

ABSTRACT
Pre-eclampsia is a life-threatening pregnancy disorder whose pathogenesis remains unclear. Plasma testosterone levels are elevated in pregnant women with pre-eclampsia and polycystic ovary syndrome, who often develop gestational hypertension. We tested the hypothesis that increased gestational testosterone levels induce hypertension via heightened angiotensin II signaling. Pregnant Sprague-Dawley rats were injected with vehicle or testosterone propionate from Gestational Day 15 to 19 to induce a 2-fold increase in plasma testosterone levels, similar to levels observed in clinical conditions like pre-eclampsia. A subset of rats in these two groups was given losartan, an angiotensin II type 1 receptor antagonist by gavage during the course of testosterone exposure. Blood pressure levels were assessed through a carotid arterial catheter and endothelium-independent vascular reactivity through wire myography. Angiotensin II levels in plasma and angiotensin II type 1 receptor expression in mesenteric arteries were also examined. Blood pressure levels were significantly higher on Gestational Day 20 in testosterone-treated dams than in controls. Treatment with losartan during the course of testosterone exposure significantly attenuated testosterone-induced hypertension. Plasma angiotensin II levels were not significantly different between control and testosterone-treated rats; however, elevated testosterone levels significantly increased angiotensin II type 1 receptor protein levels in the mesenteric arteries. In testosterone-treated rats, mesenteric artery contractile responses to angiotensin II were significantly greater, whereas contractile responses to K+ depolarization and phenylephrine were unaffected. The results demonstrate that elevated testosterone during gestation induces hypertension in pregnant rats via heightened angiotensin II type 1 receptor-mediated signaling, providing a molecular mechanism linking elevated maternal testosterone levels with gestational hypertension.
Keywords: AGTR1, angiotensin, blood pressure, losartan, mesenteric arteries, pre-eclampsia, pregnancy, testosterone, vascular function
INTRODUCTION
Pre-eclampsia is a pregnancy-specific disorder that affects 2–8% of pregnant women and is a leading cause of maternal and neonatal morbidity and mortality [1]. Pre-eclampsia is defined by the onset of hypertension and proteinuria after 20 wk of gestation and is often associated with fetal growth restriction. The pregnancy complications with abnormal fetal development not only significantly increase maternal and infant mortality and morbidity rates [2–4] but also have long-term adverse effects on adult health, predisposing individuals to cardiovascular and metabolic diseases [5–7]. The hallmark of pre-eclampsia is failure of the extravillous cytotrophoblast cells to invade and remodel the uterine spiral arterioles, leading to persistent placental hypoxia and the release of various mediators into the circulation that result in vasoconstriction and hypertension in the mother [1, 8].
Although the causes of pre-eclampsia have not been clearly defined, numerous evidence has implied a role for elevated maternal testosterone (T) levels in the pathophysiology of pre-eclampsia [9–17]. For example, increased T levels in pregnant polycystic ovary syndrome mothers increase the incidence of pre-eclampsia [18–21], and placentas of pre-eclamptic pregnancies produce increased T levels [22, 23] and express higher androgen receptor levels [22, 24]. In addition, we previously showed that increasing T levels in pregnant rats to concentrations similar to levels in pre-eclamptic pregnancies causes many of the features seen in women with pre-eclampsia, such as elevated mean arterial pressure (MAP), proteinuria, endothelial dysfunction, placental insufficiency, and reduced fetal weight [25, 26]. Increased T levels during pregnancy have also been associated with impaired placental development with significant decrease in endovascular trophoblast invasion [27] and nutrient transport capacity [26]. The molecular mechanisms by which T mediates these effects remain largely unclear.
The renin-angiotensin system (RAS) is a major determinant of blood pressure in both pregnant and nonpregnant women [28, 29]. The main effector of RAS, angiotensin II (Ang II) exerts its physiological functions through 2 7-transmembrane G protein-coupled receptors, angiotensin type 1 receptor (AGTR1) and angiotensin type 2 receptor (AGTR2) [30]. In rodents, Agtr1 exists as two isoforms, Agtr1a and Agtr1b [31], which are known to play an essential role in the elevation of blood pressure [32, 33], whereas the physiological functions of AGTR2 are not well known but are thought to exert antihypertensive effects [34].
It has been known for many years that the response to Ang II is diminished during normal pregnancy. In contrast, women who develop pre-eclampsia show an increased sensitivity to Ang II [35, 36]. In fact, increased sensitivity to Ang II is present post partum in women with a history of hypertensive pregnancy [36]. Several lines of evidence have implicated the activation of AGTR1-mediated signaling in pre-eclampsia. For example, AGTR1 is upregulated in human pre-eclamptic placentas [37]; AGTR1 expression increases in the rat placenta of endotoxin-induced pregnancy hypertension [38]; the genetic polymorphism of Agtr1, 1166C, is associated with increased risk of pre-eclampsia [39]; and Ang II hypersensitivity through AGTR1 is observed in pre-eclamptic patients [40]. These studies suggest that increased Ang II/AGTR1 may be a key molecule linking adverse stimuli with the development of pre-eclampsia.
Studies in male rats indicate that T interacts directly with the RAS, upregulating the classical constrictor pathway via upregulation of Ang II/AGTR1 pathways [41–45]. Therefore, in the present study, we investigated whether gestational increases in T induce hypertension and enhanced vascular reactivity in pregnant rats and tested the hypothesis that heightened AGTR1 signaling is a key molecular mechanism underlying T-induced increase in blood pressure.
MATERIALS AND METHODS
Animals
Pregnant Sprague-Dawley rats were purchased from Harlan-Sprague Dawley (Houston, TX) and housed in a temperature-controlled room (23°C) with a 12L:12D light-dark cycle. All experimental procedures executed in this study were in accordance with the National Institutes of Health guidelines for use and care of animals. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch. Pregnant rats were randomly divided into two groups, and T propionate (0.5 mg/kg/day; Sigma) was injected subcutaneously from Day 15 to Day 19 of gestation into one group of pregnant rats (n = 28). The control group received vehicle (sesame oil; n = 28). This dose and duration of T exposure is commonly used to mimic plasma T levels observed in pre-eclamptic women [25, 26, 46]. At Day 20 of gestation, blood pressure was measured, the animals were euthanized, plasma was separated for measurement of Ang II levels, a portion of mesenteric arteries were collected for vascular reactivity studies, and the remaining arteries were quickly frozen for RNA and protein isolation.
