 15 min) to eliminate potential genomic DNA contamination. The RNA concentration of each sample was determined at 260 nm using a Nanodrop ND1000 UV-Vis spectrophotometer (Nanodrop Technologies, Wilmington, DE). RNA samples (200 ng) were reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. Each cDNA sample was diluted 1:4 with nuclease-free water prior to analysis.
All qPCR experiments were carried out using a CFX96 Real-time System C1000 Thermal Cycler (Bio-Rad). As described previously [29], all qPCR reactions were done in triplicate, and each contained 1 μl of diluted cDNA, 1 μl of gene-specific primers (500 nM; Integrated DNA Technologies, Inc., Coralville, IA; Table 1), 3 μl of nuclease-free water, and 5 μl of SsoFast EvaGreen Supermix (Bio-Rad) for a final volume of 10 μl. As suggested by the manufacturer, the qPCR program consisted of an enzyme activation step (95°C for 1 min), an amplification and quantification program (40 cycles of 95°C for 10 sec, 60°C for 10 sec, single fluorescence reading), a step of 72°C for 5 min, a melt curve (65°C −95°C heating 0.5°C per sec with continuous fluorescence readings), and a final step at 72°C for 5 min.
TABLE 1.
Gene and qPCR primer sequence information.
Primers were designed using PrimerBLAST software [37], and sequence information is listed in Table 1. Primer specificity was assessed by 1) matching primer sequences with that of the gene of interest using PrimerBLAST or BLASTN 2.2.18+ [38, 39], 2) observing a single peak following melt curve analysis, and 3) confirming amplification of a single product of the correct size by agarose gel electrophoresis.
A standard curve was generated from five serial dilutions of a sample representing all treatment groups to calculate the amplification efficiencies of each primer. Expression data were generated using the mathematical model for relative quantification of real-time PCR data developed by Pfaffl [40]. The housekeeping gene β-actin (Actb) did not differ between treatments and thus was used as a reference gene. Reported data consist of mean relative mRNA expression ratios from three to four separate follicle culture experiments.
Flow Cytometry Analysis of Cell Cycle
Single cell suspensions were prepared by modifying a method previously used for whole ovaries [41]. Briefly, antral follicles (n = 30) were removed from culture and placed in a scintillation vial containing 1 ml of PBS. Collagenase type I/DNAse I solution was added to the antral follicle suspension and placed in an incubator at 37°C for 30 min. Following digestion, follicles were further dissociated by repeatedly passing them through a 16-gauge needle attached to a 5-ml syringe. The digested tissue was filtered through a 40-μm filter into a 50-ml conical tube followed by an additional 10 ml of PBS to wash the filter. The cell suspension was then centrifuged at 250 × g for 5 min, resuspended in 1 ml of PBS, and placed on ice. An aliquot of cells was removed, and cell numbers were determined to ensure that there were equal amounts of cells in each treatment.
Cell suspensions were then processed for propidium iodide staining by adding 3 ml of cold absolute ethanol (−20°C) while vortexing to prevent cell aggregation. Following ethanol fixation, cells were incubated overnight at 4°C and transferred to −20°C until staining. Prior to flow cytometry analysis, cells were washed twice with 0.1% BSA-PBS and, depending on cell concentration, received 0.3–1 ml of propidium iodide staining solution (3.8 mM sodium citrate; 40 μg/ml propidium iodide) and 15–50 μl of RNAse A solution (DNAse activity removed). Samples were kept at 4°C for at least 3 h in the dark prior to analysis. Flow cytometry data were obtained with an Accuri C6 flow cytometer (BD Biosciences, Santa Fe, CA) and analyzed using the MultiCycle add-on included with FCS Express 4 software (De Novo Software, Los Angeles, CA).
Statistical Analysis
All data were analyzed using SPSS statistical software (SPSS Inc., Chicago, IL). For all comparisons, statistical significance was assigned at P ≤ 0.05. Comparisons between DMSO and the different doses of DBP were conducted on data obtained from three to four experiments using ANOVA followed by Dunnett post hoc test. In cases when data were not parametric, comparisons between DMSO and the DBP groups were conducted using the Kruskal-Wallis nonparametric test followed by Mann-Whitney independent samples tests, as appropriate. Unpaired t-tests or Mann-Whitney nonparametric tests were used for data comparing DMSO and DBP 1000 μg/ml only. Additionally, linear regression analysis was used to test whether trends were statistically significant (i.e., Ccnd2, Ccne1, Ccnb1, Cdkn1a, and Bcl2 expression data).
RESULTS
Effect of DBP Treatment on In Vitro Antral Follicle Growth
We hypothesized that in vitro treatment with DBP inhibits antral follicle growth in culture. Antral follicles were treated in vitro with vehicle (DMSO) or increasing concentrations of DBP (1–1000 μg/ml) and their diameter measured every 24 h for a total culture time of 168 h (Fig. 1). Follicles treated with DMSO and DBP at 1, 10, and 100 μg/ml were able to grow over the 168-h culture period (P ≤ 0.05). DMSO-treated follicles significantly grew starting at 48 h until the end of the culture period, while follicles treated with DBP did not significantly increase in size until 72 h (1 μg/ml) or 96 h (10 and 100 μg/ml). Follicles treated with DBP at 1000 μg/ml were unable to grow over time and were significantly smaller than DMSO-treated follicles starting at 96 h and until the end of the culture period (168 h; P ≤ 0.05).
