

ABSTRACT
Diet-induced obesity induces immune cell infiltration and inflammation in peri-ovarian adipose tissue and mRNA expression of inflammatory markers in ovarian tissue. Whether these changes are associated with obesity-related ovarian dysfunction remains unknown. In the present study, qRT-PCR and Western blotting techniques were used to compare mRNA and protein abundance of ovarian immune cell and inflammation markers, along with NF-kappaB and steroidogenic pathway members in normal wild-type non-agouti (a/a; lean) and lethal yellow mice (KK.CG-Ay/J; obese) at 6, 12, 18, or 24 wk of age. Our data revealed that, beginning at 12 wk of age, NF-kappaB inflammatory signaling members were elevated (P < 0.05) in obese females. Interestingly obesity had opposing and temporal effects on the steroidogenic enzyme pathway. Obesity decreased (P < 0.05) STAR protein at 12, 18, and 24 wk of age. CYP11A1 and CYP19A1 proteins were increased (P < 0.05) at 12 wk but were decreased (P < 0.05) at 18 and 24 wk. Interestingly, CYP19A1 was increased in lethal yellow mouse ovaries at 6 wk of age, potentially indicating early puberty onset. These data demonstrate that obesity alters expression of ovarian inflammatory and steroidogenic pathway genes in ways which could adversely affect ovarian function.
Keywords: inflammation, obesity, ovary, steroidogenesis
INTRODUCTION
The mammalian ovary is essential for production of oocytes and the steroid hormones 17β-estradiol (E2) and progesterone (P4). E2 and P4 are required for reproductive function and development of female secondary sex characteristics [1–4]. Circulating lipoproteins and de novo biosynthesis provide the steroid hormone precursor cholesterol to the theca (TC) and granulosa (GC) cells under the influence of luteinizing hormone (LH) and follicle stimulating hormone (FSH), respectively [3, 4]. Steroidogenic acute regulatory protein (STAR) mediates the transfer of cholesterol from the outer to the inner mitochondrial membranes of the TC or GC, where cytochrome P450 side chain cleavage (CYP11A1) catalyzes its conversion to pregnenolone [6–8]. Pregnenolone then exits the mitochondrial matrix to the cytoplasm where 3-beta-hydroxysteroid dehydrogenase (3β-HSD) converts it to P4 [9, 10]. GC lack critical enzymes (17α-hydroxylase and C17,20-lyase) for androgen production; thus, P4 diffuses to the TC where conversion to androstenedione under the action of the enzymes 17α-hydroxylase and C17,20-lyase occurs [3, 7]. Likewise, the TC lack CYP19A1 (aromatase), required for conversation of androgens to estrogen. Therefore, androstendione diffuses into the GC where CYP19A1 catalyzes its conversion to estrone and subsequently E2 by the action of 17β-HSD. Alternatively, androstenedione can be converted to testosterone by 17β-HSD, which diffuses to the GC to be aromatized to E2 by CYP19A1 [3, 7, 10].
Obesity is a complex metabolic disorder associated with reproductive impairments [11, 12]. The deleterious effects of obesity on reproduction, including polycystic ovary syndrome (PCOS), poor oocyte quality, abridged fertility [11, 13], and increased risk of birth defects and stillbirth [14–16], have been long appreciated, but the underlying molecular mechanisms involved are unclear. Additionally, obesity alters ovarian steroid hormone synthesis and metabolism [17] potentially by modifying the expression and activity of steroidogenic enzymes. Obesity is linked with low-grade systematic inflammation, which is also implicated in infertility [12, 18, 19]. Macrophages and inflammatory pathways can serve as modulators of ovarian function [20–23]. The chemoattractants interleukin 8 (IL-8) and chemokine (C-C motif) ligand 2 (Ccl2), are major inflammatory response mediators and are involved in ovarian follicular development, steroidogenesis, ovulation, and atresia [23]. Furthermore, IL-6, through its ability to modulate cyclic AMP (cAMP) [23], potentially also regulates ovarian steroid production. Similarly, tumor necrosis factor (TNF) and IL-1 can stimulate ovarian P4 production [23]. The proinflammatory cytokines TNF-α and interleukins have been implicated in a number of ovarian pathologies [20] including PCOS [24], ovarian hyperstimulation [25, 26], ovarian cancer [27–29], endometriosis [30], and impaired steroid production [21]. Therefore it is possible that derangements in ovarian proinflammatory cytokine cues could compromise ovarian function and consequently lead to suboptimal fertility.
The lethal yellow mouse carries a deletion mutation in the normal wild-type non-agouti (a/a) background which results in ectopic expression of agouti in various tissues including hypothalamus and adipose tissue [31–33]. Hypothalamic agouti overexpression interferes with alpha-melanocyte-stimulating hormone (α-MSH) and cocaine- and amphetamine-regulated transcript (CART) by inhibiting the MSH receptor [34], leading to hyperphagia [32, 34]. Hence this serves as a useful model for understanding the effects of progressive obesity on ovarian function. The main objective of the present study was to determine the effects of obesity onset and progression on expression of ovarian proinflammatory and steroidogenesis pathway members in mice. To achieve this objective, changes in gene expression between the normal wild-type non-agouti a/a (lean) and lethal yellow KK.CG-Ay/J (obese) mice at 6, 12, 18, and 24 wk of age were compared using qRT-PCR and Western blotting techniques.
MATERIALS AND METHODS
Reagents
Custom-designed primers were obtained from the DNA facility of the Office of Biotechnology at Iowa State University. d-Glucose, 2-β-mercaptoethanol, 30% acrylamide/0.8% bis-acrylamide, ammonium persulfate, glycerol, N′,N′,N′,N′-tetramethylethylenediamine (TEMED), Tris base, Tris HCl, sodium chloride, Tween-20, bovine serum albumin (BSA), ascorbic acid (vitamin C), phosphatase inhibitor, protease inhibitor, and transferrin were purchased from Sigma-Aldrich, Inc. (St. Louis, MO). Contour model blood glucose meter and glucose strips were purchased from Bayer Ag (Leverkusen, Germany). RNAlater was obtained from Ambion, Inc. (Austin, TX). Hanks' balanced salt solution (without CaCl2, MgCl2, or MgSO4) and Superscript III one-step RT-PCR system were obtained from Invitrogen, Co. (Carlsbad, CA). RNeasy mini-kit, QIAshredder kit, RNeasy MinElute kit, and Quantitect SYBR Green PCR kit were purchased from Qiagen, Inc. (Valencia, CA). Ponceau S was purchased from Fisher Scientific (Waltham, MA). Goat anti-rabbit (anti-rabbit immunoglobulin G [IgG], horseradish peroxidase [HRP]-linked antibody 7074), and horse anti-mouse (anti-mouse IgG, HRP-linked antibody 7076) secondary antibodies along with anti-STAR ([D10H12] XP rabbit monoclonal antibody [mAb] 8449), anti-CYP11A1 (CYP11A1 antibody 12491), anti-CYP19A1 (aromatase antibody 8799), anti-pIκBαSer32/36 (phospho-IκBα [Ser32/36] [5A5] mouse mAb 9246), anti-pIKKα/βSer176/180 (phospho-IKKα/β [Ser176/180] [16A6] rabbit mAb 2697) anti-pNFκBp65Ser536 (phospho-NF-κB p65 [Ser536] [93H1] rabbit mAb 3033) primary antibodies and SignalFire ECL reagent (product 6883) were from Cell Signaling Technology (Danvers, MA). The anti-3β-HSD (product [P-18]: sc-30820) antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Donkey anti-goat secondary antibody was purchased from Pierce Biotechnology (product SA5-10085; Rockford, IL). Restore PLUS Western blot stripping buffer was purchased from Thermo Scientific (Rockford, IL).
Animal Procedures
All experimental protocols and procedures were approved by the Iowa State University Animal Care Committee (IACUC). At 4 wk of age, female normal wild-type non-agouti (a/a; designated lean; n = 20) and agouti lethal yellow (KK.Cg-Ay/J; designated obese; n = 20) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed at the animal facility at Iowa State University under controlled room temperature (21°–22°C) and lighting (12L:12D) cycle. All animals had ad libitum access to feed and water. Body weight was measured once weekly. Glucose tolerance testing followed by euthanasia and ovary collection were performed at 6, 12, 18, or 24 wk of age (n = 5 per group).