Experimental Procedures
Mean arterial pressure and losartan treatment.
Under isoflurane anesthesia on Day 19 of gestation, carotid arterial catheters were inserted for blood pressure measurements. The catheters inserted for this procedure were PE50 tubing, which is tunneled to the back of the neck and exteriorized. On Day 20 of gestation, MAP was analyzed after placing the rats in individual restraining cages. MAP was monitored with a pressure transducer (Cobe III transducer; CDX Sema) and recorded continuously after a 1-h stabilization period. A set of control (n = 8) and T-treated rats (n = 8) were given AGTR1 antagonist losartan (20 mg·kg−1·day−1; Sigma) by gavage [47] from day 15 to day 19 of gestation. Following losartan treatment, changes in arterial pressure were recorded as described above, using an indwelling arterial catheter.
Ang II enzyme immunoassay.
An Ang II enzyme immunoassay kit (Phoenix Pharmaceutical Inc.) was used to measure plasma Ang II concentrations. A total of 50 μl of plasma in duplicate was used for this assay. All procedures were conducted according to the assay kit instructions.
Western blotting.
Arteries were homogenized in ice-cold Radioimmunoprecipitation assay buffer (Cell Signaling Technology) containing a protease inhibitor tablet (Roche) and phosphatase inhibitor cocktail-2 and −3 (Sigma). Tissue lysates were centrifuged (14 000 × g for 10 min at 4°C), and the protein content was measured by using the BCA protein assay kit (Pierce; Thermo Scientific). The supernatant was resuspended in NuPAGE lithium dodecyl sulfate sample buffer and reducing agent (Invitrogen). Proteins (30 μg) alongside Precision Plus Standard (Kaleidoscope; Bio-Rad Laboratories) were resolved on 4–12% gradient NuPAGE Bis-Tris gels (Invitrogen) at 100 V for 2 h at room temperature and then transferred onto Immobilon-P membranes (Millipore Inc.) at 100 V for 1.5 h. The membranes were blocked with 5% bovine serum albumin (BSA) for 1 h and then incubated overnight at 4°C with primary antibodies. The primary antibodies were mouse monoclonal AGTR1 (1:500 dilutino; BD Transduction Labs) and β-actin (1:5000 dilution; Cell Signaling). After being washed, the membranes were incubated with secondary antibodies (anti-rabbit or -mouse conjugated with horseradish peroxidase) at 1:10 000 dilution and detected with the ECL detection kits (Thermo Scientific). Densitometric measurement was done using AlphaEase Flurochem 8000 software (Alpha Innotech). Results were expressed as ratios of the intensity of a specific band to that of β-actin.
Quantitative real-time PCR.
Mesenteric arteries were processed for the total RNA extraction (TRIzol; Invitrogen, Carlsbad, CA). All RNA isolates were made DNA-free by treatment with DNase and further purified with RNeasy clean-up kit (QIAGEN Inc.). Total RNA concentration and integrity were determined using a spectrophotometer (ND-1000 Nanodrop; Thermo Fisher Scientific). One microgram of total RNA was reverse transcribed using a modified Maloney murine leukemia virus-derived RT (New England Biolabs Inc.) and a blend of oligo(dT) and random hexamer primers (Invitrogen). The reaction was carried out at 28°C for 15 min and at 42°C for 50 min, then stopped by heating at 94°C for 5 min, followed by 4°C before storage at −20°C until further analysis. One microliter of the diluted cDNA corresponding to 100 ng of RNA was amplified by quantitative real-time (qRT)-PCR using FAM (Invitrogen) as the fluorophore in a CFX96 real-time thermal cycler (Bio-Rad). PCR conditions used were 2 min at 50°C for 1 cycle; 10 min at 95°C, 15 sec at 95°C, and 1 min at 60°C for 40 cycles; and a final dissociation step (0.05 sec at 65°C and 0.5 sec at 95°C). Results were calculated using the 2–ΔΔCT method and expressed in fold increase/decrease of the gene of interest in T-treated versus control rats. All reactions were performed in duplicate, and β-actin was used as an internal control. TaqMan assays were carried out in 10-μl volumes for real-time PCR at a final concentration of 250 nM TaqMan probe and 900 nM of each primer. Agtr1a (Rn01435427_m1) and Agtr1b (Rn02132799_s1) analyses were obtained by Assay-on-Demand (Applied Biosystems).
Ex vivo vascular reactivity studies.
Mesenteric arteries were removed and dissected free of adherent connective tissues. Arterial rings of 2-mm length were suspended in a Halpern-Mulvany myograph (model 610 M; Danish Myo Technology A/S) using 25-μm stainless steel wires for recording isometric contractile force (PowerLab; ADInstruments). Preparations were immersed in Krebs-Ringer bicarbonate solution (37°C, aerated with a 95% O2 /5% CO2 gas mixture, pH 7.4) of the following composition: NaCl, 118 mM; KCl, 4.7 mM; CaCl2, 2.5 mM; MgSO4, 1.2 mM; KH2PO4, 1.2 mM; NaHCO3, 25 mM; and glucose, 11.1 mM. Because we were primarily interested in studying differences in vascular smooth muscle force generation, endothelium was removed by gently rubbing the intimal surface of rings with a tungsten wire. Endothelium removal was verified by the absence of relaxation to acetylcholine (10 μM) in rings precontracted with phenylephrine (PE; 3μM). It should be noted that we have previously shown that pregnant rats with elevated T levels have endothelial dysfunction; thus, leaving the endothelium intact would have complicated any assessment of vascular smooth muscle function. The rings were allowed to equilibrate in Krebs-Ringer solution for 1 h at a resting tension. After stabilization, the rings were normalized to an internal diameter of 0.9 of L13.3kPa using a normalization software package (Myodata; Danish Myotechnology). Dose-response curves were constructed using cumulative doses of KCl (10–120 mM; Sigma), phenylephrine (10−9 to 10−5 M; Sigma) and Ang II (10−13 to 10−8M; Sigma). Ang II dose responses were also measured in the presence of losartan (10 μM). Antagonist was added to the bath 30 min before cumulative dose-response curves. Only one cumulative dose-response curve to Ang II was obtained per arterial ring, as tachyphylaxis developed if a second dose response was constructed.