FIG. 1.

Effect of DBP on in vitro antral follicle growth. Antral follicles were mechanically isolated, placed in culture for 168 h, and growth was assessed as described in Materials and Methods. Data represent means ± SEM from four separate experiments, each with 8–16 follicles per treatment. Asterisks (*) indicate statistically significant differences (P < 0.05) between DBP-treated follicles and vehicle-treated follicles (DMSO).
Effect of DBP Treatment on Expression of Cell Cycle Regulators
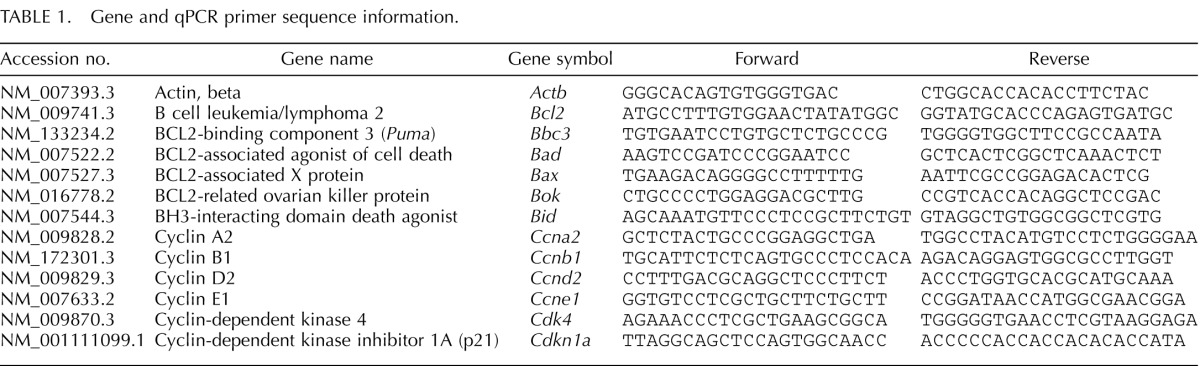
We next hypothesized that DBP-induced inhibition of antral follicle growth is caused by alterations in expression of genes that regulate the cell cycle. Therefore, at the end of 168 h, follicles were processed for analysis of gene expression by real-time qPCR. We first elected to focus on cyclin D2 (Ccnd2), cyclin-dependent kinase 4 (Cdk4), and cyclin E1 (Ccne1) because they are important regulators of the progression through G1 in the cell cycle (Fig. 2). Although there was no difference in the expression of Cdk4 between follicles treated with DMSO and DBP at 1000 μg/ml, follicles treated with DBP had significantly lower expression of Ccnd2 compared to DMSO (P ≤ 0.05). Expression of Ccne1, which is involved in the G1/S transition and S phase of the cell cycle, was not changed by DBP treatment at 168 h (data not shown).
FIG. 2.
Effect of in vitro treatment with DBP on the expression of Ccnd2 mRNA in mouse antral follicles treated in vitro for 168 h. Antral follicles were mechanically isolated and placed in culture for 168 h as described in Materials and Methods. At the end of 168 h, follicles were pooled per treatment group and processed for qPCR analysis of gene expression as described in Materials and Methods. All gene expression data were normalized to the expression of Actb. Data represent means ± SEM from three separate experiments each with 8–16 follicles per treatment. The asterisk (*) indicates a statistically significant difference (P ≤ 0.05) between DBP-treated follicles and vehicle-treated follicles (DMSO).
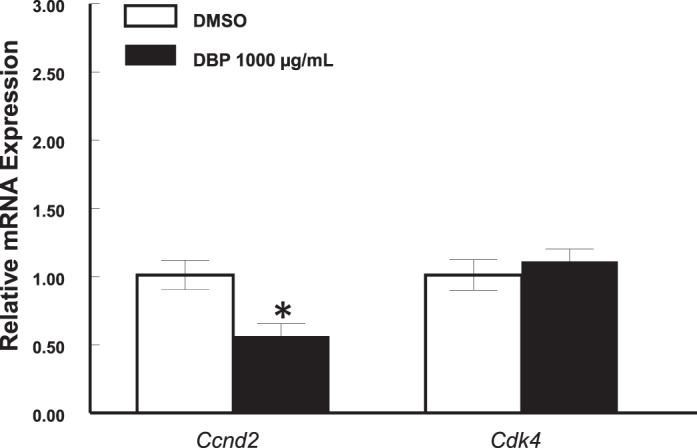
Although down-regulation of Ccnd2 may be an indication that DBP treatment may result in cell cycle arrest, whether this observation is the earliest event leading to inhibited follicle growth was not clear. Therefore, we determined whether down-regulation of Ccnd2 and other cell cycle genes occurs in the first 24 h of culture. We focused on cyclins involved in each stage of the cell cycle and compared the expression of Ccnd2, Ccne1, cyclin A2 (Ccna2; S and G2 phases), and cyclin B1 (Ccnb1; M phase) in control and DBP-treated follicles. We also determined the expression of cyclin-dependent kinase inhibitor 1A (Cdkn1a; also referred to as p21), which is a transcriptionally activated gene that leads to cell cycle arrest at the G1 to S checkpoint [42]. Interestingly, expression of all genes tested was altered by DBP treatment after a 24-h exposure. Specifically, expression of Ccnd2 (Fig. 3A) and Ccna2 (Fig. 3C) was significantly reduced in follicles treated with DBP at 100 and 1000 μg/ml (P ≤ 0.05), while Ccne1 (Fig. 3B) expression was significantly decreased by DBP in follicles treated with 10, 100, and 1000 μg/ml (P ≤ 0.05). Further, DBP at 100 (P ≤ 0.05) and 1000 μg/ml (P = 0.07, trend) resulted in reduced expression of Ccnb1 (Fig. 3D). Finally, increasing concentrations of DBP treatment resulted in increased expression of Cdkn1a (linear regression, P ≤ 0.05), but statistically significant differences compared to vehicle were observed only in follicles treated with DBP at 10 and 1000 μg/ml (P ≤ 0.05; Fig. 4).
