

ABSTRACT
Formation of the inner cell mass (ICM) and trophectoderm (TE) marks the first differentiation event in mammalian development. These two cell types have completely divergent fates for the remainder of the developmental process. The molecular mechanisms that regulate ICM and TE formation are poorly characterized in horses. The objective of this study was to establish the transcriptome profiles of ICM and TE cells from horse blastocysts using RNA sequencing (RNA-seq). A total of 12 270 genes were found to be expressed in either lineage. Global analysis of the transcriptome profiles by unsupervised clustering indicated that ICM and TE samples presented different gene expression patterns. Statistical analysis indicated that 1662 genes were differentially expressed (adjusted P < 0.05 and fold change > 2) between ICM and TE. Genes known to be specific to the ICM and TE were expressed primarily in their respective tissue. Transcript abundance for genes related to biological processes important for horse blastocyst formation and function is presented and discussed. Collectively, our data and analysis serve as a valuable resource for gene discovery and unraveling the fundamental mechanisms of early horse development.
Keywords: blastocyst, embryonic capsule, horse, inner cell mass, pluripotency, RNA-seq, transcriptome, trophectoderm
INTRODUCTION
During embryo preimplantation development, the first differentiation event occurs at the morula stage, leading to the formation of the blastocyst and the establishment of two distinct cell lineages, the inner cell mass (ICM) and the trophectoderm (TE). The ICM then contributes to form the fetus, whereas the TE will contribute only to placental tissues. The ICM thus constitutes a pluripotent tissue, possessing the capacity to differentiate into all three developmental layers—ectoderm, mesoderm, and endoderm—and some extra embryonic tissues, including the primitive endoderm (PE). Distinct developmental fates of the ICM and TE are regulated and controlled by their respective developmental programs and expressed molecular functions.
To better understand the process of early embryonic development, it is necessary to decipher the molecular events and regulatory networks that are the basis of early differentiation. Comparative global gene expression profiling of the ICM and TE could help to reconstruct the molecular basis for the fundamental differences between the ICM and TE. In the mouse, microarray studies have been conducted comparing ICM and TE transcriptomes and differences between IVF and in vivo-produced embryos identified [1]. Also, bovine IVF-derived ICM and TE have been profiled using microarray [2] and RNA-seq [3] technologies. It has been established that differences between species exist in terms of the molecular determination of the ICM and TE. Horse blastocysts have some unique morphological characteristics when compared to embryos of other widely studied mammalian species, such as humans, mice and cattle. For instance, horse blastocysts achieve a larger size, have a substantially higher number of cells, and form a distinctive glycoprotein protective layer between the zona pellucida and the embryo called the embryonic capsule [4]. Studying the horse blastocyst transcriptome will not only provide general insights into early molecular events of ICM and TE determination but also help uncover the molecular basis of these unique characteristics. Microarray analyses of whole horse embryos have identified some of the basis for these differences [5]; however, analyses of ICM and TE have not been performed in the horse.
In monovulatory species, in order to get sufficient amounts of sample necessary for transcriptome profiling, it is common to use IVF embryos. Recent advances in transcriptome amplification and next-generation sequencing allow for performing whole-genome studies with limited amounts of materials, including single embryos [6]. This provides the possibility of using in vivo-derived embryos and allows discrimination of interindividual variability in transcription patterns. In the current study, we measured the global gene expression profile and compared underlying differences in gene expression of various cellular and physiological processes of individual ICM and TE samples from in vivo-produced blastocysts using RNA-seq.
MATERIALS AND METHODS
Sample Preparation and RNA Extraction
Animal experiments were reviewed and approved by the Animal Care and Use Committee at the University of California, Davis. Equine blastocysts were harvested from superovulated mares by uterine flush on Day 8 after ovulation as previously described [7]. On average, superovulation treatment resulted in 4.9 ± 0.9 ovulations per mare with 2 ± 0.4 embryos resulting per flush. Six expanded blastocysts were used for this study (Fig. 1A). Recovered blastocysts were treated for 10 sec with Pronase (10 mg/ml) to remove the zona pellucida and/or capsule. ICM or TE cells were isolated using immunosurgery and microsurgery, respectively (Fig. 1B). Immunosurgery was carried out by incubating the embryo with 2% anti-horse whole serum (1 h at 37°C; Jackson Immunoresearch, West Grove, PA), followed by repeated washes and incubation with 20% guinea pig complement (1 h at 37°C; Innovative Research, Novi, MI). For microsurgical TE isolation, a microblade connected to micromanipulation equipment was used to mechanically separate the TE from the ICM (Fig. 1B). ICM and TE were individually stored in 20 μl of extraction buffer from the PicoPure RNA Isolation kit (Applied Biosystems, Carlsbad, CA) at −80°C. RNA was extracted using the PicoPure RNA Isolation Kit (Applied Biosystems), including DNAse (Qiagen, Valencia, CA) treatment, following the manufacturer's instructions but with a modified RNA elution step. In order to achieve a high concentration of RNA, elution was performed using 7 μl of diethyl pyrocarbonate-treated water, and the eluate was run through the column one more time after the initial elution. RNA quantification and quality control were performed using a high-sensitivity RNA chip with the Experion microfluidic gel system (Bio-Rad, Hercules, CA). RNA quality was evaluated by examining the 18S and 28S ribosomal subunits, quantity, and RNA quality indicator score.