Statistical Analysis
For comparison of arterial pressure levels, analysis was performed using ANOVA, with adjustments for multiple comparisons. For comparison of genes and proteins expressed between the control and T-treated groups, unpaired Student t-test was used. Cumulative concentration-response curves were analyzed by computer fitting to a 4-parameter sigmoid curve, using Prism 6 software (GraphPad, San Diego, CA) to evaluate the half-maximal effective concentration (EC50) and Emax, the maximum asymptote of the curve. All values are means ± SEM. A P value of <0.05 was considered significant.
RESULTS
Plasma T levels
Plasma T levels were 2-fold increased in T-injected rats (2.0 ± 0.27 ng/ml; n = 7) compared to those in controls (1.0 ± 0.23 ng/ml; n = 7; P < 0.05).
Mean Arterial Pressure
MAP was increased significantly in T-exposed rats (104 ± 4.4 mm Hg; n = 7) compared with control rats (90 ± 2.4 mm Hg; n = 11) (Fig. 1; P < 0.05).
FIG. 1.
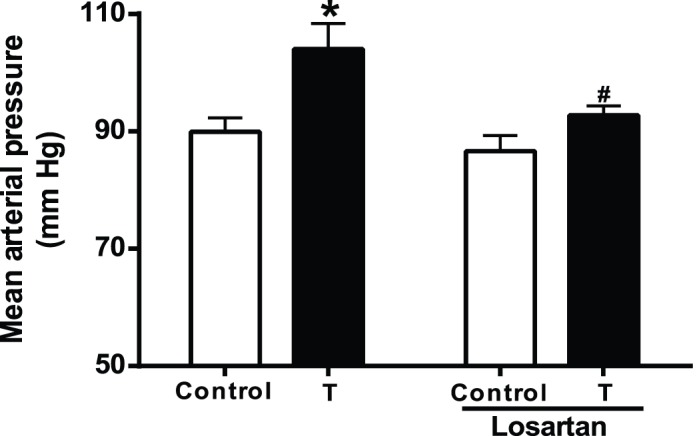
Changes in blood pressure in control and T-exposed pregnant rats. Mean arterial pressure was measured in conscious, free-moving control (n = 11) and T-treated rats (0.5 mg/kg/day, subcutaneously from Gestational Day 15–19; n = 7) through carotid arterial catheters. Another set of control T rats was treated with losartan (20 mg·kg−1·day−1; n = 6 in each group) by gavage during the course of T exposure, and then mean arterial pressure was recorded. Data points means ± SEM. *P < 0.05 versus untreated control. #P < 0.05 versus untreated T group.
Losartan administration attenuated the blood pressure increase associated with elevated T levels. MAP levels were no different in losartan-treated T-exposed rats than in losartan-treated control rats. MAP was 93 ± 1.6 mm Hg (n = 8) in losartan control pregnant rats versus 87 ± 2.7 mm Hg (n = 8) in losartan-treated T-exposed pregnant rats (Fig. 1, P < 0.05).
Plasma Ang II Levels
To determine whether circulating Ang II levels were correlated with increased blood pressure in T-treated rats, plasma Ang II levels were determined with enzyme immunoassay. As shown in Figure 2, plasma Ang II levels were not significantly different between control and T-treated rats (n = 8 in each group).
FIG. 2.
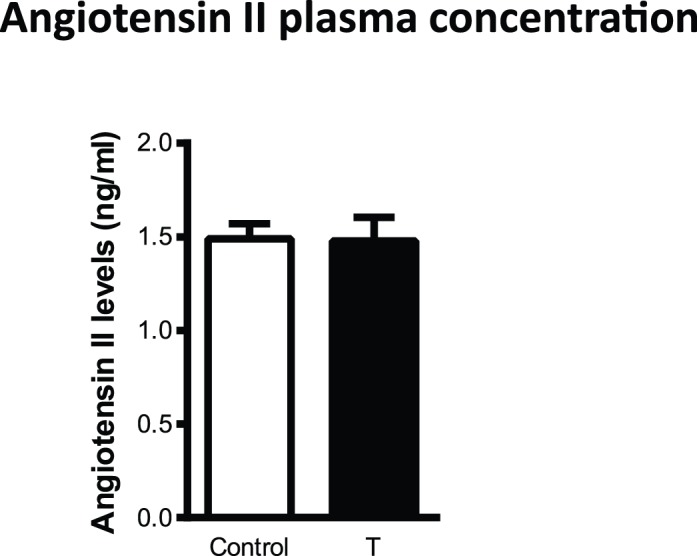
No changes in plasma angiotensin II levels were detected between control and T-exposed pregnant rats. Plasma angiotensin II levels were measured in control (n = 8) and T-treated rats (n = 8) by using enzyme immunoassay. Data points are means ± SEM.
Arterial AGTR1 Expression
We next determined whether AGTR1 expression correlated with the increased blood pressure in T-treated rats. As shown in Figure 3, Western blotting showed that AGTR1 protein levels were significantly increased in the mesenteric arteries from T-treated rats (n = 6) compared with those from the control (n = 6) (Fig. 3, P < 0.05).
FIG. 3.