Estrous Cyclicity Monitoring
To determine any changes in the estrous cycle, fresh, wet unstained vaginal smears were examined daily using a model DMI300B fluorescent microscope (Leica, Vienna, Austria) for 14 consecutive days prior to the end of each time point (at 12, 18, and 24 wk). Classification of estrous cycle stages was based on the presence or absence of leucocytes and nucleated and cornified epithelial cells as previously described [35]. Briefly, a stage was classified as proestrous if the vaginal smears were characterized by mostly small, round nucleated epithelial cells, some cornified epithelial cells, and no to few leucocytes. Estrus was when vaginal smears were characterized by the presence of numerous large, cornified cells with degenerate nuclei. During metestrous, vaginal smears were characterized primarily by the presence of many leucocytes and a few cornified cells. In diestrous, vaginal smears were populated largely by many polymorphonuclear leucocytes and few nucleated epithelial cells.
Glucose Tolerance Testing
At the end of each experimental time point (6, 12, 18, and 24 wk of age; n = 4 per treatment per time point), a glucose tolerance test was performed. Mice were weighed and placed in separate cages with access to water but not feed for 16 h (overnight) prior to the test. Glucose solution (0.25 g/ml; 2.5 g of d-glucose in 10 ml of purified water [Milli-Q; Millipore Corp., Billerica, MA]) was prepared the day before the test and left to stand at room temperature overnight. Blood from the lateral saphenous vein was collected to determine fasting blood glucose (at time designated 0 min), followed by intraperitoneal injection (1g/kg of body weight) of glucose solution. Blood from the lateral saphenous vein was used to measure blood glucose concentration at 30, 60, 90, and 120 min after the injection, using a hand-held glucometer.
Tissue Collection
At the end of each experimental time point (6, 12, 18, and 24 wk of age; n = 4 per treatment per time point), mice were euthanized in their proestrous phase by CO2 asphyxiation. Ovaries were collected, trimmed of excess fat and weighed. One ovary was fixed in 4% paraformaldehyde for histology and follicle counts while the other ovary was stored in RNAlater at −80°C for RNA and protein expression studies.
RNA Isolation
Total RNA was isolated from one half of each sample ovary, using the RNeasy mini kit (n = 4 per treatment per time point [Qiagen]) according to the manufacturer's procedure. Briefly, tissues were lysed and homogenized using a hand-held homogenizer. The homogenate was then applied to a QIAshredder column placed in a collection tube and centrifuged at 10 000 rpm (12 197 × g) for 2 min at room temperature. The flow-through was then applied to an RNeasy Mini column, allowing RNA to bind to the filter cartridge. Following washing, RNA was eluted from the filter, using RNase-free water, and concentrated by using an RNeasy MinElute kit. The concentration was determined using an ND-1000 spectrophotometer (λ = 260/280 nm [NanoDrop Technologies, Inc., Wilmington, DE]).
First-Strand cDNA Synthesis and Quantitative Real-Time PCR
An equal amount (0.5 μg) of total RNA was reverse transcribed to cDNA by using Superscript III reverse transcriptase (Invitrogen) according to the manufacturer's protocol. Two microliters of diluted cDNA (1:25) were amplified with a Mastercycler ((Eppendorf AG, Hamburg, Germany) using Quantitect SYBR Green PCR kit and primers specific for mouse Tnfa, Tnfr1 (p55), Tnfr2 (p75), Il1b, Il6, Nos2, Cd3e, Cd4, Cd8b, Emr1, Ccl2, Ccl5, Chuk, Ikbkb, Rela (for sequences see ref. [36]), Gapdh, Il1rn, Il10, Ikbkg, Nfkb1, Star, Cyp11a1, Cyp19a1, Erα, and Erβ (for sequences see Table 1). The PCR cycling program consisted of a 15-min hold at 95°C and 40 cycles of denaturing for 15 sec at 95°C, annealing for 15 sec at 58°C, and an extension at 72°C for 20 sec. Product melt conditions were determined by using a temperature gradient from 72°C to 99°C with a 1°C increase at each step. Three replicates of each sample (n = 4) were included. The relative mRNA expression for each of the above-mentioned genes was normalized using the Gapdh housekeeping gene, and relative fold change calculated using the 2−ΔΔCT method. Results are presented as mean fold change ± standard error relative to that of the lean matched control group.
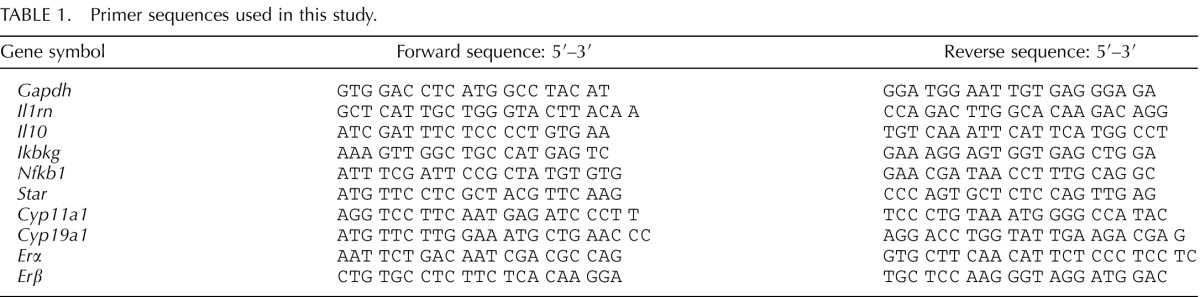
TABLE 1.
Primer sequences used in this study.
Protein Isolation and Western Blot Analysis
Total ovarian protein was isolated from all 4 animals per group, and Western blotting was performed as previously described with 4 randomly selected samples per time point [36]. Briefly, ovaries were homogenized in 300 μl of ice-cold extraction buffer followed by two rounds of centrifugation each for 15 min at 9300 relative centrifugal force (RCF) at 4°C. Protein concentration was determined using a standard BCA protocol, and absorbance values were detected with a λ = 560 nm excitation on a Synergy HT multidetection microplate reader using KC4 software (Bio-Tek Instruments Inc. Winooski, VT). Total protein (25 μg) was separated using 10%–12% SDS-PAGE, and electrotransfer of proteins from the gel to a nitrocellulose membrane was performed for 1.2 h at 100 V. The membranes were stained with Ponceau S to visualize the amount of total protein transferred in each lane. To reduce nonspecific binding, membranes were preincubated for 2 h on a rocker at room temperature in a blocking buffer containing 5% non-fat dried milk, 5 M NaCl, 20 mM Tris-HCl, and 0.15% Tween-20, pH 8. Membranes were probed using a specific primary antibody against anti-STAR (1:1000 dilution), anti-CYP11A1 (1:1000 dilution), anti-3β-HSD (1:500 dilution), anti-aromatase (1:500 dilution), anti-pIκBαSer32/36 (1:500 dilution), anti-pIKKα/βSer176/180 (1:250 dilution), anti-pNFκBp65Ser536 (1:100 dilution) diluted in 5% BSA in Tris-buffered saline with Tween-20 (TTBS) overnight at 4°C. After being washed three times (5 min each) in TTBS, membranes were incubated at room temperature for 1 h with HRP-conjugated suitable secondary antibodies (1:10 000) against the primary antibodies. Membranes were washed three times in TTBS followed by a single wash in Tris-buffered saline (TBS). Autoradiograms were visualized on X-ray film in a dark room following a 5- to 10-min incubation of membranes with 1× SignalFire ECL reagent (Cell Signaling Technology). Densitometry of the appropriate sized bands was measured using molecular imaging software (version 5.0; Carestream Health Inc., Rochester, NY), which eliminates background noise. Values were normalized to Ponceau S staining.
Histology and Follicle Counting
Ovaries were fixed in 4% paraformaldehyde overnight, transferred to 70% ethanol, and processed at the Iowa State University veterinary medicine histopathology laboratory. Each ovary was dehydrated and embedded in paraffin blocks. The entire ovary was serially sectioned (5 μM), and every sixth section was mounted and stained with hematoxylin and eosin (H&E). The number of healthy follicles in every sixth section throughout the entire ovary was determined using direct counts on all consecutive 5-μm sections in both lean and obese female mice. using a DMI300B fluorescent microscope (Leica). A follicle was considered healthy if it contained a distinct oocyte nucleus. A follicle was classified as a primordial follicle if its nucleated oocyte was surrounded by a partial or complete layer of squamous granulosa cells. A primary follicle contained a single layer of cuboidal granulosa cells surrounding its oocyte. A follicle whose nucleated oocyte was surrounded by multiple layers of granulosa cells was classified as a secondary follicle. A follicle was classified as a preovulatory follicle if its nucleated oocyte was surrounded with at least two layers of granulosa cells and a fluid-filled antrum.