FIG. 3.

Effect of in vitro treatment with DBP on transcript levels of four major cyclin mRNAs in mouse antral follicles treated in vitro for 24 h: A) Ccnd2. B) Ccne1. C) Ccna2. D) Ccnb1. Antral follicles were mechanically isolated and placed in culture for 24 h as described in Materials and Methods. At the end of culture, follicles were pooled per treatment group and processed for qPCR analysis of gene expression as described in Materials and Methods. All gene expression data were normalized to the expression of Actb. Data represent means ± SEM from three to four separate experiments, each with 8–16 follicles per treatment. Asterisks (*) indicate statistically significant differences (P < 0.05) between DBP-treated follicles and vehicle-treated follicles (DMSO).
FIG. 4.
Effect of in vitro treatment with DBP on the expression of Cdkn1a mRNA in mouse antral follicles treated in vitro for 24 h. Antral follicles were mechanically isolated and placed in culture for 24 h and processed for qPCR as described in Materials and Methods. All gene expression data were normalized to the expression of Actb. Data represent means ± SEM from three to four separate experiments, each with 8–16 follicles per treatment. Asterisks (*) indicate statistically significant differences (P < 0.05) between DBP-treated follicles and vehicle-treated follicles (DMSO).
Effect of DBP Treatment on the Cell Cycle Staging of Follicular Cells
Based on follicular growth and gene expression data, we hypothesized that DBP suppresses antral follicle growth by causing cell cycle arrest at the G1 phase of the cell cycle. To test this hypothesis, following treatment with DBP (1000 μg/ml) for 24 h, antral follicles were dispersed by gentle enzymatic digestion and follicular cells stained with propidium iodide for cell cycle staging by flow cytometry. Representative images of flow cytometry data are shown in Figure 5, A and 5B, and a summary of all data is shown in Figure 5C. We observed a significant accumulation of DBP-treated follicular cells in G1 (90.9 ± 0.6% gated cells; P ≤ 0.05) compared to cells from follicles treated with vehicle (82.2 ± 2.7% gated cells). Further, this accumulation resulted in decreased numbers of cells in S phase between DBP-treated follicles (6.6 ± 0.9% gated cells; P ≤ 0.05) and vehicle-treated follicles (13.5 ± 3.1% gated cells). Although not statistically significant, there was a trend for reduced number of cells in G2 between DBP (2.5 ± 0.9% gated cells) and vehicle-treated follicles (4.3 ± 0.4% gated cells; P > 0.05).
FIG. 5.

Effect of in vitro treatment with DBP on cell cycle staging of follicular cells. Antral follicles were mechanically isolated and placed in culture for 24 h and processed for flow cytometry as described in Materials and Methods. Data represent means ± SEM obtained from three separate experiments each with ≥30 follicles per treatment. Representative histograms from vehicle-treated follicles (A) and DBP-treated follicles (B). Summary data from all four experiments (C). Asterisks (*) indicate statistically significant differences (P < 0.05) between DBP-treated follicles and vehicle-treated follicles (DMSO).
Effect of DBP Treatment on Antral Follicle Atresia and Regulators of Apoptosis
We hypothesized that another consequence of treatment with DBP is follicular death by apoptosis. Cultures were stopped after 24 or 168 h and antral follicles processed for morphological evaluation of follicular atresia or gene expression analysis by qPCR. After 24 h, there were no differences in the number of apoptotic bodies (indicators of follicle atresia) present in vehicle- and DBP-treated follicles (P > 0.05; Fig. 6A). At 168 h, although there were no differences between follicles treated with DMSO and DBP at 1, 10, and 100 μg/ml, follicles treated with DBP at 1000 μg/ml had significantly more apoptotic bodies (P ≤ 0.05; Fig. 6B).
FIG. 6.

Effect of in vitro treatment with DBP on the expression of Bid and Bcl2 mRNAs and on follicular atresia in mouse antral follicles treated in vitro. Antral follicles were mechanically isolated and placed in culture for 24 h and processed for qPCR or histological evaluation of atresia as described in Materials and Methods. All gene expression data were normalized to the expression of Actb. Data represent means ± SEM from three to four separate experiments each with 8–16 follicles (qPCR) or three to four follicles (follicle atresia) per treatment. Asterisks (*) indicate statistically significant differences (P ≤ 0.05) between DBP-treated follicles and vehicle-treated follicles (DMSO). A, B) Data for atresia rating at 24 and 168 h, respectively. C, D) mRNA expression data for Bid and Bcl2 at 24 h. Atresia rating scale: 1–4 for the presence of apoptotic bodies: 1 for healthy, 2 for 1%–10%, 3 for 10%–30%, and 4 for greater than 30% apoptotic bodies.