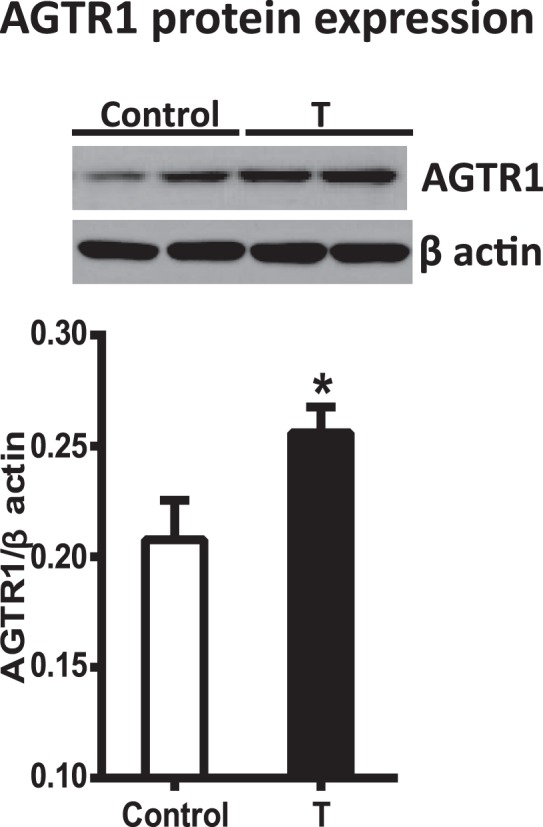
Changes in angiotensin receptor protein levels in mesenteric arteries from control and T-exposed pregnant rats. Representative Western blots for AGTR1 are shown at the top; blot density obtained from densitometric scanning of AGTR1 normalized to β-actin is shown at the bottom. Values are means ± SEM of 6 rats in each group. *P ≤ 0.05 versus control.
At the mRNA level, rodents possess two Agtr1 receptor isoforms, designated Agtr1a and Agtr1b. Because no specific antibodies are available to detect these Agtr1 isoforms, we used qRT-PCR to measure the mRNA levels of Agtr1a and Agtr1b in the mesenteric arteries. The expression of Agtr1a mRNA in mesenteric arteries in control was comparable to that in T-treated rats (Fig. 4; n = 7 in each group). However, expression of Agtr1b mRNA in the mesenteric arteries was 2.8-fold increased in T-treated rats (n = 7) compared to that in controls (Fig. 4; n = 7; P < 0.05).
FIG. 4.
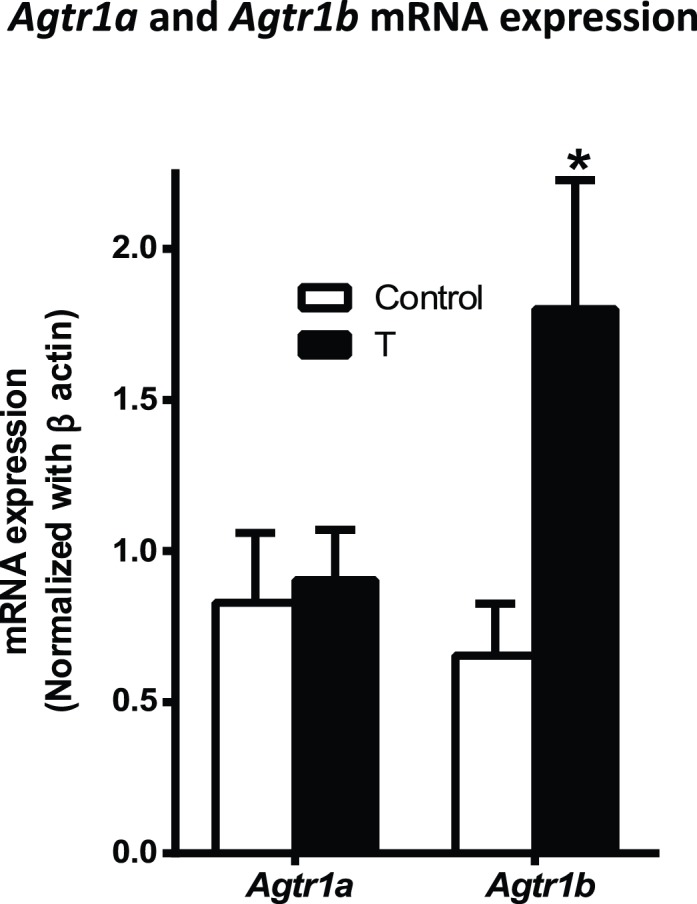
Changes in mRNA levels of angiotensin type 1 receptor isoforms in mesenteric arteries from control and T-exposed pregnant rats. Real-time PCR was used to assess vascular Agtr1a and Agtr1b mRNA expression levels. Quantitation of vascular Agtr1a and Agtr1b mRNA expression was normalized relative to that of β-actin. Values are means ± SEM of 7 rats in each group. *P < 0.05 versus control.
Vasoconstrictor Response
Vascular contractile responses to cumulative doses of KCl, a determination of depolarization-induced vessel contraction, and PE, a measurement of α1-adrenoceptor-induced contraction, in arterial segments from T rats were similar to those in controls (Fig. 5, A and B; n = 8 in each group). However, presence of elevated T levels exaggerated the Ang II-induced contractile responses with a leftward shift in the dose-response curves (pD2 = 8.8 ± 0.05 vs. 8.4 ± 0.02 in controls) as well as an increase in maximal responses (Emax = 54.1 ± 3.52% vs. 41.6 ± 3.65% in controls) (Fig. 5C; n = 8 in each group; P < 0.05). Pretreatment of the vascular rings with losartan completely inhibited the Ang II-induced vasoconstriction in arterial segments from T-treated rats and controls (Fig. 5C; n = 5).
FIG. 5.

Contractile responses in mesenteric arterial rings of control and T-exposed pregnant rats. Endothelium-denuded mesenteric arterial rings were incubated in Krebs buffer, and then vascular contractile responses were taken to cumulative additions of KCl (A), phenylephrine (B), and Ang II (C). Contractions to Ang II were also taken in the presence of losartan (10 μM). K+ depolarizing contractions are presented as absolute tension (mN/mm) and as the percentage of maximal KCl contractions. Phenylephrine and angiotensin II contractions are presented as percentages of their maximal contractions, as well as percentages of 80 mM KCl contractions. Values are means ± SEM. Mesenteric arterial rings (n = 10 to 12) from 8 rats of each group were used.