Statistical Analysis
Statistical analysis was performed using the unpaired t-test function of Prism version 5.5 software (GraphPad, LaJolla, CA) with a statistical significance level set at P < 0.05.
RESULTS
Obesity Increases Body Weight and Fasting Blood Glucose in the Lethal Yellow Mice Relative to That of Their Controls
There were no differences in body weight between control and lethal yellow mice at 6 wk. From 12 wk of age onward, a progressive increase (P < 0.05) in body weight was observed in the lethal yellow mice compared to their controls (Table 2); thus, from this point onward, the controls are designated lean and the lethal yellow mice are designated obese. Increased (P < 0.05) fasting blood glucose in lean females was compared to that in obese females from 12 wk onward (Table 2). In addition, glucose tolerance testing revealed that relative to lean mice, obese mice had reduced clearance of glucose following 2 h of a glucose bolus challenge at 12, 18, and 24 wk (data not shown). There was no effect of obesity on ovarian weight across the time points of this study (Table 2).
TABLE 2.
Progressive obesity effects on fasting blood glucose and ovarian weight.a
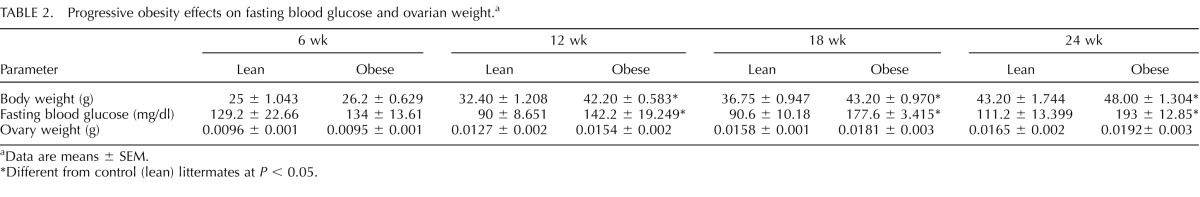
Data are means ± SEM.
Different from control (lean) littermates at P < 0.05.
Progressive Obesity Alters Estrous and Diestrous Phases Without Altering Overall Length of the Estrous Cycle
To evaluate the impact of progressive obesity on estrous cyclicity, fresh, wet unstained vaginal smears obtained daily for 14 consecutive days prior to the end of each time point (12, 18, and 24 wk) from both lean and obese mice were examined (Table 3). There was no impact of obesity on proestrus or metestrus or on the overall length of the estrous cycle; however, at both 18 and 24 wk, obese mice displayed a decrease (P < 0.05) in the length of the estrous phase and an increase (P < 0.01) in length of the diestrous phase relative to their lean counterparts (Table 3).
TABLE 3.
Impact of progressive obesity on estrous cyclicity.a
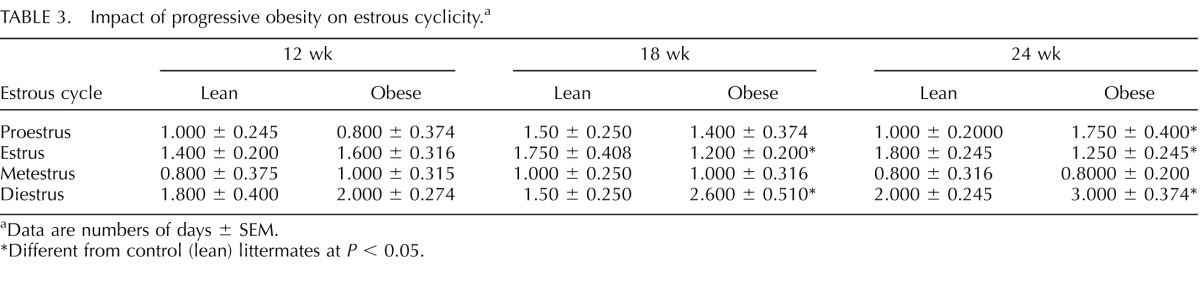
Data are numbers of days ± SEM.
Different from control (lean) littermates at P < 0.05.
Obesity Decreases the Number of Healthy Primordial and Primary Follicles in Female Mice
The impact of obesity advancement on the number of ovarian healthy follicle populations was determined (Fig. 1). Ovaries from obese mice had decreased (P < 0.05) numbers of healthy primordial (Fig. 1A) and primary (Fig. 1B) follicles at 12, 18, and 24 wk of age as compared to ovaries from their lean matched controls. Although there was no impact of obesity on secondary follicles across time points, at 18 wk, there was a tendency (P = 0.07) for an increased number of healthy secondary follicles in ovaries of obese mice relative to their lean counterparts (Fig. 1C). Interestingly, obesity increased (P < 0.05) the number of antral follicles at 12 and 24 wk but not at 18 wk (Fig. 1D). To rule out any impact of genotype on follicle number, ovaries examined at 6 wk of age, when there are no differences in body weight, and no differences in any follicle type number were observed between the two strains of mice (Fig. 1, A–D). Interestingly, the numbers of primordial and small primary follicles were also decreased in lean mice with aging (Fig. 1, A and B).
FIG. 1.

Effect of obesity on follicle number. At 6, 12, 18, or 24 wk of age, mice were euthanized and ovaries collected. One ovary was fixed in 4% paraformaldehyde, complete serial sections were mounted, and healthy follicles were classified and counted in both lean and obese mice (A–D); healthy primordial follicle number (A); healthy primary follicle number (B); healthy secondary follicle number (C); and healthy antral follicle number (D). Bars represent means ± SEM. *Significant (P < 0.05; † = P < 0.1; n = 4) differences from respective lean group.
Obesity Alters mRNA Expression of Ovarian Steroidogenic Enzymes in Mice
The impact of continuing obesity on mRNA expression of genes encoding enzymes involved in ovarian steroidogenesis (Fig. 2) was examined. At 6 and 24 wk, there were no difference in Star mRNA levels between ovaries from lean and obese mice; however, at both 12 (P < 0.0001) and 18 (P < 0.05) wk, obesity reduced ovarian Star mRNA abundance (Fig. 2A). There was no impact of obesity on ovarian Cyp11a1 mRNA levels at any time point tested in this study (Fig. 2B). We observed a decrease in Cyp19a1 mRNA levels in ovaries obtained from obese mice relative to their matched controls across all time points (Fig. 2C). We further investigated the impact of progressive obesity on the two estrogen receptors (Fig. 1, D and E). At 6 and 12 but not at 18 or 24 wk of age, ovaries from obese mice had increased (P < 0.05) Erα mRNA levels compared to their respective lean controls (Fig. 2D). Furthermore, progressive obesity increased (P ≤ 0.01) Erβ mRNA expression from 12 wk onward (Fig. 2E).
FIG. 2.
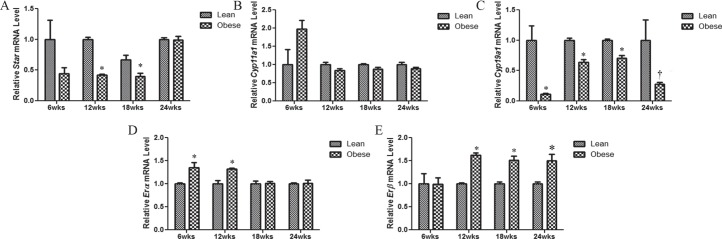
Obesity alters mRNA expression of ovarian steroidogenic enzymes in mice. At the end of each experimental time point (6, 12, 18, and 24 wk of age), ovaries were removed from lean or obese mice, and total RNA was isolated (n = 4 per treatment per time point) and reverse transcribed to cDNA. mRNA levels of Star (A), Cyp11a1 (B), Cyp19a1 (C), Erα (D), and Erβ (E) were quantified by quantitative RT-PCR. After normalization to Gapdh, relative fold change was calculated using the 2−ΔΔCT method. Results are given as mean fold change ± SE relative to that of lean matched control value of 1 ± SE. *Different from control at P < 0.05; †P < 0.1.