FIG. 1.

ICM and TE isolation from horse blastocyst. A) Embryos used for ICM and TE isolation. B) Processing of embryos for isolation of TE and ICM. Bar = 100 μm.
RNA Amplification and Sequencing Library Preparation
Total RNA from isolated single ICM (n = 3) and TE (n = 3) was used as input for the Ovation RNA-Seq V2 kit (NuGen, San Carlos, CA). Output of cDNA was analyzed for correct size distribution with an Experion Standard Sensitivity RNA chip (Bio-Rad) and quantified using a Qubit Fluorometer (Invitrogen, Carlsbad, CA). Amplified cDNA with a size distribution of ∼200–500 bp was considered acceptable. Next, 1 μg of cDNA from each sample was used with the TruSeq DNA Sample Prep Kit V1 (Illumina, San Diego, CA) to produce sequencing libraries sized between 200 and 400 bp. Libraries were sequenced at the Vincent J. Coates Genomics Sequencing Laboratory at the University of California, Berkeley, on a HiSeq 2000 apparatus (Illumina) as 100-bp single reads. Quality control of reads was performed using CLC Genomics Workbench (CLCbio, Aarhus, Denmark). Reads with >2 ambiguous nucleotides or low-quality bases were removed based on Illumina quality scores. Also, 9 bp from the 5′ end of each read were trimmed based on altered GC ratios, as we have previously indicated [6]. Sequenced reads are available under GEO accession number GSE51431.
Read Mapping and Gene Expression Analysis
Read mappings were done using the RNA-Seq Analysis tool of CLC Genomics Workbench 6.0 with default settings (CLCbio). Reads were first mapped to the ENSEMBL annotated genes (EquCab2.73). Unaligned reads were then mapped to the NCBI annotation (Equus caballus 2.0, GenBank; ftp://ftp.ncbi.nlm.nih.gov/genomes/Equus_caballus; Supplemental Table S1; all Supplemental Data are available online at www.biolreprod.org). Finally, remaining unaligned reads were mapped to putative genes discovered using the ab-initio transcript discovery tool from CLC Genomics Workbench (using default parameters). To identify de novo annotated genes, their transcript sequences were BLASTed to the Human RefSeq mRNA database, and official gene names were given to matched transcripts (E-value < 10−4). All mappings were combined and reads per kilobase of exon model per million mapped reads (RPKM) values calculated for each gene. Genes with an RPKM >0.4 in at least two samples within a sample type were considered expressed. DESeq was used for differential gene expression analysis using total exon-mapped reads as input and total mapped reads for normalization of read counts across samples [8]. Hierarchical clustering and heat maps were constructed in R using the heatmap2 package and the variance stabilized data file from DESeq as input.
Real-Time PCR
Real-time quantitative PCR was performed using SSOFast EvaGreen Supermix (Bio-Rad) with the following cycling parameters: 30 sec at 94°C followed by 40 repeats of 5 sec at 94°C, 10 sec at 60°C, and finally 15 sec at 72°C. Primers were designed using Primer Express (Applied Biosystems). All reactions were performed in duplicate using 20-μl volumes containing 1 ng of cDNA used for RNA-seq and 0.3 μM of each primer. The ΔΔCt method was used for relative quantification and data analysis. The melting curves of each primer pair were evaluated to ensure the absence of nonspecific products and primer dimer formation. RPL15 was used to normalize gene expression levels. Data were then reported as mean fold change (FC). The genes analyzed by qPCR were NANOG, CYP1B1, GPC4, PDGFRA, SPP1, SOX2, and OCT4 and were chosen based on differential gene expression and specificity for the ICM cell lineage. A Student t-test was used to compare log2 FC values across tissue types.