We next determined the expression of two genes known to code proteins involved in the ovarian apoptotic pathway: BCL2-associated X protein (Bax; proapoptotic) and B-cell leukemia/lymphoma 2 (Bcl2; antiapoptotic) factor at 168 h. Follicles treated with DBP at all doses had similar expression of Bax and Bcl2 compared to vehicle-treated follicles (data not shown). Interestingly, data obtained from follicles treated with DBP at 1000 μg/ml showed a trend for increased expression of Bcl2 (data not shown).
As with expression of cell cycle genes, we elected to determine the expression of Bax, Bcl2, and other apoptosis related genes in samples exposed to DBP for 24 h. We specifically determined the expression of BCL2-associated agonist of cell death (Bad), BCL2-related ovarian killer protein (Bok), BH3-interacting domain death agonist (Bid), and BCL2 binding component 3 (Bbc3), previously known as p53 up-regulated modulator of apoptosis (Puma). Expression of Bax, Bad, and Bok was not affected by DBP at all doses tested relative to vehicle-treated follicles (p > 0.05; data not shown). Expression of Bid was not affected by DBP at 1, 10, and 100 μg/ml but was significantly increased in follicles treated with DBP at 1000 μg/ml (P ≤ 0.05; Fig. 6C). Interestingly, expression of Bbc3 was not detectable, or levels were very low in all treatments (data not shown). Finally, follicles treated with DBP showed overall lower levels of Bcl2 (linear regression, P ≤ 0.05), and this difference was statistically significant at the 10- and 1000-μg/ml doses versus vehicle-treated follicles (P ≤ 0.05; Fig. 6D).
Effect of DBP on Production of E2 and Progesterone by Isolated Antral Follicles
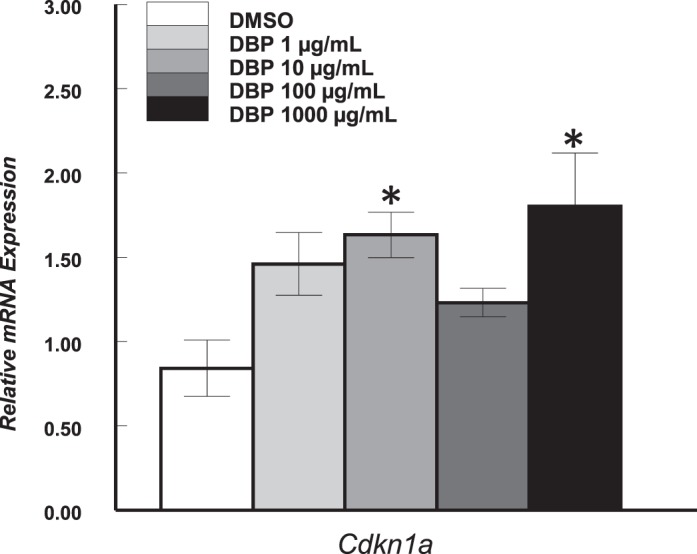
We also hypothesized that by compromising their health, DBP may decrease the production of E2 and progesterone by cultured antral follicles. Levels of E2 and progesterone were determined in media samples collected from follicles treated for 24 or 96 h in culture, normalized to follicle size, and compared between groups. After 24 h, there were no differences in the amount of E2 accumulated in the media of vehicle and DBP-treated follicles (P > 0.05; Fig. 7A). However, at 96 h, follicles treated with DBP at 1000 μg/ml had accumulated significantly less E2 in media than vehicle-treated follicles (P ≤ 0.05; Fig. 7B). There were no significant differences between controls and DBP-treated follicles in levels of progesterone at 24 h (DMSO: 0.014 ± 0.006 ng/ml; DBP1: 0.018 ± 0.009 ng/ml; DBP10: 0.017 ± 0.002 ng/ml; DBP100: 0.020 ± 0.006 ng/ml; DBP1000: 0.015 ± 0.002 ng/ ml; P > 0.05) and 96 h (DMSO: 0.048 ± 0.002 ng/ml; DBP1: 0.044 ± 0.002 ng/ml; DBP10: 0.050 ± 0.006 ng/ml; DBP100: 0.049 ± 0.007 ng/ml; DBP1000: 0.080 ± 0.018 ng/ml; P > 0.05).
FIG. 7.
Effect of in vitro treatment with DBP on levels of E2 produced by mouse antral follicles. Antral follicles were mechanically isolated and placed in culture for 24 or 168 h and E2 levels measured as described in Materials and Methods. Media samples from individual follicles were obtained at the end of the 24-h cultures (A) or during the 96-h media change of the 168-h cultures (B). Data represent means ± SEM from three to four separate experiments for a total of 12–18 follicles per treatment. The asterisk (*) indicates a statistically significant difference (P < 0.05) between DBP-treated follicles and vehicle-treated follicles (DMSO).
DISCUSSION
We used an isolated follicle culture system to study direct effects of DBP exposure on mouse ovarian antral follicles. Our main findings suggest that the effect of DBP on mouse antral follicle physiology is concentration dependent and involves alterations in transcripts that regulate cell cycle and apoptotic pathways. Specifically, we show that DBP at 1, 10, and 100 μg/ml does not alter the ability of mouse antral follicles to grow and survive in vitro. However, when given at 1000 μg/ml, DBP significantly suppresses the growth of antral follicles, causes cell cycle arrest by altering the expression of key cell cycle regulator transcripts, and induces follicular atresia by disrupting the balance between pro- and antiapoptotic transcripts.