Progressive Obesity Alters Protein Expression of Ovarian Steroidogenic Enzymes in Mice
No differences in STAR protein expression between the two groups were observed at 6 wk of age; however, ovaries from obese mice had decreased STAR protein levels compared to ovaries from their matched lean controls at 12 (P < 0.01), 18 (P < 0.05), and 24 (P < 0.01) wk of age (Fig. 3A). Although there were no differences in CYP11A1 protein expression between the 2 groups at 6 or 18 wk, a decrease (P < 0.01) in CYP11A1 protein levels was observed at 24 wk of age in ovaries from obese females (Fig. 3B). In contrast, at 12 wk of age, ovaries from obese mice had increased (P < 0.001) CYP11A1 protein levels compared to those from lean mice (Fig. 3B). We did not find any differences between lean and obese females in 3β-HSD protein levels for all time points tested during this study (Fig. 3C). Interestingly, at 6 wk of age, increased (P < 0.05) ovarian CYP19A1 protein levels were observed in the lethal yellow mice relative to the lean group (Fig. 3D). Although there was no impact of obesity on CYP19A1 protein level at 12 wk; there was a tendency (P < 0.1) for decreased CYP19A1 protein expression in the obese mice compared to their lean counterparts at 18 and 24 wk (Fig. 3D).
FIG. 3.

Obesity impacts protein abundance of ovarian steroidogenic enzymes. At the end of each experimental time point (6, 12, 18, and 24 wk of age), ovaries were removed from lean (L) or obese (O) female mice, and total ovarian protein was isolated (n = 3–4 per treatment group per time point). Western blotting was performed to measure STAR (A), CYP11A1 (B), 3β-HSD (C), and CYP19A1 (D) protein levels. Densitometry of the appropriate bands was performed using Carestream molecular imaging software and normalized to Ponceau S staining prior to statistical analysis. Bars represent means ± SEM; *significant differences from respective lean group at P < 0.05; † = P < 0.1. Ponceau S loading control is indicated by LC.
Impact of Progressive Obesity on Relative mRNA Expression of Ovarian Immune Cells, Inflammation, and NF-κB Pathway Members in Mice
The expression profiles of immune cell infiltration and inflammatory marker genes were investigated over the temporal pattern of obesity onset and advancement (Table 4). The mRNA levels of Tnfα, p55, p75, Cd3e, Cd8eb1, F4-80, Ikbkb, Ikbkg, and RelA were not affected by obesity (Table 4). However, the mRNA levels of Nos2, a marker for oxidative stress, was increased at 6 (P < 0.01), 18 (P < 0.01), and 24 (P < 0.001) wk of age (Table 4). Additionally, the mRNA expression of the anti-inflammatory gene Il10 was increased (P < 0.05), with obesity at both 18 and 24 wk (Table 4). Further still, the mRNA expression of Il1b was decreased at 12 and 24 (P < 0.05) wk but increased at 18 (P < 0.01) wk with obesity. Ovaries from obese mice also showed a marked decrease in Il1ra mRNA at 12 (P < 0.0001), 18 (P < 0.0001), and 24 (P < 0.01) wk compared to ovaries from lean matched mice. Cd3 mRNA was increased (P < 0.05) by obesity only at 24 wk. At 12 wk, obesity decreased (P < 0.01) Cd4 mRNA, but at 18 wk, Cd4 mRNA was increased (P < 0.05) with obesity. Although Mcp1 (P < 0.001) and Ccl5 (P < 0.01) mRNA levels were decreased by obesity at 12 wk, at 24 wk only Mcp1 was decreased (P < 0.001) by obesity. Of the NF-κB pathway members tested in this study, only Ikaka (P < 0.001) and Nfkb1 (P < 0.05) mRNA levels were decreased by obesity at 18 wk (Table 4).
TABLE 4.
Impact of progressive obesity on mRNA level of markers of immune cells, inflammation, and NF-κB pathway members.a
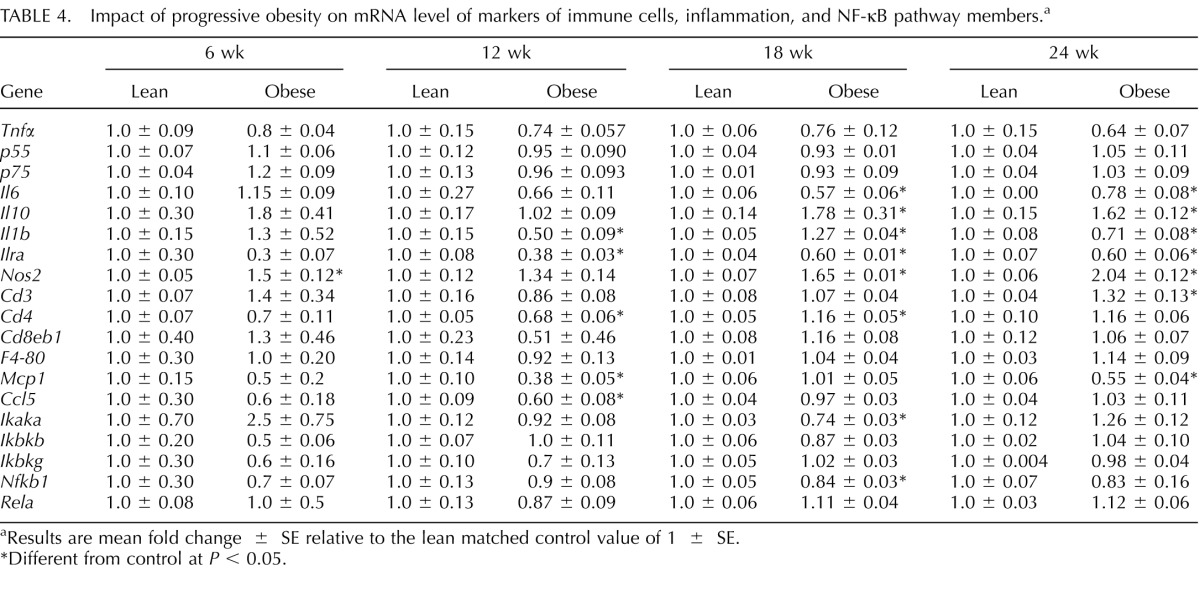
Results are mean fold change ± SE relative to the lean matched control value of 1 ± SE.
Different from control at P < 0.05.
Progressive Obesity Increases Ovarian Protein Expression of NF-κB Pathway Members in Mice
There was no effect of progressive obesity on phosphorylated IKKα/β Ser176/180 protein levels across time points (Fig. 4A). However, phosphorylated IκBSer32/36 protein level was decreased in lethal yellow mice (P < 0.01) at 6 wk but increased at 12 (P < 0.01) and 24 (P < 0.05) wk in obese females relative to their lean littermates (Fig. 4B). Similarly, ovarian pNF-κBp65Ser536 was reduced in the lethal yellow mice at 6 wk (P < 0.05) but increased (P < 0.01) by obesity from 12 wk onward (Fig. 4C).
FIG. 4.

Ovarian NF-κB pathway member protein levels are affected by obesity. At each time point (6, 12, 18, and 24 wk of age), total ovarian protein was isolated from both lean (L) and obese (O) female mice. Western blotting was performed to quantify pIKKα/βSer176/180 (A), pIκBSer32/36 (B), and pNF-κB p65Ser536 (C) protein levels. Densitometry of the appropriate bands was performed using Carestream molecular imaging software and normalized to Ponceau S staining prior to statistical analysis. Bars represent means ± SEM; *significant differences from respective lean group at P < 0.05. Ponceau S loading control is indicated by LC.
DISCUSSION
Obesity is associated with impaired fertility [13, 37, 38], which often phenotypically presents as higher incidences of poor oocyte quality [39, 40], irregular menstrual cycles [41, 42], anovulation [43], and increased miscarriage rates [44, 45]; suggestive of derangements in ovarian folliculogenesis and steroidogenesis. Additionally, obesity is associated with low-grade inflammation, which if not properly regulated, can lessen reproductive function [46, 47]. The main objective of this study was to determine the impact of obesity onset and progression on the expression profiles of immune cell and inflammation markers as well as the NF-κB and steroidogenic pathway members in murine ovaries. The lethal yellow mouse, a model of progressive obesity was used to compare gene expression beginning at 6 wk of age when there is no reported difference in body weight between genotypes [48], which we confirmed in this study.
Beginning at 12 wk of age, a progressive increase in body weight among the lethal yellow mice compared to their a/a matched controls was observed. Increased fasting blood glucose was also evident as body weight increased from 12 wk onward. Glucose tolerance testing revealed that relative to their age matched controls, obese mice had reduced clearance of glucose following a glucose bolus challenge at 12, 18, and 24 wk (data not shown). We did not measure blood insulin levels in these animals because previous data showed that the lethal yellow mice have increased circulating insulin and leptin levels compared to those of age-matched lean controls from 12 wk onward [48], matching our phenotypic data. Thus, elevated fasting blood glucose, insulin levels [48], and reduced glucose clearance rate indicate that these mice have compromised peripheral insulin sensitivity.