Gene Ontology Analysis
Gene classifications and biological functional annotations were done using the online DAVID database v6.7 [9, 10]. Gene symbols of differentially expressed (adjusted P ≤ 0.05) genes in ICM and TE were submitted to DAVID. The human database was used for gene ontology (GO) biological function annotation and reference. After functional annotation clustering using default parameters, the most significant category within each of the most significant clusters was selected.
RESULTS
Overview of RNA-Seq Results
Transcript abundance and differential gene expression were determined in individual ICM (n = 3) and TE (n = 3) samples by RNA-seq. An average of 5.5 ± 0.4 and 3.2 ± 0.5 ng of total RNA were obtained from single ICM and TE samples, respectively. After single-primer isothermal amplification (Ovation RNA-seq System V2), more than 5 μg of cDNA per sample were available for Illumina Truseq library preparation and sequencing. After read processing, a total of 193 million high-quality reads (32 million per sample) were used for analysis (Supplemental Table S1).
De novo transcript discovery was performed to enhance the equine genome annotation. A total of 571 newly annotated genes were expressed in equine embryos, of which 306 had a human ortholog. This gene group was enriched for the GO biological processes transcription and response to estrogen stimulus.
Mapping sequentially to ENSEMBL, NCBI and de novo transcripts resulted in 77% of the reads (approximately 25 million reads per sample) assigned to genes. This represented 12 270 genes expressed at RPKM > 0.4 in at least two samples within each tissue. The highest RPKM value was 115 666, indicating a dynamic range of expression spanning seven orders of magnitude. Of all expressed genes, 10 364 were present in both ICM and TE, whereas 1201 and 705 genes were exclusively expressed in ICM and TE, respectively (Fig. 2A). Unsupervised hierarchical clustering using all the expressed genes segregated ICM and TE samples into two separate groups (Fig. 2B), suggesting that each compartment has a distinct gene expression signature. Overall, these results show that global transcriptome analysis of ICM and TE from individual blastocysts by RNA-seq is possible.
FIG. 2.

Transcriptome analysis of equine ICM and TE samples. A) Venn diagram indicating the number of expressed genes detected (RPKM > 0.4) in ICM and TE samples. B) Unsupervised hierarchical clustering based on global gene expression of ICM and TE samples.
Differential Gene Expression Between ICM and TE Samples and qPCR Validation of RNA-Seq Results
A total of 2040 genes were differentially expressed between ICM and TE samples (Fig. 3; adjusted P-value < 0.05). Among these, 1145 were up-regulated (FC > 2) in the ICM, and 517 were up-regulated (FC > 2) in the TE (Table 1). Lists of all genes up-regulated in either ICM or TE samples are presented in Supplemental Table S2.
FIG. 3.

ICM and TE differential gene expression. Heat map and hierarchical cluster of samples based on differentially expressed genes (adjusted P < 0.05).
TABLE 1.
Number of genes differentially expressed between ICM and TE (adjusted P < 0.05; DESeqa).

Only genes with RPKM > 0.4 were included in analysis.
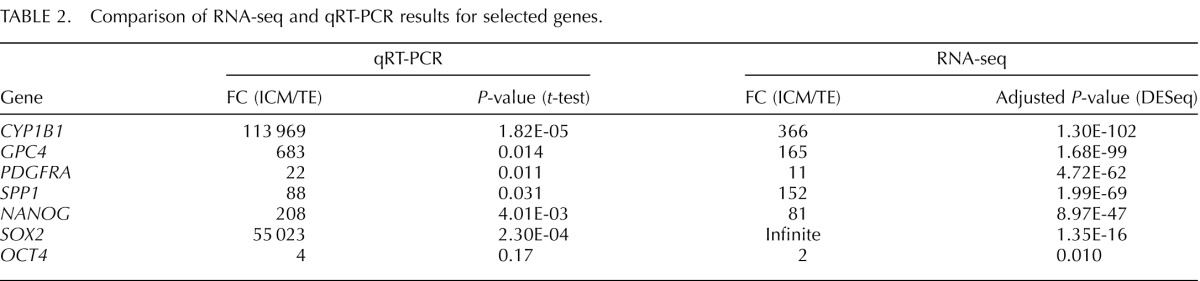
A set of genes found differentially expressed based on RNA-seq results were selected for analysis using quantitative real-time RT-PCR (qRT-PCR). A similar trend in expression levels and differential expression between ICM and TE was observed when comparing qRT-PCR and RNA-seq results (Table 2), validating the RNA-seq approach for determining single-embryo ICM and TE gene expression profiles.