Although this is the first study evaluating the effects of DBP on cell cycle and apoptotic pathways in antral follicles, others have reported similar effects in reproductive tissues exposed to other endocrine disruptors. In mice, DEHP has been shown to decrease Ccnd2 mRNA and protein in both male germ cells [43] and ovarian antral follicles [14]. Further, MEHP has been shown to increase apoptosis in male germ cells [44] and decrease Ccnd2 and Cdk4 mRNA in mouse antral follicles [14]. Another endocrine disruptor present in consumer products, bisphenol A (BPA), has been shown to disrupt cell cycle and apoptosis gene expression in mouse antral follicles [35] and cause apoptosis and G2/M cell cycle arrest in mouse granulosa cells [45]. In cell lines, exposure to brominated BPA leads to decreased cell viability, marked G2/M cell cycle arrest, and alterations in MAPK signaling pathways [46]. In rat granulosa cells, dihydrotestosterone treatment leads to decreased proliferation, decreased Ccnd2 mRNA, and induced cell cycle arrest in G1 [47]. Also, hexavalent chromium decreased proliferation, decreased expression of cyclins and CDKs, increased expression of CDK inhibitors [48], and induced apoptosis in rat granulosa cells [49].
In the present study, we did not expect to observe mRNA expression changes in doses that do not disrupt in vitro antral follicle growth. To our surprise, we observed that DBP exposure, regardless of concentration and effect on follicle growth, resulted in decreased expression of four major cyclin mRNAs. Therefore, it is possible that DBP causes cell cycle arrest at all doses but that somehow antral follicles treated with lower doses of DBP are capable of escaping arrest. Further, we think that antral follicles exposed to 1000 μg/ml undergo cell cycle arrest but are unable to exit arrest and subsequently undergo apoptosis. These ideas are supported by our findings showing that all DBP-treated follicles, regardless of dose, expressed greater levels of the cell cycle arrest marker Cdkn1a than controls and that, although all DBP follicles had reduced levels of the antiapoptotic gene Bcl2, only the follicles treated with 1000 μg/ml had increased transcript for the proapoptotic gene Bid when compared to vehicle-treated controls. Another finding that supports the idea of cell cycle arrest followed by apoptosis is the observation that follicles treated with DBP at 1000 μg/ml did not show histological signs of follicular atresia at 24 h when Cdkn1a was increased in all doses but had a significantly greater incidence of apoptotic bodies after 168 h in culture.
We also hypothesized that DBP-treated follicles would contain more follicular cells arrested early in the cell cycle. Along with our gene expression data, which suggested that cell cycle arrest at G1 occurs at 24 h, we observed a significant accumulation of follicular cells from DBP-treated follicles in G1 at that time point. Further, we observed that accumulation in G1 was accompanied by reductions in the number of cells in S and G2. Although no other studies have evaluated the effect of DBP on cell cycle staging in ovarian cells, Xu et al. [45] evaluated the effect of BPA on granulosa cells and observed cell cycle arrest at G2/M. These findings highlight the idea that endocrine disruptors can alter common parameters (e.g., cell cycle gene expression, follicle growth) but do so by acting via different mechanisms (e.g., G1/S vs. G2/M arrest).
Finally, we also used accumulation of E2 and progesterone as measurements of antral follicle function and health and observed that at 24 h all DBP-treated follicles had accumulated levels of both steroids that were similar to those observed in control follicles. However, at 96 h, follicles exposed to DBP at 1000 μg/ml had reduced levels of E2 in media. There were no statistically significant differences in progesterone levels at 96 h between treatments, but there was a trend for higher levels in DBP-treated follicles (1000 μg/ml). These data suggest that impaired E2 production, potentially as a result of decreased follicle health, occurs sometime after 24 h but before 96 h. Further, the lack of an inhibitory effect on progesterone production suggests that DBP may target E2 production directly. The present study constitutes the first report of DBP-induced effects on in vitro ovarian antral follicle E2 production, and it is consistent with previous studies reporting decreased in vitro antral follicle steroidogenesis following exposure to methoxychlor and its metabolites [29, 50], BPA [51], and DEHP and MEHP [14]. Further, various studies have described the ability of DBP to disrupt steroidogenesis in rodent animal models. In utero exposure to DBP decreased testicular steroidogenic enzyme activity and serum testosterone in F1 rats [52], decreased expression of proteins required for cholesterol transport and steroidogenesis [53, 54], and decreased steroidogenesis by fetal-type Leydig cells in primates and rodents [55]. In female rats, oral exposure to DBP from weaning through mating and gestation resulted in decreased ex vivo ovarian hormone production [32]. Similarly, recent studies have described the detrimental effects of DEHP and its metabolite on steroidogenesis by cultured rat fetal testis [56] and adult human testis explants [57]. Therefore, evidence exists that DBP as well as other phthalates that alter cell cycle and apoptosis regulation also affect the ability of the gonad to synthesize steroids.