Obesity impacted ovarian cyclicity by decreasing the length of time spent in estrus with a concomitant increase in the length of the diestrous phase. The impact of obesity on the estrous cycle has been previous reported in rats [49, 50]. In agreement with our results, high-fat diet (HFD)-fed obese rats exhibited longer diestrous but shorter estrous stages compared to chow fed lean animals [50]. Physiologically, the ovarian steroids predominantly associated with estrous and diestrous phases are E2 and P4, respectively [51]. Therefore, shorter estrous stages may imply lower E2 levels whereas longer diestrous stages could imply higher P4 levels in obese females. In premenopausal females, an inverse relationship between E2 and body mass index has been reported [52–54], and higher P4 levels have been observed in obese rodent models [50]. Taken together, these data suggest that alterations in the estrous cycle during obesity could impact ovulation and subsequently compromise the reproductive capacity of obese females.
In order to separate the phenotypic effects of obesity from the Ay/J genotype on folliculogenesis, we included a 6-wk-old group in which we observed no differences in body weight from the a/a genotype mice. Importantly, also at this time point, there were no differences in the number of any follicular stage, and numbers were in a similar range as we have previously reported in these two strains of mice [55]. However, despite a lack of any impact of progressive obesity on ovarian weight, fewer primordial and primary follicles, and increased numbers of tertiary follicle subtypes was present as body mass increased. These data are in agreement with those of our previous study in which 20-wk-old obese mice which were found to have reduced primordial and small primary but increased numbers of secondary and antral follicles [55]. Furthermore, Sprague-Dawley rats fed a HFD for 18 wk demonstrated a decreased number of primordial follicles but increased number of developing and atretic follicles [56]. This difference in follicle populations could indicate increased activation of primordial follicles. This scenario is possible because obesity is associated with elevated insulin levels, obese females have higher levels of insulin in their follicular fluid [57], and insulin administration promoted primordial to primary transition in neonatal rat ovaries [58].
Several studies have reported an inverse relationship between steroid hormone level and primordial follicle activation [59–62]. E2 levels are lower in HFD-induced obese rats [49] and obese females [52–54] and reduced E2 levels stimulated primordial follicle activation from the resting into the growing follicular pool [60, 61]. In mice [60, 62] and in neonatal rats [59], administration of E2 or P4 was associated with impaired primordial follicle assembly and decreased numbers of both primordial and primary follicles. Further still, increased activation of ovarian phosphatidylinositol-3 kinase (PI3K) signaling [1, 63], which we have previously reported to be altered during HFD-induced obesity [64], is involved in early stages of folliculogenesis. Therefore, it is possible that the observed low numbers of primordial follicles and higher numbers of growing follicles observed in ovaries from obese relative to lean mice is indicative that obesity triggers changes in the intrinsic ovarian signals responsible for initiation of follicle activation and recruitment.
Ovarian steroidogenesis is mediated by FSH and LH through the action of several enzymes including STAR, CYP11A1, 3β-HSD, and CYP19A1. In the testes, during PI3K activation, insulin inhibited cAMP-induced STAR, CYP11A1, and 3β-HSD gene expression [65]. However, conversely, insulin induces steroidogenesis in ovarian somatic cells cultured in vitro [66, 67]. We found that obesity has differential effects of the expression profiles of genes involved in ovarian steroidogenesis. With the exception of CYP19A1 protein expression, which was increased at 6 wk of age in the lethal yellow mice, potentially indicating early onset of puberty, there was no impact of genotype (6 wk of age) on levels of ovarian steroidogenic enzymes (mRNA or protein). Progressive obesity decreased both Star mRNA and protein levels, indicating that the rate-limiting step in steroidogenesis could be compromised during an obese state. Additionally, despite its increase in the early stages of obesity (at 12 wk), CYP11A1 protein expression was markedly downregulated with progressive obesity, reaching a 50% reduction by 24 wk of age. Erα mRNA levels were increased at 6 and 12 in obese females, while Erβ mRNA levels were elevated from 12 onward due to the obese phenotype, suggesting a temporal pattern of E2 receptor response to increased body mass. 3β-HSD protein levels were unchanged, but Cyp19a1 mRNA and protein were both negatively impacted by obesity. Whether, these alterations result in increased levels of androgens cannot be answered by the current study, but taken together, these results indicate that obesity may alter ovarian steroidogenesis, contributing to the reproductive disorders experienced by obese females. Our data also reveal a temporal effect of obesity on ovarian steroidogenic genes underscoring the importance of considering the time point at which data is collected when interpreting obesity-associated ovarian impacts.
Ovarian steroids may vary depending on energetic conditions of the cell [68] and inflammatory changes [69], both of which are effected by obesity. Elevated TNF-α and IL-6 levels have been reported in the follicular fluid of obese women [70], whereas obesity-induced increased Tnfα expression has been observed in human adipose tissue [71, 72], muscle [73], rat adipose tissue [74], mouse muscle, and adipose tissues [75]. IL-6 , through its ability to modulate cAMP, can regulate ovarian steroid production [23] and ovulation [76, 77]. TNF-α and IL-1 can also stimulate ovarian P4 production [21, 23] and regulate ovarian cell differentiation, proliferation, and apoptosis [21] as well as inflammation [78]. Derangements in these inflammatory pathways have been linked to various reproductive disorders [20, 79]. For example, elevated levels of IL-6 have been associated with ovarian hyperstimulation syndrome [25, 26], tumor cell activation [29], and chemotherapeutic resistance [80]. Furthermore, IL-6, in conjunction with TNF-α and IL-1β, has been associated with ovarian neoplasia [27].
Changes to all immune cell infiltration and inflammation markers were not observed. Similar to previously reported elevations in nitric oxide synthase 2 (Nos2) mRNA in animal models of obesity [81, 82], we observed a temporal increase in Nos2 mRNA in ovaries of obese mice compared to their lean littermates. Nos2 is a marker for oxidative stress, dysregulation of which has been linked to impaired ovulation [83], ovarian cancer [84], endometriosis [85], and infertility [86]. Similarly, ovarian mRNA expression of the anti-inflammatory gene Il10 was increased by obesity. In contrast, obesity decreased Il1ra mRNA, a modulator of the proinflammatory actions of IL-1. Interestingly, Il1b mRNA was decreased at 12 and 24 wk, but subsequently increased at 18 wk with obesity. Also, although both Mcp1 and Ccl5 mRNA levels were decreased by obesity, only the decrease in Mcp1 was maintained. MCP1 is involved in ovarian follicular development, steroidogenesis, ovulation, and atresia [23]. Interpreted locally, these mRNA results would suggest a downward trend in expression profiles of both immune cell infiltration and inflammatory markers. Despite no observed alteration to Tnfα, it is possible that the protein level may have been altered. Taken together, these results indicate that progressive obesity alters inflammatory members and that these changes may have negative consequences for ovarian function.
The mammalian NF-κB family, is made of five proteins: RelA (p65), RelB, c-Rel, NF-κB1 and NF-κB2 carrying a Rel-homology domain [87]. Currently, there are two known activation pathways for the NF-κB activity: the canonical pathway which is activated by proinflammatory cytokines including TNF-α and other TNF cytokines [87]. Before activation, the inactive NF-κB complex is localized in the cytoplasm, bound to IκB proteins such as IκBα, IκBβ, and IκBγ, which block its nuclear localization sequence function of the Rel-homology domain [87]. During the canonical activation pathway, proinflammatory cytokines, work through different TNF receptors (TNFR) and Toll-like receptor-interleukin-1 receptor (IL-1R) superfamilies to activate the IκB kinases (IKKs) [87]. Like the IκB complex, the IKK complex is mainly made up of two catalytic subunits, IKKα and IKKβ and a regulatory subunit IKKγ. Activated IKK proteins can catalyze the phosphorylation of IκB proteins mainly at the serine 32 and 36 sites of IκBα, leading to proteasome degradation of the IκB complex and subsequent release and translocation of the NF-κB proteins to the nucleus [87, 88]. Once in the nucleus, the NF-κB proteins will bind and activate transcription of several genes particularly those involved in immune and inflammatory cascades [87, 88].
Although progressive obesity did not impact the mRNA expression profiles of TNF-α-NF-κB pathway members as previously observed during HFD-induced obesity [36], we found that obesity advancement increased abundance of protein expression of the NF-κB pathway member proteins pIκBSer32/36 and pNF-κB p65Ser536 , indicating increased activation of NF-κB signaling pathway in ovaries of obese mice. Surprisingly, before the onset of obesity (at 6 wk of age), ovaries from the lethal yellow mice displayed a marked decrease in pIκBSer32/36 and pNF-κB p65Ser536 proteins compared to non-agouti (a/a) mice, potentially indicating that the Ay/J genotype has some culpability; it is more likely, however, that even though no differences in body weight are detectable at this age point, changes in central metabolism have been activated; this is strengthened by the fact that the lethal yellow mice displayed increased CYP19A1, a marker that puberty onset is impending at an earlier time than the control genotype.