TABLE 2.
Comparison of RNA-seq and qRT-PCR results for selected genes.
One advantage of single-embryo transcriptome determination is the possibility of identifying the sex of the embryo [6]. We explored this possibility by comparing the levels of XIST expression between samples, given that XIST is expressed during X-chromosome inactivation in female cells. ICM_22, ICM_25, and TE_16 presented high levels of XIST expression (RPKM: 71.04, 69.64, and 53.67, respectively) and were likely derived from female embryos. On the other hand, ICM_15, TE_23, and TE_24 showed reduced levels of expression (RPKM: 0.70, 0.03, and 0.35, respectively) and were thus likely derived from male embryos.
Functional Annotation of ICM and TE Differentially Expressed Genes
Biological processes enriched in genes overexpressed in ICM were related mainly to phosphorylation, cytoskeleton organization, regeneration, cell morphogenesis involved in differentiation, and regulation of cell migration (Table 3). In contrast, the TE was enriched mainly in biological processes involving cellular transport, such as sodium ion, solute, and carboxylic acid transport (Table 4).
TABLE 3.
GO biological processes enriched among genes overexpressed in ICM compared to TE (adjusted P < 0.05).

TABLE 4.
GO biological processes enriched among genes overexpressed in TE compared to ICM (adjusted P < 0.05).

Expression of Genes Related to Biological Processes Relevant to Horse Embryonic Development
RNA-seq results were mined to determine the expression of genes known to be associated with biological processes associated with horse blastocyst formation and early development. These included expression of lineage specific markers and epigenetic regulators, blastocyst expansion, and capsule formation. Results for selected genes are presented in Supplemental Tables S3–S6 and discussed in the following section.
DISCUSSION
This study represents the first comprehensive global characterization of gene expression of individual in vivo ICM and TE compartments from equine blastocysts. Use of RNA-seq technology in ICM and TE allowed not only the comprehensive characterization of gene expression in these cell lineages but also the annotation of novel horse genes. The newly annotated genes were involved mainly in biological processes, such as transcription. Previous transcriptome studies of ICM and TE in other species used samples from multiple pooled blastocysts for analysis [1, 3]. Pooling methods mask individual embryo variability and gene expression heterogeneity, which is of increasing importance in basic research and medical sciences. Clustering samples based on their global gene expression patterns separated each sample to its corresponding type; however, more variability was observed among ICM samples, with ICM25 being the most deviant, while TEs clustered closer together. Higher variability in ICM may be contributed by differences in the differentiation state of the embryo or sample preparation. Considering that sample ICM25 could be an outlier, we also analyzed the data excluding this sample (not shown), and while the specific numbers of differentially expressed genes changed, the overall biological conclusions remained the same.
In an earlier bovine single-embryo study [6], as well as in this one, we showed that single-embryo transcriptome analysis allows for the determination of embryonic sex. This is an important consideration given that significant differences in gene expression patterns have been reported for male and female embryos [6, 11]. However, the low number of samples within each group prevented us from incorporating embryo gender as a covariate. It is also important to recognize that some of the changes reported here could be due to the use of embryos obtained from superovulated mares, as it has previously been reported in mice that superovulation can affect the blastocyst transcriptome [12].
Functional annotation of genes differentially expressed in ICM and TE showed that the developmental fate of each cell type is reflected by the respective transcriptome [13]. Major processes active in ICM were protein phosphorylation related to signaling pathways and morphogenesis/regeneration related to growth factors and signaling, whereas TE was enriched in cellular transport, placental development, and polymer glycosylation.