DISCUSSION
The major finding of this study was that at clinically relevant concentrations, the elevated maternal T levels during pregnancy led to increased blood pressure in pregnant rats. Our study demonstrates for the first time that elevated maternal T upregulates mesenteric vascular Agtr1b mRNA transcripts relating to the increase in blood pressure. Blockade of AGTR1 with losartan reversed blood pressure increase in pregnant rats with elevated T, suggesting that RAS is a cause and not a consequence of blood pressure increase. The increased Agtr1b expression in the mesenteric arteries was associated with exaggerated vascular contractile responses in an agonist-specific manner. The mesenteric vasomotor response mediated by Ang II was greater, and responses to other vasoconstrictors, KCl and phenylephrine, were not increased in pregnant rats with elevated T compared to that in controls. Therefore, we suggest that increases in vascular AGTR1- and Ang II-stimulated responses may mediate the development and maintenance of hypertension in pregnant rats exposed to elevated T levels.
Although pre-eclampsia is one of the major causes of maternal and perinatal mortality and morbidity, the pathophysiology of this disease has yet to be completely understood. The initiating event of the disease is associated with abnormal cytotrophoblast invasion, resulting in inadequate remodeling of the uterine spiral arteries and reduced blood flow to the uteroplacental unit [1, 8]. The poorly perfused and hypoxic placenta is thought to release factors that result in vasoconstriction and hypertension in the mother. Placentas of pre-eclamptic pregnancies contribute increased T production [22, 23]. In addition, placental androgen receptor levels are upregulated in pre-eclamptic pregnancies [22, 24]. We previously reported that elevated maternal T levels are an important stimulus for increased blood pressure in pregnant rats [25]. We not only confirmed that elevated T levels in pregnant rats increase arterial pressure, we also provided novel data showing that treatment with a selective AGTR1 antagonist, losartan, abolished the hypertensive response to T during pregnancy. This suggests that AGTR1 activation contributes, in part, to the increase in blood pressure in T-exposed hypertensive rats. However, plasma Ang II level, the main effector of RAS, was not significantly altered by gestational T exposure, suggesting that circulating Ang II may not contribute to the increased AGTR1 activation. The present study demonstrated AGTR1 protein expression increased in mesenteric arteries of T rats. This is likely to contribute to the increased blood pressure response to Ang II in T rats.
At the mRNA level, Agtr1 exists as two isoforms, Agtr1a and Agtr1b [31]. In the present study, we demonstrated that both receptor subtypes are present in abundance in the mesenteric arteries; however, T selectively upregulates the expression of Agtr1b receptors, in contrast to the reported specific effect of androgen to regulate Agtr1a mRNA abundance in abdominal aortas [48]. These results suggest that T exhibits tissue- and/or cell-specific regulation of Agtr1. Interestingly, recent studies demonstrated that smooth muscle cells of the mesentery artery are serosal mesothelium-derived, whereas smooth muscle cells of the abdominal aorta are somite-derived [49]. These differences in smooth muscle embryonic origins may have contributed to regional differences in Agtr1a regulation by T. Given that receptor subtypes Agtr1a and Agtr1b exhibit differential cell and tissue distribution, differences in the promoters of these distinct genes may contribute to T-specific effects to increase Agtr1b mRNA abundance. Because Agtr1b has been demonstrated to play a prominent role in Ang II-induced contractile responses [32, 33], the finding that gestational elevation in T levels causes selective upregulation of Agtr1b in mesenteric arteries indicates that Agtr1b may contribute to T-induced increases in vasoconstriction and blood pressure. Indeed, the present finding that treatment with the AGTR1-selective antagonist losartan during the course of T exposure abrogated T-induced blood pressure increases in pregnant rats, indicating a causative role for heightened Ang II/AGTR1-mediated signaling in the pathogenesis of T-induced vasoconstriction and blood pressure increases in pregnant rats. Possible mechanisms of T-dependent upregulation of Agtr1b receptor mRNA expression include modulation of receptor transcription through androgen response elements. Preliminary analysis of the Agtr1 promoter reveals the presence of partial androgen response elements. T may also activate Agtr1 transcription from alternative response elements such as the glucocorticoid response element [50, 51], by enhancing assembly of general transcription factors including TATA-box-binding protein [52], activating activator protein-1, nuclear factor-kappa β [53, 54], or increasing mRNA stability [55]. In addition, T-induced activation of Rho kinase [56], mitogen-activated protein kinases [57], and phosphatidylinositol-4,5-bisphosphate 3-kinase [58] could also contribute to Agtr1 upregulation [59, 60]. The exact mechanism by which T regulates Agtr1 remains to be elucidated.
Consistent with increases in mesenteric vascular Agtr1b expression, other studies also show an AGTR1 increase in kidney and brain. To dissect out the contribution of the vasculature to contractile responses and to blood pressure increase, we examined vascular reactivity to Ang II. In the present study, Ang II-induced contractile responses were significantly increased in T-treated rats. The ED50 and Emax were greater in the T-treated rats and were related to the blood pressure increases. The inhibition of Ang II-induced arterial contractions by losartan in both control and T-treated animals indicated a primary role for AGTR1 in vasoconstrictions. Because elevated T caused increases in Ang II-induced contractions in the absence of functional endothelium, we suggest the enhanced arterial sensitivity to Ang II occurred primarily in the vascular smooth muscle cells. Other studies in male hypertensive rats have also reported exaggerated vasomotor responses to Ang II in renal arteries [56, 61, 62]. Interestingly, the vasomotor response to other potent constrictors, such as K+ depolarization and phenylephrine, was not enhanced in T-treated rats. Thus, it is likely that the effect of elevated T on vasoconstrictors is agonist specific. These findings suggest that elevated T-mediated increases in vasoconstriction and blood pressure occur at the agonist-specific level rather than at common intracellular signaling pathways. Although we focused on the vasculature in this study, enhanced Ang II pathway can also contribute to hypertension through sympathoadrenal-mediated mechanism. This warrants further investigation.