In summary, we have shown that in vitro exposure to DBP alters the levels of key transcripts that regulate progression through the cell cycle, inhibition of proliferation by cell cycle arrest, and activation of apoptosis in mouse antral follicles. Further, we demonstrated that these alterations occur at DBP concentrations that do not alter antral follicle growth and thus may remain undetected. These findings are significant because they provide evidence that the ovary is a target for DBP toxicity, that DBP toxicity to antral follicles occurs at concentrations that do not cause disruptions in in vitro growth, and that mechanisms exist for antral follicles to undergo cell cycle arrest and repair or cell cycle arrest followed by apoptosis. Now that the current study has identified direct effects of DBP on ovarian antral follicles and identified key end points and pathways that become affected, subsequent studies will be aimed at conducting environmental risk assessments and further elucidating the molecular mechanisms involved.
ACKNOWLEDGMENT
The authors thank Timothy DelValle and Dr. Liying Gao for technical help and Barbara Pilas (University of Illinois, Flow Cytometry Core) for technical training.
Footnotes
Supported by National Institute on Environmental Health NIH grants K99ES021467 (Z.R.C.), R01ES019178 (J.A.F.), and T32ES007326 (W.W.); an Environmental Toxicology Fellowship (A.Z.G.); and the Billie A. Field Fellowship in Reproductive Biology (Z.R.C., W.W.). Presented, in part, at the 45th Annual Meeting of the Society for the Study of Reproduction, 12–15 August 2012, State College, Pennsylvania.
REFERENCES
- Center for the Evaluation of Risks to Human Reproduction. NTP-CERHR Expert Panel Report on Di(2-Ethylhexyl) Phthalate Alexandria, VA: Science International, Inc.; 2000. [Google Scholar]
- Third National Report on Human Exposure to Environmental Chemicals NCEH Publication No. 05–0570 Washington, DC: Department of Health and Human Services, Centers for Disease Control and Prevention; 2005. [Google Scholar]
- National Toxicology Program Report on Carcinogens, 8th ed Research Triangle Park, NC: National Toxicology Program; 1998. [Google Scholar]
- Hernández-Díaz S, Mitchell AA, Kelley KE, Calafat AM, Hauser R. Medications as a potential source of exposure to phthalates in the U.S. population. Environ Health Perspect. 2009;117:185–189. doi: 10.1289/ehp.11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldyreva MV, Klimova TS, Iziumova AS, Timofeevskaia LA. The effect of phthalate plasticizers on the generative function. Gig Tr Prof Zabol. 1975;19:25–29. [PubMed] [Google Scholar]
- Tabacova S, Little R, Balabaeva L. Maternal exposure to phthalates and complications of pregnancy. Epidemiology. 1999;10((suppl)):S127. [Google Scholar]
- Hauser R, Calafat A. Phthalates and human health. Occup Environ Med. 2005;62:806–818. doi: 10.1136/oem.2004.017590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines C, Hopf N, Deddens J, Silva M, Calafat A. Estimated daily intake of phthalates in occupationally exposed groups. J Expo Sci Environ Epidemiol. 2011;21:133–141. doi: 10.1038/jes.2009.62. [DOI] [PubMed] [Google Scholar]
- Silva M, Barr D, Reidy J, Kato K, Malek N, Hodge C, Hurtz D III, Calafat A, Needham L, Brock J. Glucuronidation patterns of common urinary and serum monoester phthalate metabolites. Arch Toxicol. 2003;77:561–567. doi: 10.1007/s00204-003-0486-3. [DOI] [PubMed] [Google Scholar]
- Qiao L, Cai D, Shen J, Gu A. Dectective of serum di-n-butyl phthalate and di-2-ethylhexyl phthalate level in normal children. Wei Sheng Yan Jiu. 2010;39:501–503. [PubMed] [Google Scholar]
- Blount B, Silva M, Caudill S, Needham L, Pirkle J, Sampson E, Lucier G, Jackson R, Brock J. Levels of seven urinary phthalate metabolites in a human reference population. Environ Health Perspect. 2000;108(10):979–982. doi: 10.1289/ehp.00108979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota K, Nishimura H. Teratogenicity of di(2-ethylhexyl) phthalate (DEHP) and di-n-butyl phthalate (DBP) in mice. Environ Health Perspect. 1982;45:65–70. doi: 10.1289/ehp.824565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BJ, Maronpot RR, Heindel JJ. Di-(2-ethylhexyl) phthalate suppresses estradiol and ovulation in cycling rats. Toxicol Appl Pharmacol. 1994;128:216–223. doi: 10.1006/taap.1994.1200. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Singh JM, Leslie TC, Meachum S, Flaws JA, Yao HH. Di-(2-ethylhexyl) phthalate and mono-(2-ethylhexyl) phthalate inhibit growth and reduce estradiol levels of antral follicles in vitro. Toxicol Appl Pharmacol. 2010;242:224–230. doi: 10.1016/j.taap.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Craig Z, Basavarajappa M, Gupta R, Flaws J. Di (2-ethylhexyl) phthalate inhibits growth of mouse antral follicles through an oxidative stress pathway. Toxicol Appl Pharmacol. 2012;258:288–295. doi: 10.1016/j.taap.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Craig Z, Basavarajappa M, Hafner K, Flaws J. Mono (2-ethylhexyl) phthalate induces oxidative stress and inhibits growth of mouse ovarian antral follicles Biol Reprod 2012. 87 152, 1 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt K, Saunders M, McPherson S, Misso M, Simpson E, Findlay J. Estrogen actions on follicle formation and early follicle development. Biol Reprod. 2004;71:1712–1723. doi: 10.1095/biolreprod.104.028175. [DOI] [PubMed] [Google Scholar]