NF-κB pathway is activated in response to inflammatory responses [87, 89]; therefore, its sustained activation in ovaries from obese mice is indicative of aggravated inflammatory signaling, which in turn is associated with a number of ovarian pathologies [30, 90] including polycystic ovary syndrome, endometriosis [30, 91], and premature ovarian failure [22]. Additionally, because inflammatory signals can influence generation of reactive oxygen species and oxidative stress [20], ovarian steroid production and ovulation [21, 22, 76, 77] as well as cell differentiation, proliferation and apoptosis [21], deregulation of inflammatory signals in the ovary may adversely impact tissue function and pose dire consequences for female fertility.
In conclusion, our results indicate that progressive obesity alters the expression of inflammatory and steroidogenesis signaling pathway members in ways which could alter optimal ovarian function. The data herein are novel in that physiological changes that occur at both the onset and progression of obesity were studied, and dietary composition was not altered between the mouse strains. In addition, the importance of studying ovarian changes during obesity progression is demonstrated by the dynamic alterations to signaling pathways observed. Manipulation of dietary treatments to lessen obesity presents as an attractive intervention approach for future experiments. These observations underscore the importance of the temporal pattern of ovarian signaling pathway changes that are induced by increasing body mass and the accompanying metabolic changes.
Footnotes
Supported by National Institute of Environmental Health Science award number R00ES016818 to A.F.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health.
REFERENCES
- Edson MA, Nagaraja AK, Matzuk MM. The mammalian ovary from genesis to revelation. Endocr Rev. 2009;30:624–712. doi: 10.1210/er.2009-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutilainen R, Tapanainen J, Chung BC, Matteson KJ, Miller WL. Hormonal regulation of P450scc (20,22-desmolase) and P450c17 (17 alpha-hydroxylase/17, 20-lyase) in cultured human granulosa cells. J Clin Endocrinol Metab. 1986;63:202–207. doi: 10.1210/jcem-63-1-202. [DOI] [PubMed] [Google Scholar]
- Jamnongjit M, Hammes SR. Ovarian steroids: the good, the bad, and the signals that raise them. Cell Cycle. 2006;5:1178–1183. doi: 10.4161/cc.5.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquette GN, LaPolt PS, Oikawa M, Hsueh AJ. Regulation of luteinizing hormone receptor messenger ribonucleic acid levels by gonadotropins, growth factors, and gonadotropin-releasing hormone in cultured rat granulosa cells. Endocrinol. 1991;128:2449–2456. doi: 10.1210/endo-128-5-2449. [DOI] [PubMed] [Google Scholar]
- Mason JI, Rainey WE. Steroidogenesis in the human fetal adrenal: a role for cholesterol synthesized de novo. J Clin Endocrinol Metab. 1987;64:140–147. doi: 10.1210/jcem-64-1-140. [DOI] [PubMed] [Google Scholar]
- Miller WL. StAR search–what we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol Endocrinol. 2007;21:589–601. doi: 10.1210/me.2006-0303. [DOI] [PubMed] [Google Scholar]
- Hall PF. Cytochromes P-450 and the regulation of steroid synthesis. Steroids. 1986;48:131–196. doi: 10.1016/0039-128x(86)90002-4. [DOI] [PubMed] [Google Scholar]
- Simpson ER. Cholesterol side-chain cleavage, cytochrome P450, and the control of steroidogenesis. Mol Cell Endocrinol. 1979;13:213–227. doi: 10.1016/0303-7207(79)90082-0. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Auchus RJ. Minireview: cellular redox state regulates hydroxysteroid dehydrogenase activity and intracellular hormone potency. Endocrinol. 2005;146:2531–2538. doi: 10.1210/en.2005-0061. [DOI] [PubMed] [Google Scholar]
- Lee TC, Miller WL, Auchus RJ. Medroxyprogesterone acetate and dexamethasone are competitive inhibitors of different human steroidogenic enzymes. J Clin Endocrinol Metab. 1999;84:2104–2110. doi: 10.1210/jcem.84.6.5646. [DOI] [PubMed] [Google Scholar]
- Rachoń D, Teede H. Ovarian function and obesity—interrelationship, impact on women's reproductive lifespan and treatment options. Mol Cell Endocrinol. 2010;316:172–179. doi: 10.1016/j.mce.2009.09.026. [DOI] [PubMed] [Google Scholar]
- Ogbuji QC. Obesity and reproductive performance in women. Afr J Reprod Health. 2010;14:143–151. [PubMed] [Google Scholar]
- Brewer CJ, Balen AH. The adverse effects of obesity on conception and implantation. Reproduction. 2010;140:347–364. doi: 10.1530/REP-09-0568. [DOI] [PubMed] [Google Scholar]
- Blencowe H, Cousens S, Oestergaard MZ, Chou D, Moller AB, Narwal R, Adler A. Vera Garcia C, Rohde S, Say L, Lawn JE. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet. 2012;379:2162–2172. doi: 10.1016/S0140-6736(12)60820-4. [DOI] [PubMed] [Google Scholar]
- Gilboa SM, Correa A, Botto LD, Rasmussen SA, Waller DK, Hobbs CA, Cleves MA, Riehle-Colarusso TJ, Study NBDP. Association between prepregnancy body mass index and congenital heart defects. Am J Obstet Gynecol. 2010;202:51.e51–51.e10. doi: 10.1016/j.ajog.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Rasmussen KM, Kjolhede CL. Maternal obesity: a problem for both mother and child. Obesity. 2008;16:929–931. doi: 10.1038/oby.2008.36. [DOI] [PubMed] [Google Scholar]
- Pasquali R, Pelusi C, Genghini S, Cacciari M, Gambineri A. Obesity and reproductive disorders in women. Hum Reprod Update. 2003;9:359–372. doi: 10.1093/humupd/dmg024. [DOI] [PubMed] [Google Scholar]
- Nestler JE. Obesity, insulin, sex steroids and ovulation Int J Obes Relat Metab Disord 2000. 24 (suppl 2): S71 73 [DOI] [PubMed] [Google Scholar]
- Pasquali R, Gambineri A, Pagotto U. Review article: The impact of obesity on reproduction in women with polycystic ovary syndrome. Br J Obstet Gynecol. 2006;113:1148–1159. doi: 10.1111/j.1471-0528.2006.00990.x. [DOI] [PubMed] [Google Scholar]
- Jabbour HN, Sales KJ, Catalano RD, Norman JE. Inflammatory pathways in female reproductive health and disease. Reproduction. 2009;138:903–919. doi: 10.1530/REP-09-0247. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Keren-Tal I, Aharoni D, Dantes A, Land-Bracha A, Rimon E, Sasson R, Hirsh L. Steroidogenesis and apoptosis in the mammalian ovary. Steroids. 2003;68:861–867. doi: 10.1016/j.steroids.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Wu R, Van der Hoek KH, Ryan NK, Norman RJ, Robker RL. Macrophage contributions to ovarian function. Hum Reprod Update. 2004;10:119–133. doi: 10.1093/humupd/dmh011. [DOI] [PubMed] [Google Scholar]
- Bornstein SR, Rutkowski H, Vrezas I. Cytokines and steroidogenesis. Mol Cell Endocrinol. 2004;215:135–141. doi: 10.1016/j.mce.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Qiao J, Feng HL. Extra- and intra-ovarian factors in polycystic ovary syndrome: impact on oocyte maturation and embryo developmental competence. Hum Reprod Update. 2011;17:17–33. doi: 10.1093/humupd/dmq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loret de Mola JR, Flores JP, Baumgardner GP, Goldfarb JM, Gindlesperger V, Friedlander MA. Elevated interleukin-6 levels in the ovarian hyperstimulation syndrome: ovarian immunohistochemical localization of interleukin-6 signal. Obstet Gynecol. 1996;87:581–587. doi: 10.1016/0029-7844(95)00456-4. [DOI] [PubMed] [Google Scholar]
- Loret de Mola JR, Baumgardner GP, Goldfarb JM, Friedlander MA. Ovarian hyperstimulation syndrome: pre-ovulatory serum concentrations of interleukin-6, interleukin-1 receptor antagonist and tumour necrosis factor-alpha cannot predict its occurrence. Hum Reprod. 1996;11:1377–1380. doi: 10.1093/oxfordjournals.humrep.a019403. [DOI] [PubMed] [Google Scholar]
- Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, Gould D, Ayhan A, Balkwill F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007;67:585–592. doi: 10.1158/0008-5472.CAN-06-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D, Matte I, Rancourt C, Piché A. Prognostic significance of IL-6 and IL-8 ascites levels in ovarian cancer patients. BMC Cancer. 2011;11:210. doi: 10.1186/1471-2407-11-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CH, Wei LH, Kuo ML, Huang YJ, Lai KP, Chen CA, Hsieh CY. Up-regulation of interleukin-6 in human ovarian cancer cell via a Gi/PI3K-Akt/NF-kappaB pathway by lysophosphatidic acid, an ovarian cancer-activating factor. Carcinogenesis. 2005;26:45–52. doi: 10.1093/carcin/bgh301. [DOI] [PubMed] [Google Scholar]
- Carlberg M, Nejaty J, Fröysa B, Guan Y, Söder O, Bergqvist A. Elevated expression of tumour necrosis factor alpha in cultured granulosa cells from women with endometriosis. Hum Reprod. 2000;15:1250–1255. doi: 10.1093/humrep/15.6.1250. [DOI] [PubMed] [Google Scholar]
- Michaud EJ, Bultman SJ, Klebig ML, van Vugt MJ, Stubbs LJ, Russell LB, Woychik RP. A molecular model for the genetic and phenotypic characteristics of the mouse lethal yellow (Ay) mutation. Proc Natl Acad Sci U S A. 1994;91:2562–2566. doi: 10.1073/pnas.91.7.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebig ML, Wilkinson JE, Geisler JG, Woychik RP. Ectopic expression of the agouti gene in transgenic mice causes obesity, features of type II diabetes, and yellow fur. Proc Natl Acad Sci U S A. 1995;92:4728–4732. doi: 10.1073/pnas.92.11.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll L, Voisey J, van Daal A. Mouse models of obesity. Clin Dermatol. 2004;22:345–349. doi: 10.1016/j.clindermatol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Lu D, Willard D, Patel I, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik R, Wilkison W. Agouti protein is an antagonist of the melanocyte-stimulating hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- Byers SL, Wiles MV, Dunn SL, Taft RA. Mouse estrous cycle identification tool and images. PLoS One. 2012;7:e35538. doi: 10.1371/journal.pone.0035538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nteeba J, Ortinau LC, Perfield JW, Keating AF. Diet-induced obesity alters immune cell infiltration and expression of inflammatory cytokine genes in mouse ovarian and peri-ovarian adipose depot tissues. Mol Reprod Develop. 2013;80:948–958. doi: 10.1002/mrd.22231. [DOI] [PubMed] [Google Scholar]
- Zain MM, Norman RJ. Impact of obesity on female fertility and fertility treatment. Womens Health (Lond Engl) 2008;4:183–194. doi: 10.2217/17455057.4.2.183. [DOI] [PubMed] [Google Scholar]
- Clark AM, Thornley B, Tomlinson L, Galletley C, Norman RJ. Weight loss in obese infertile women results in improvement in reproductive outcome for all forms of fertility treatment. Hum Reprod. 1998;13:1502–1505. doi: 10.1093/humrep/13.6.1502. [DOI] [PubMed] [Google Scholar]
- Depalo R, Garruti G, Totaro I, Panzarino M, Vacca MP, Giorgino F, Selvaggi LE. Oocyte morphological abnormalities in overweight women undergoing in vitro fertilization cycles. Gynecol Endocrinol. 2011;27:880–884. doi: 10.3109/09513590.2011.569600. [DOI] [PubMed] [Google Scholar]
- Jungheim ES, Schoeller EL, Marquard KL, Louden ED, Schaffer JE, Moley KH. Diet-induced obesity model: abnormal oocytes and persistent growth abnormalities in the offspring. Endocrinol. 2010;151:4039–4046. doi: 10.1210/en.2010-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Martinez L, Lopez-Alvarenga JC, Villa AR, Gonzalez-Barranco J. Menstrual cycle length disorders in 18- to 40-y-old obese women. Nutrition. 2003;19:317–320. doi: 10.1016/s0899-9007(02)00998-x. [DOI] [PubMed] [Google Scholar]
- Hartz AJ, Barboriak PN, Wong A, Katayama KP, Rimm AA. The association of obesity with infertility and related menstrual abnormalities in women. Int J Obes. 1979;3:57–73. [PubMed] [Google Scholar]
- Clark AM, Ledger W, Galletly C, Tomlinson L, Blaney F, Wang X, Norman RJ. Weight loss results in significant improvement in pregnancy and ovulation rates in anovulatory obese women. Hum Reprod. 1995;10:2705–2712. doi: 10.1093/oxfordjournals.humrep.a135772. [DOI] [PubMed] [Google Scholar]
- Bellver J, Pellicer A, Garcia-Velasco JA, Ballesteros A, Remohi J, Meseguer M. Obesity reduces uterine receptivity: clinical experience from 9, 587 first cycles of ovum donation with normal weight donors. Fertil Steril. 2013;100:1050–1058. doi: 10.1016/j.fertnstert.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Jungheim ES, Schon SB, Schulte MB, DeUgarte DA, Fowler SA, Tuuli MG. IVF outcomes in obese donor oocyte recipients: a systematic review and meta-analysis. Hum Reprod. 2013;28:2720–2727. doi: 10.1093/humrep/det292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Ferrante AW. Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J Intern Med. 2007;262:408–414. doi: 10.1111/j.1365-2796.2007.01852.x. [DOI] [PubMed] [Google Scholar]
- Yang Z, Norwood KA, Smith JE, Kerl JG, Wood JR. Genes involved in the immediate early response and epithelial-mesenchymal transition are regulated by adipocytokines in the female reproductive tract. Mol Reprod Dev. 2012;79:128–137. doi: 10.1002/mrd.22006. [DOI] [PubMed] [Google Scholar]
- Balasubramanian P, Jagannathan L, Mahaley RE, Subramanian M, Gilbreath ET, Mohankumar PS, Mohankumar SM. High fat diet affects reproductive functions in female diet-induced obese and dietary resistant rats. J Neuroendocrinol. 2012;24:748–755. doi: 10.1111/j.1365-2826.2011.02276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akamine EH, Marcal AC, Camporez JP, Hoshida MS, Caperuto LC, Bevilacqua E, Carvalho CR. Obesity induced by high-fat diet promotes insulin resistance in the ovary. J Endocrinol. 2010;206:65–74. doi: 10.1677/JOE-09-0461. [DOI] [PubMed] [Google Scholar]
- McCann JP, Reimers TJ. Effects of obesity on insulin and glucose metabolism in cyclic heifers. J Anim Sci. 1986;62:772–782. doi: 10.2527/jas1986.623772x. [DOI] [PubMed] [Google Scholar]
- Freeman EW, Sammel MD, Lin H, Gracia CR. Obesity and reproductive hormone levels in the transition to menopause. Menopause. 2010;17:718–726. doi: 10.1097/gme.0b013e3181cec85d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HI, Sammel MD, Freeman EW, Lin H, DeBlasis T, Gracia CR. Body size affects measures of ovarian reserve in late reproductive age women. Menopause. 2008;15:857–861. doi: 10.1097/gme.0b013e318165981e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph JF, Sowers M, Gold EB, Mohr BA, Luborsky J, Santoro N, McConnell DS, Finkelstein JS, Korenman SG, Matthews KA, Sternfeld B, Lasley BL. Reproductive hormones in the early menopausal transition: relationship to ethnicity, body size, and menopausal status. J Clin Endocrinol Metab. 2003;88:1516–1522. doi: 10.1210/jc.2002-020777. [DOI] [PubMed] [Google Scholar]
- Nteeba J, Ganesan S, Keating AF. Impact of obesity on ovotoxicity induced by 7, 12-dimethylbenz[a]anthracene in mice. Biol Reprod. 2014;90:68. doi: 10.1095/biolreprod.113.114215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Luo LL, Xu JJ, Xu MY, Zhang XM, Zhou XL, Liu WJ, Fu YC. Obesity accelerates ovarian follicle development and follicle loss in rats. Metabolism. 2014;63:94–103. doi: 10.1016/j.metabol.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Robker RL, Akison LK, Bennett BD, Thrupp PN, Chura LR, Russell DL, Lane M, Norman RJ. Obese women exhibit differences in ovarian metabolites, hormones, and gene expression compared with moderate-weight women. J Clin Endocrinol Metab. 2009;94:1533–1540. doi: 10.1210/jc.2008-2648. [DOI] [PubMed] [Google Scholar]