In conclusion, results from this study demonstrate that T positively regulates Agtr1b mRNA abundance in mesenteric arteries and that this effect relates to development of hypertension in pregnant dams. The exaggerated vasoconstriction in pregnant rats with elevated T appears to be specific to Ang II. This suggests that vascular RAS, in addition to central and intrarenal RAS, may play an important role in the development and maintenance of hypertension during pregnancy. Therefore, an adequate repression of the vascular AGTR1-mediated pathway in pregnancy might be effective for improving pregnancy-induced hypertension.
Growing evidence indicates a role for elevated T levels in pre-eclampsia, yet whether T is a causal factor and is involved in the pathogenesis of pre-eclampsia remains unclear. The present study demonstrates that elevated T levels at concentrations found under clinical conditions during gestation induce exaggerated vasoconstriction and hypertension in pregnant rats via heightened AGTR1-mediated signaling, providing a molecular mechanism linking gestational T excess with increased risk of pre-eclampsia. Although caution should always be observed in extrapolating the findings of animal studies directly to humans, the present finding has translational potential and provides a mechanistic understanding worthy of investigation in humans. Strategies that target excessive androgen or AGTR1 action in the systemic circulation could have important therapeutic potential in treatment of pregnancies complicated by hypertension.
Footnotes
Supported by National Institutes of Health grants HD069750 and HL119869 to K.S. and HL102866 and HL58144 to C.Y. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH.
REFERENCES
- Steegers EA. von DP, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376:631–644. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- Lackman F, Capewell V, Richardson B, daSilva O, Gagnon R. The risks of spontaneous preterm delivery and perinatal mortality in relation to size at birth according to fetal versus neonatal growth standards. Am J Obstet Gynecol. 2001;184:946–953. doi: 10.1067/mob.2001.111719. [DOI] [PubMed] [Google Scholar]
- Morsing E, Marsal K. Pre-eclampsia—an additional risk factor for cognitive impairment at school age after intrauterine growth restriction and very preterm birth. Early Hum Dev. 2014;90:99–101. doi: 10.1016/j.earlhumdev.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Romo A, Carceller R, Tobajas J. Intrauterine growth retardation (IUGR): epidemiology and etiology. Pediatr Endocrinol Rev. 2009;6((suppl 3)):332–336. [PubMed] [Google Scholar]
- Fraser A, Nelson SM, Macdonald-Wallis C, Sattar N, Lawlor DA. Hypertensive disorders of pregnancy and cardiometabolic health in adolescent offspring. Hypertension. 2013;62:614–620. doi: 10.1161/HYPERTENSIONAHA.113.01513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnold M, Tamayo E, Kechichian T, Gamble P, Longo M, Hankins GD, Saade GR, Costantine MM. The effect of prenatal pravastatin treatment on altered fetal programming of postnatal growth and metabolic function in a preeclampsia-like murine model. Am J Obstet Gynecol. 2014;210:542.e1–7. doi: 10.1016/j.ajog.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Ryckman KK, Borowski KS, Parikh NI, Saftlas AF. Pregnancy complications and the risk of metabolic syndrome for the offspring. Curr Cardiovasc Risk Rep. 2013;7:217–223. doi: 10.1007/s12170-013-0308-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumwell S, Karumanchi SA. Pre-eclampsia: clinical manifestations and molecular mechanisms. Nephron Clin Pract. 2007;106:c72–c81. doi: 10.1159/000101801. [DOI] [PubMed] [Google Scholar]
- Atamer Y, Erden AC, Demir B, Kocyigit Y, Atamer A. The relationship between plasma levels of leptin and androgen in healthy and preeclamptic pregnant women. Acta Obstet Gynecol Scand. 2004;83:425–430. doi: 10.1111/j.0001-6349.2004.00276.x. [DOI] [PubMed] [Google Scholar]
- Ficicioglu C, Kutlu T. The role of androgens in the aetiology and pathology of pre-eclampsia. J Obstet Gynaecol. 2003;23:134–137. doi: 10.1080/0144361031000074637. [DOI] [PubMed] [Google Scholar]
- Ghorashi V, Sheikhvatan M. The relationship between serum concentration of free testosterone and pre-eclampsia. Endokrynol Pol. 2008;59:390–392. [PubMed] [Google Scholar]
- Lorzadeh N, Kazemirad S. The effects of fetal gender on serum human chorionic gonadotropin and testosterone in normotensive and preeclamptic pregnancies J Pregnancy 2012. 2012 874290. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Salamalekis E, Bakas P, Vitoratos N, Eleptheriadis M, Creatsas G. Androgen levels in the third trimester of pregnancy in patients with preeclampsia. Eur J Obstet Gynecol Reprod Biol. 2006;126:16–19. doi: 10.1016/j.ejogrb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Serin IS, Kula M, Basbug M, Unluhizarci K, Gucer S, Tayyar M. Androgen levels of preeclamptic patients in the third trimester of pregnancy and six weeks after delivery. Acta Obstet Gynecol Scand. 2001;80:1009–1013. doi: 10.1034/j.1600-0412.2001.801107.x. [DOI] [PubMed] [Google Scholar]
- Sharifzadeh F, Kashanian M, Fatemi F. A comparison of serum androgens in pre-eclamptic and normotensive pregnant women during the third trimester of pregnancy. Gynecol Endocrinol. 2012;28:834–836. doi: 10.3109/09513590.2012.683061. [DOI] [PubMed] [Google Scholar]
- Miller NR, Garry D, Cohen HW, Figueroa R. Serum androgen markers in preeclampsia. J Reprod Med. 2003;48:225–229. [PubMed] [Google Scholar]
- Jirecek S, Joura EA, Tempfer C, Knofler M, Husslein P, Zeisler H. Elevated serum concentrations of androgens in women with pregnancy-induced hypertension. Wien Klin Wochenschr. 2003;115:162–166. doi: 10.1007/BF03040303. [DOI] [PubMed] [Google Scholar]
- Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Perez-Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002;17:2573–2579. doi: 10.1093/humrep/17.10.2573. [DOI] [PubMed] [Google Scholar]
- Naver K, Grinsted J, Larsen S, Hedley P, Jorgensen F, Christiansen M, Nilas L. Increased risk of preterm delivery and pre-eclampsia in women with polycystic ovary syndrome and hyperandrogenaemia. BJOG. 2014;30:311–315. doi: 10.1111/1471-0528.12558. [DOI] [PubMed] [Google Scholar]
- Qin JZ, Pang LH, Li MJ, Fan XJ, Huang RD, Chen HY. Obstetric complications in women with polycystic ovary syndrome: a systematic review and meta-analysis Reprod Biol Endocrinol 2013. 11 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjerulff LE, Sanchez-Ramos L, Duffy D. Pregnancy outcomes in women with polycystic ovary syndrome: a metaanalysis. Am J Obstet Gynecol. 2011;204:558.e1–558.e6. doi: 10.1016/j.ajog.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Sathishkumar K, Balakrishnan M, Chinnathambi V, Chauhan M, Hankins GD, Yallampalli C. Fetal sex-related dysregulation in testosterone production and their receptor expression in the human placenta with preeclampsia. J Perinatol. 2011;32:328–335. doi: 10.1038/jp.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertig A, Liere P, Chabbert-Buffet N, Fort J, Pianos A, Eychenne B, Cambourg A, Schumacher M, Berkane N, Lefevre G, Uzan S, Rondeau E, et al. Steroid profiling in preeclamptic women: evidence for aromatase deficiency. Am J Obstet Gynecol. 2010;203:477–479. doi: 10.1016/j.ajog.2010.06.011. [DOI] [PubMed] [Google Scholar]
- Hsu TY, Lan KC, Tsai CC, Ou CY, Cheng BH, Tsai MY, Kang HY, Tung YH, Wong YH, Huang KE. Expression of androgen receptor in human placentas from normal and preeclamptic pregnancies. Taiwan J Obstet Gynecol. 2009;48:262–267. doi: 10.1016/S1028-4559(09)60301-6. [DOI] [PubMed] [Google Scholar]
- Chinnathambi V, Balakrishnan M, Ramadoss J, Yallampalli C, Sathishkumar K. Testosterone alters maternal vascular adaptations: role of the endothelial NO system. Hypertension. 2013;61:647–654. doi: 10.1161/HYPERTENSIONAHA.111.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Chinnathambi V, Gao H, Hankins GD, Yallampalli C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod Biol Endocrinol. 2011;9:1–12. doi: 10.1186/1477-7827-9-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomba S, Russo T, Falbo A, Di CA, Amendola G, Mazza R, Tolino A, Zullo F, Tucci L, La Sala GB. Decidual endovascular trophoblast invasion in women with polycystic ovary syndrome: an experimental case-control study. J Clin Endocrinol Metab. 2012;97:2441–2449. doi: 10.1210/jc.2012-1100. [DOI] [PubMed] [Google Scholar]
- August P, Lenz T, Ales KL, Druzin ML, Edersheim TG, Hutson JM, Muller FB, Laragh JH, Sealey JE. Longitudinal study of the renin-angiotensin-aldosterone system in hypertensive pregnant women: deviations related to the development of superimposed preeclampsia. Am J Obstet Gynecol. 1990;163:1612–1621. doi: 10.1016/0002-9378(90)90639-o. [DOI] [PubMed] [Google Scholar]
- Brown MA, Nicholson E, Gallery ED. Sodium-renin-aldosterone relations in normal and hypertensive pregnancy. Br J Obstet Gynaecol. 1988;95:1237–1246. doi: 10.1111/j.1471-0528.1988.tb06812.x. [DOI] [PubMed] [Google Scholar]
- Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JA, Smith RD. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- Yoshida H, Kakuchi J, Guo DF, Furuta H, Iwai N. van der Meer-de Jong, Inagami T, Ichikawa I. Analysis of the evolution of angiotensin II type 1 receptor gene in mammals (mouse, rat, bovine and human) Biochem Biophys Res Commun. 1992;186:1042–1049. doi: 10.1016/0006-291x(92)90852-c. [DOI] [PubMed] [Google Scholar]
- Poduri A, Owens AP, III, , Howatt DA, Moorleghen JJ, Balakrishnan A, Cassis LA, Daugherty A. Regional variation in aortic AT1b receptor mRNA abundance is associated with contractility but unrelated to atherosclerosis and aortic aneurysms PLoS One 2012. 7 e48462 1- e48462 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swafford AN, Jr, , Harrison-Bernard LM, Dick GM. Knockout mice reveal that the angiotensin II type 1B receptor links to smooth muscle contraction. Am J Hypertens. 2007;20:335–337. doi: 10.1016/j.amjhyper.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Coffman TM. Gene targeting in physiological investigations: studies of the renin-angiotensin system. Am J Physiol. 1998;274:F999–1005. doi: 10.1152/ajprenal.1998.274.6.F999. [DOI] [PubMed] [Google Scholar]
- Gant NF, Daley GL, Chand S, Whalley PJ, MacDonald PC. A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Invest. 1973;52:2682–2689. doi: 10.1172/JCI107462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena AR, Karumanchi SA, Brown NJ, Royle CM, McElrath TF, Seely EW. Increased sensitivity to angiotensin II is present postpartum in women with a history of hypertensive pregnancy. Hypertension. 2010;55:1239–1245. doi: 10.1161/HYPERTENSIONAHA.109.147595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PS, Tsai SJ, Wallukat G, Leung TN, Lau TK. The upregulation of angiotensin II receptor AT(1) in human preeclamptic placenta. Mol Cell Endocrinol. 2001;184:95–102. doi: 10.1016/s0303-7207(01)00637-2. [DOI] [PubMed] [Google Scholar]
- Doering TP, Haller NA, Montgomery MA, Freeman EJ, Hopkins MP. The role of AT1 angiotensin receptor activation in the pathogenesis of preeclampsia. Am J Obstet Gynecol. 1998;178:1307–1312. doi: 10.1016/s0002-9378(98)70337-0. [DOI] [PubMed] [Google Scholar]
- Nalogowska-Glosnicka K, Lacka BI, Zychma MJ, Grzeszczak W, Zukowska-Szczechowska E, Poreba R, Michalski B, Kniazewski B, Rzempoluch J. Angiotensin II type 1 receptor gene A1166C polymorphism is associated with the increased risk of pregnancy-induced hypertension. Med Sci Monit. 2000;6:523–529. [PubMed] [Google Scholar]
- Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Kost CK, Jr., , Martin DS. Androgens augment renal vascular responses to ANG II in New Zealand genetically hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1608–R1615. doi: 10.1152/ajpregu.00364.2005. [DOI] [PubMed] [Google Scholar]
- Ojeda NB, Royals TP, Black JT, Dasinger JH, Johnson JM, Alexander BT. Enhanced sensitivity to acute angiotensin II is testosterone dependent in adult male growth-restricted offspring. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1421–R1427. doi: 10.1152/ajpregu.00096.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freshour JR, Chase SE, Vikstrom KL. Gender differences in cardiac ACE expression are normalized in androgen-deprived male mice. Am J Physiol Heart Circ Physiol. 2002;283:H1997–H2003. doi: 10.1152/ajpheart.01054.2001. [DOI] [PubMed] [Google Scholar]
- Hilliard LM, Sampson AK, Brown RD, Denton KM. The “his and hers” of the renin-angiotensin system. Curr Hypertens Rep. 2013;15:71–79. doi: 10.1007/s11906-012-0319-y. [DOI] [PubMed] [Google Scholar]
- Leung PS, Wong TP, Chung YW, Chan HC. Androgen dependent expression of AT1 receptor and its regulation of anion secretion in rat epididymis. Cell Biol Int. 2002;26:117–122. doi: 10.1006/cbir.2001.0830. [DOI] [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Yallampalli U, Balakrishnan M, Yallampalli C. Fetal programming of adult hypertension in female rat offspring exposed to androgens in utero. Early Hum Dev. 2011;87:407–414. doi: 10.1016/j.earlhumdev.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalk P, Sharkovska Y, Kashina E, von Websky K, Relle K, Pfab T, Alter M, Guillaume P, Provost D, Hoffman K, Fischer Y, Hocher B. Endothelin-converting enzyme/neutral endopeptidase inhibitor SLV338 prevents hypertensive cardiac remodeling in a blood pressure-independent manner. Hypertension. 2011;57:755–763. doi: 10.1161/HYPERTENSIONAHA.110.163972. [DOI] [PubMed] [Google Scholar]
- Henriques T, Zhang X, Yiannikouris FB, Daugherty A, Cassis LA. Androgen increases AT1a receptor expression in abdominal aortas to promote angiotensin II-induced AAAs in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1251–1256. doi: 10.1161/ATVBAHA.107.160382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasteson P, Johansson BR, Jukkola T, Breuer S, Akyurek LM, Partanen J, Lindahl P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development. 2008;135:1823–1832. doi: 10.1242/dev.020958. [DOI] [PubMed] [Google Scholar]
- Adler AJ, Danielsen M, Robins DM. Androgen-specific gene activation via a consensus glucocorticoid response element is determined by interaction with nonreceptor factors. Proc Natl Acad Sci U S A. 1992;89:11660–11663. doi: 10.1073/pnas.89.24.11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdarina IG, King PJ, Clark AJ. Characterization of the angiotensin (AT1b) receptor promoter and its regulation by glucocorticoids. J Mol Endocrinol. 2009;43:73–80. doi: 10.1677/JME-09-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan IJ, Gustafsson J. Interaction of the human androgen receptor transactivation function with the general transcription factor TFIIF. Proc Natl Acad Sci U S A. 1997;94:8485–8490. doi: 10.1073/pnas.94.16.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DF, Inagami T. The genomic organization of the rat angiotensin II receptor AT1B. Biochim Biophys Acta. 1994;1218:91–94. doi: 10.1016/0167-4781(94)90105-8. [DOI] [PubMed] [Google Scholar]
- Lee DK, Chang C. Molecular communication between androgen receptor and general transcription machinery. J Steroid Biochem Mol Biol. 2003;84:41–49. doi: 10.1016/s0960-0760(03)00005-0. [DOI] [PubMed] [Google Scholar]
- Nickenig G, Strehlow K, Wassmann S, Baumer AT, Albory K, Sauer H, Bohm M. Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation. 2000;102:1828–1833. doi: 10.1161/01.cir.102.15.1828. [DOI] [PubMed] [Google Scholar]
- Song J, Kost CK, Jr, , Martin DS. Androgens potentiate renal vascular responses to angiotensin II via amplification of the Rho kinase signaling pathway. Cardiovasc Res. 2006;72:456–463. doi: 10.1016/j.cardiores.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Fix C, Jordan C, Cano P, Walker WH. Testosterone activates mitogen-activated protein kinase and the cAMP response element binding protein transcription factor in Sertoli cells. Proc Natl Acad Sci U S A. 2004;101:10919–10924. doi: 10.1073/pnas.0404278101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Yang L, Feldman RI, Sun XM, Bhalla KN, Jove R, Nicosia SV, Cheng JQ. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278:42992–43000. doi: 10.1074/jbc.M306295200. [DOI] [PubMed] [Google Scholar]
- Bagi Z, Feher A, Cassuto J, Akula K, Labinskyy N, Kaley G, Koller A. Increased availability of angiotensin AT 1 receptors leads to sustained arterial constriction to angiotensin II in diabetes - role for Rho-kinase activation. Br J Pharmacol. 2011;163:1059–1068. doi: 10.1111/j.1476-5381.2011.01307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol. 2009;296:H1425–H1433. doi: 10.1152/ajpheart.00942.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Eyster KM, Kost CK, Jr, , Kjellsen B, Martin DS. Involvement of protein kinase C-CPI-17 in androgen modulation of angiotensin II-renal vasoconstriction. Cardiovasc Res. 2010;85:614–621. doi: 10.1093/cvr/cvp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM, Endemann D, He G, Li JS, Schiffrin EL. Role of AT2 receptors in angiotensin II-stimulated contraction of small mesenteric arteries in young SHR. Hypertension. 1999;33:366–372. doi: 10.1161/01.hyp.33.1.366. [DOI] [PubMed] [Google Scholar]