- Hirsfield A. Development of follicles in the mammalian ovary. Int Rev Cytol. 1991;124:43–101. doi: 10.1016/s0074-7696(08)61524-7. [DOI] [PubMed] [Google Scholar]
- Levi AJ, Widra EA. Basic infertility: etiology and therapy In Seifer DB, Samuels P, Kniss DA. (eds.), The Physiologic Basis of Gynecology and Obstetrics Philadelphia: Lippincott Williams & Wilkins; 1994. 245 263 [Google Scholar]
- Bagur AC, Mautzlen C. Risk for developing osteoporosis in untreated premature menopause. Calcif Tiss Int. 1992;51:4–7. doi: 10.1007/BF00296207. [DOI] [PubMed] [Google Scholar]
- Dennerstein L, Lehert P, Burger H, Mood Dudley E. and the menopausal transition. J Nerv Ment Dis. 1999;187(11):685–691. doi: 10.1097/00005053-199911000-00006. [DOI] [PubMed] [Google Scholar]
- Hu FB, Grodstein F, Hennekens CH, Colditz GA, Johnson M, Manson JE. Age at natural menopause and risk of cardiovascular disease. Arch Intern Med. 1999;159(10):1061–1066. doi: 10.1001/archinte.159.10.1061. [DOI] [PubMed] [Google Scholar]
- Graña X, Reddy E. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs) Oncogene. 1995;11:211–219. [PubMed] [Google Scholar]
- Robker R, Richards J. Hormone-induced proliferation and differentiation of granulosa cells: a coordinate balance of the cell cycle regulators cyclin D2 and p27Kip1. Mol Endocrinol. 1998;12:924–940. doi: 10.1210/mend.12.7.0138. [DOI] [PubMed] [Google Scholar]
- Hirsfield A. Size-frequency analysis of atresia in cycling rats. Biol Reprod. 1988;38:1181–1188. doi: 10.1095/biolreprod38.5.1181. [DOI] [PubMed] [Google Scholar]
- Hengartner M. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Quirk S, Cowan R, Harman R, Hu C-L, Porter D. Ovarian follicular growth and atresia: the relationship between cell proliferation and survival. J Anim Sci. 2004;82:E40–E52. doi: 10.2527/2004.8213_supplE40x. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources. Guide for the Care and Use of Laboratory Animals Washington, DC: National Academies Press; 1996. [Google Scholar]
- Craig Z, Leslie T, Hatfield K, Gupta R, Flaws J. Mono-hydroxy methoxychlor alters levels of key sex steroids and steroidogenic enzymes in cultured mouse antral follicles. Toxicol Appl Pharmacol. 2010;249:107–113. doi: 10.1016/j.taap.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao A, Man X, Guo X, Dong H, Wang F, Sun H, Wang Y, Zhou Z, Sha J. Effects of di-n-butyl phthalate on male rat reproduction following pubertal exposure. Asian J Androl. 2011;13:702–709. doi: 10.1038/aja.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Sun W, Jing Y, Liu S, Ma Z, Hong Y, Ma L, Qin C, Liu Q, Stratton H, Xia S. Prenatal exposure to di-n-butyl phthalate induces anorectal malformations in male rat offspring. Toxicology. 2011;290:322–326. doi: 10.1016/j.tox.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Gray LE, Laskey J, Ostby J. Chronic di-n-butyl phthalate exposure in rats reduces fertility and alters ovarian function during pregnancy in female Long Evans Hooded Rats. Toxicol Sci. 2006;93:189–195. doi: 10.1093/toxsci/kfl035. [DOI] [PubMed] [Google Scholar]
- Cortvrindt R, Smitz J. Follicle culture in reproductive toxicology: a tool for in-vitro testing of ovarian function. Hum Reprod Update. 2002;8:243–254. doi: 10.1093/humupd/8.3.243. [DOI] [PubMed] [Google Scholar]
- Paulose T, Hannon P, Peretz J, Craig Z, Flaws J. Estrogen receptor alpha overexpressing mouse antral follicles are sensitive to atresia induced by methoxychlor and its metabolites. Reprod Toxicol. 2012;33:353–360. doi: 10.1016/j.reprotox.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz J, Craig Z, Flaws J. Bisphenol A inhibits follicle growth and induces atresia in cultured mouse antral follicles independently of the genomic estrogenic pathway Biol Reprod 2012. 87 63, 1 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KP, Gupta RK, Flaws JA. Methoxychlor metabolites may cause ovarian toxicity through estrogen-regulated pathways. Toxicol Sci. 2006;93:180–188. doi: 10.1093/toxsci/kfl034. [DOI] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden T. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134–145. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers E, Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden T, Schäffer A, Zhang J, Zhang J, Zhang Z, Miller W, Lipman D., Gapped BLAST. and PSI-BLAST: a new generation of protein database searcg programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley O, Kim H, El-Amouri I, Lin P, Cho J, Bani-Ahmad M, Ko C. Periovulatory leukocyte infiltration in the rat ovary. Endocrinology. 2010;151:4551–4559. doi: 10.1210/en.2009-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J, Adami G, Wei N, Keyomarsi K, Elledge S. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Li L, Jester W, Laslett A, Orth J. A single dose of di-(2-ethylhexyl) phthalate in neonatal rats alters gonocytes, reduced sertoli cell proliferation, and decreases cyclin D2 expression. Toxicol Appl Pharmacol. 2000;166:222–229. doi: 10.1006/taap.2000.8972. [DOI] [PubMed] [Google Scholar]