- Kezele PR, Nilsson EE, Skinner MK. Insulin but not insulin-like growth factor-1 promotes the primordial to primary follicle transition. Mol Cell Endocrinol. 2002;192:37–43. doi: 10.1016/s0303-7207(02)00114-4. [DOI] [PubMed] [Google Scholar]
- Kezele P, Skinner MK. Regulation of ovarian primordial follicle assembly and development by estrogen and progesterone: endocrine model of follicle assembly. Endocrinol. 2003;144:3329–3337. doi: 10.1210/en.2002-0131. [DOI] [PubMed] [Google Scholar]
- Britt KL, Drummond AE, Dyson M, Wreford NG, Jones ME, Simpson ER, Findlay JK. The ovarian phenotype of the aromatase knockout (ArKO) mouse. J Steroid Biochem Mol Biol. 2001;79:181–185. doi: 10.1016/s0960-0760(01)00158-3. [DOI] [PubMed] [Google Scholar]
- Britt KL, Saunders PK, McPherson SJ, Misso ML, Simpson ER, Findlay JK. Estrogen actions on follicle formation and early follicle development. Biol Reprod. 2004;71:1712–1723. doi: 10.1095/biolreprod.104.028175. [DOI] [PubMed] [Google Scholar]
- Chen Y, Jefferson WN, Newbold RR, Padilla-Banks E, Pepling ME. Estradiol, progesterone, and genistein inhibit oocyte nest breakdown and primordial follicle assembly in the neonatal mouse ovary in vitro and in vivo. Endocrinol. 2007;148:3580–3590. doi: 10.1210/en.2007-0088. [DOI] [PubMed] [Google Scholar]
- Liu K. Stem cell factor (SCF)-kit mediated phosphatidylinositol 3 (PI3) kinase signaling during mammalian oocyte growth and early follicular development. Front Biosci. 2006;11:126–135. doi: 10.2741/1785. [DOI] [PubMed] [Google Scholar]
- Nteeba J, Ross JW. Perfield Ii JW, Keating AF. High fat diet induced obesity alters ovarian phosphatidylinositol-3 kinase signaling gene expression. Reprod Toxicol. 2013;42:68–77. doi: 10.1016/j.reprotox.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SW, Gang G-T, Kim YD, Ahn R-S, Harris RA, Lee C-H, Choi H-S. Insulin directly regulates steroidogenesis via induction of the orphan nuclear receptor DAX-1 in testicular Leydig cells. J Biol Chem. 2013;288:15937–15946. doi: 10.1074/jbc.M113.451773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani T, Maruo T, Yukimura N, Mochizuki M. Effect of insulin on porcine granulosa cells: implications of a possible receptor mediated action. Acta Endocrinol (Copenh) 1985;108:104–110. doi: 10.1530/acta.0.1080104. [DOI] [PubMed] [Google Scholar]
- Spicer LJ, Echternkamp SE. The ovarian insulin and insulin-like growth factor system with an emphasis on domestic animals. Domest Anim Endocrinol. 1995;12:223–245. doi: 10.1016/0739-7240(95)00021-6. [DOI] [PubMed] [Google Scholar]
- Ellison PT. Energetics and reproductive effort. Am J Hum Biol. 2003;15:342–351. doi: 10.1002/ajhb.10152. [DOI] [PubMed] [Google Scholar]
- Clancy KB, Klein LD, Ziomkiewicz A, Nenko I, Jasienska G, Bribiescas RG. Relationships between biomarkers of inflammation, ovarian steroids, and age at menarche in a rural Polish sample. Am J Hum Biol. 2013;25:389–398. doi: 10.1002/ajhb.22386. [DOI] [PubMed] [Google Scholar]
- La Vignera S, Condorelli R, Bellanca S, La Rosa B, Mousaví A, Busà B, Vicari LO, Vicari E. Obesity is associated with a higher level of pro-inflammatory cytokines in follicular fluid of women undergoing medically assisted procreation (PMA) programs. Eur Rev Med Pharmacol Sci. 2011;15:267–273. [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. 1995;95:2111–2119. doi: 10.1172/JCI117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghizadeh M, Ong JM, Garvey WT, Henry RR, Kern PA. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J Clin Invest. 1996;97:1111–1116. doi: 10.1172/JCI118504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Hofmann C, Lorenz K, Braithwaite SS, Colca JR, Palazuk BJ, Hotamisligil GS, Spiegelman BM. Altered gene expression for tumor necrosis factor-alpha and its receptors during drug and dietary modulation of insulin resistance. Endocrinol. 1994;134:264–270. doi: 10.1210/endo.134.1.8275942. [DOI] [PubMed] [Google Scholar]
- Mikuni M. [Effect of interleukin-2 and interleukin-6 on ovary in the ovulatory period–establishment of the new ovarian perfusion system and influence of interleukins on ovulation rate and steroid secretion] Hokkaido Igaku Zasshi. 1995;70:561–572. [PubMed] [Google Scholar]
- Van der Hoek KH, Woodhouse CM, Brännström M, Norman RJ. Effects of interleukin (IL)-6 on luteinizing hormone- and IL-1beta-induced ovulation and steroidogenesis in the rat ovary. Biol Reprod. 1998;58:1266–1271. doi: 10.1095/biolreprod58.5.1266. [DOI] [PubMed] [Google Scholar]
- Roby KF, Weed J, Lyles R, Terranova PF. Immunological evidence for a human ovarian tumor necrosis factor-alpha. J Clin Endocrinol Metab. 1990;71:1096–1102. doi: 10.1210/jcem-71-5-1096. [DOI] [PubMed] [Google Scholar]
- Andreani CL, Payne DW, Packman JN, Resnick CE, Hurwitz A, Adashi EY. Cytokine-mediated regulation of ovarian function. Tumor necrosis factor alpha inhibits gonadotropin-supported ovarian androgen biosynthesis. J Biol Chem. 1991;266:6761–6766. [PubMed] [Google Scholar]
- Duan Z, Foster R, Bell DA, Mahoney J, Wolak K, Vaidya A, Hampel C, Lee H, Seiden MV. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin Cancer Res. 2006;12:5055–5063. doi: 10.1158/1078-0432.CCR-06-0861. [DOI] [PubMed] [Google Scholar]
- Agarwal A, Gupta S, Sharma RK. Role of oxidative stress in female reproduction. Reprod Biol Endocrinol. 2005;3:28. doi: 10.1186/1477-7827-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Joo BS, Na YJ, Yoon MS, Choi OH, Kim WW. Relationships between concentrations of tumor necrosis factor-alpha and nitric oxide in follicular fluid and oocyte quality. J Assist Reprod Genet. 2000;17:222–228. doi: 10.1023/A:1009495913119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefler LA, Gregg AR. Inducible and endothelial nitric oxide synthase: genetic background affects ovulation in mice. Fertil Steril. 2002;77:147–151. doi: 10.1016/s0015-0282(01)02952-1. [DOI] [PubMed] [Google Scholar]
- Hefler LA, Ludwig E, Lampe D, Zeillinger R, Leodolter S, Gitsch G, Koelbl H, Tempfer CB. Polymorphisms of the endothelial nitric oxide synthase gene in ovarian cancer. Gynecol Oncol. 2002;86:134–137. doi: 10.1006/gyno.2002.6749. [DOI] [PubMed] [Google Scholar]
- Szczepańska M, Koźlik J, Skrzypczak J, Mikołajczyk M. Oxidative stress may be a piece in the endometriosis puzzle. Fertil Steril. 2003;79:1288–1293. doi: 10.1016/s0015-0282(03)00266-8. [DOI] [PubMed] [Google Scholar]
- Agarwal A, Gupta S, Sharma R. Oxidative stress and its implications in female infertility - a clinician's perspective. Reprod Biomed Online. 2005;11:641–650. doi: 10.1016/s1472-6483(10)61174-1. [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Manning AM. NF-kappaB as a primary regulator of the stress response. Oncogene. 1999;18:6163–6171. doi: 10.1038/sj.onc.1203174. [DOI] [PubMed] [Google Scholar]
- Ma CH, Yan LY, Qiao J, Sha W, Li L, Chen Y, Sun QY. Effects of tumor necrosis factor-alpha on porcine oocyte meiosis progression, spindle organization, and chromosome alignment. Fertil Steril. 2010;93:920–926. doi: 10.1016/j.fertnstert.2009.01.131. [DOI] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]