- Suominen J, Linderborg J, Nikula H, Hakovirta H, Parvinen M, Toppari J. The effects of mono-2-ethylhexyl phthalate, adriamycin and N-ethyl-N-nitrosourea on stage-specific apoptosis and DNA synthesis in the mouse spermatogenesis. Toxicol Lett. 2003;143:163–173. doi: 10.1016/s0378-4274(03)00170-x. [DOI] [PubMed] [Google Scholar]
- Xu J, Osuga Y, Yano T, Morita Y, Tang X, Fujiwara T, Takai Y, Matsumi H, Koga K, Taketani Y, Tsutsumi O. Bisphenol A induces apoptosis and G2-to-M arrest of ovarian granulosa cells. Biochem Biophys Res Commun. 2002;292:456–462. doi: 10.1006/bbrc.2002.6644. [DOI] [PubMed] [Google Scholar]
- Strack S, Detzel T, Wahl M, Kuch B, Krug H. Cytotoxicity of TBBPA and effects on proliferation, cell cycle and MAPK pathways in mammalian cells. Chemosphere. 2007;67:S405–S411. doi: 10.1016/j.chemosphere.2006.05.136. [DOI] [PubMed] [Google Scholar]
- Pradeep P, Li X, Peegel H, Menon K. Dihydrotestosterone inhibits granulosa cell proliferation by decreasing the cyclin D2 mRNA expression and cell cycle arrest at G1 phase. Endocrinology. 2002;143:2930–2935. doi: 10.1210/endo.143.8.8961. [DOI] [PubMed] [Google Scholar]
- Stanley J, Lee J, Nithy T, Arosh J, Burghardt R, Banu S. Chromium-VI arrests cell cycle and decreases granulosa cell proliferation by down-regulating cyclin-dependent kinases (CDK) and cyclins and up-regulating CDK-inhibitors. Reprod Toxicol. 2011;32:112–123. doi: 10.1016/j.reprotox.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu S, Stanley J, Lee J, Stephen S, Arosh J, Hoyer P, Burghardt R. Hexavalent chromium-induced apoptosis of granulosa cells involves selective sub-cellular translocation of Bcl-2 members, ERK1/2 and p53. Toxicol Appl Pharmacol. 2011;251:253–266. doi: 10.1016/j.taap.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa M, Craig Z, Hernández-Ochoa I, Paulose T, Leslie T, Flaws J. Methoxychlor reduces estradiol levels by altering steroidogenesis and metabolism in mouse antral follicles in vitro. Toxicol Appl Pharmacol. 2011;253:161–169. doi: 10.1016/j.taap.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz J, Gupta R, Singh J, Hernández-Ochoa I, Flaws J. Bisphenol A impairs follicle growth, inhibits steroidogenesis, and downregulates rate-limiting enzymes in the estradiol biosynthesis pathway. Toxicol Sci. 2011;119:209–217. doi: 10.1093/toxsci/kfq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giribabu N, Sainath S, Sreenivasula Reddy P. Prenatal di-n-butyl phthalate exposure alters reproductive functions at adulthood in male rats Environ Toxicol 2013. ( in press). Published online ahead of print 4 April 2012; doi: 10.1002/tox.21779. [DOI] [PubMed] [Google Scholar]
- Lehman K, Phillips S, Sar M, Foster P, Gaido K. Dose-dependent alterations in gene expression and testosterone synthesis in the fetal testes of male rats exposed to di(n-butyl) phthalate. Toxicol Sci. 2004;81:60–68. doi: 10.1093/toxsci/kfh169. [DOI] [PubMed] [Google Scholar]
- Thompson C, Ross S, Gaido K. Di(n-butyl) phthalate impairs cholesterol transport and steroidogenesis in the fetal rat testis through a rapid and reversible mechanism. Endocrinology. 2004;145:1227–1237. doi: 10.1210/en.2003-1475. [DOI] [PubMed] [Google Scholar]
- Hallmark N, Walker M, McKinnell C, Mahood I, Scott H, Bayne R, Coutts S, Anderson R, Greig I, Morris K, Sharpe R. Effects of monobutyl and di(n-butyl) phthalate in vitro on steroidogenesis and Leydig cell aggregation in fetal testis explants from the rat: comparison with effects in vivo in the fetal rat and neonatal marmoset and in vitro in the human. Environ Health Perspect. 2007;115:390–396. doi: 10.1289/ehp.9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvigné F, Plummer S, Lesné L, Cravedi J, Dejucq-Rainsford N, Fostier A, Jéqou B. Mono-(2-ethylhexyl) phthalate directly alters the expression of Leydig cell genes and CYP17 lyase activity in cultured rat fetal testis. PLoS One. 2011;6:e27172. doi: 10.1371/journal.pone.0027172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desdoits-Lethimonier C, Albert O, Le Bizec B, Perdu E, Zalko D, Courant F, Lesné L, Guillé F, Dejucq-Rainsford N, Jéqou B. Human testis steroidogenesis is inhibited by phthalates. Hum Reprod. 2012;27:1451–1459. doi: 10.1093/humrep/des069. [DOI] [PubMed] [Google Scholar]