Expression of Pluripotency and Lineage Specific Markers
ICM and TE cell type-specific markers were directly explored in the RNA-seq transcriptome data to help elucidate mechanistic aspects of lineage determination in the early horse embryo (Fig. 4). The ICM molecular signature included genes commonly associated with pluripotency and ICM specification, such as POU5F1 (OCT4), NANOG, SOX2, KLF4, LIN28B, DNMT3B, SMARCA2, UTF1, ID2, and SPP1. DPPA4 and SALL4 were also numerically higher in the ICM but did not reach statistical significance. SOX2, NANOG, SMARCA2, and SPP1 were exclusively expressed in ICM samples. In general, these expression patterns agree with what is commonly reported for other species [2, 3, 14]. Interestingly, ID2 was expressed higher in the ICM, similar to what has been recently reported for cattle embryos [2] but in contrast to reports in mice [14]. It is also worth noting that POU5F1 (OCT4) is expressed in both ICM and TE of horse blastocysts. In the RNAseq data, a significant 2× higher expression in the ICM was observed, while qPCR analysis revealed a 4FC, but because of high variability between samples, the difference was not statistically significant. Overall, these results are in agreement with reported immunostaining for OCT4 in in vivo-derived Day 7–8 horse embryos in which nuclear localization of the protein is observed in both ICM and TE, although with a reduced level in the TE [15].
FIG. 4.

Expression of lineage-specific genes in individual horse ICM and TE samples. Cell color indicates relative level of expression within gene, with red and green indicating high and low expression, respectively. RPKM, reads per kilobase of exon model per million mapped reads; FC, fold change; a DESeq analysis.
For Day 8–14 conceptuses, SPP1 has been shown to be expressed highest in Day 8 embryos [5]. SPP1 is controlled by upstream factor OCT4 in mouse ICM, implicating the role of SPP1 in ICM pluripotency. Exclusive expression of SPP1 solely in the ICM and downstream of OCT4 makes it a strong pluripotency marker candidate for horse ES cells. Furthermore, SOX2, SMARCA2, and NANOG appear as better ICM and pluripotency markers in these samples compared to other commonly used genes, such as POU5F1 (OCT4), due to the exclusivity of expression in the ICM.
We also investigated the expression of components of signaling pathways generally associated with pluripotent cells and ICM determination. FGF2 signaling has been shown to support pluripotency and self-renewal of human embryonic stem cells [11], which have epiblast characteristics. FGF2 induces biological responses by binding to and activating fibroblast growth factor receptor (FGFR). Genes encoding FGF2 ligand and receptors, such as FGFR1, FGFR4, and GAB1, were selectively and significantly up-regulated in the ICM (FGFR1 and FGFR4; P < 0.05; Supplemental Table S3), indicating a potential role in determining the pluripotent nature of the ICM, whereas GRB2 and FGFBP1 were expressed higher in TE (Supplemental Table S3). Transmembrane heparan sulfate proteoglycans, such as Syndecan-2 (SDC2), are able to bind FGF2 to heparan sulfate chains and present it to the FGFR1 [12]. SDC2 was 16.8× higher in the ICM as compared to TE.
Major downstream components of FGF signaling include mitogen-activated protein kinase (MAPK) pathway and phosphatidylinositol-3-kinase kinase (PI3K) stimulated by GAB1. MAPK1 and 2 (MEK1/2) and MAPK1 and 3 (ERK1/2) components were expressed in both compartments, with some up-regulated in the ICM (Supplemental Table S3). PI3K genes expressed in blastocyst included PIK3C2B, PIK3C3, PIK3CA, PIK3CB, PIK3R1, and PIK3R4, with PIK3C2A expressed 2.3× significantly higher in the ICM (Supplemental Table S3). Also, downstream effectors of these pathways, such as AKT and KRAS, were also expressed.
Active FGF signaling is essential for segregation of the ICM lineage from the PE lineage during blastocyst development [16]. Selective up-regulation of FGF signaling components in the ICM may promote the formation of PE in horse blastocysts at early developmental stages. Previous attempts to generate horse and other ungulate ES cells from the dissected ICM failed because the ICM grows into trophectodermal and PE-like cells instead of ES cells, which can still form embryoid-like bodies similar to those formed by ESCs [17]. High FGF pathway activity in the horse ICM, as shown by our data, may interfere with the successful derivation of pluripotent horse ES cells.
In mouse stem cells and embryos, LIF signaling is required for maintaining pluripotency. We observed expression of genes associated with LIF signaling, such as receptors LIFR and IL (gp130), and downstream effectors and targets, such as JAK2, STAT3, and ID, that were significantly up-regulated (LIFR, IL, JAK2, and ID2; Supplemental Table S3) in the horse ICM compared to the TE. STAT1 and STAT3 were expressed in both ICM and TE, while the LIF ligand was expressed only in TE (Supplemental Table S3). These data support an active LIF signaling pathway in horse ICM cells and is reinforced by horse iPSC data in which LIF was used as a supplemental factor [18, 19].
BMP signaling is also important for mouse ESC pluripotency. Ligands and receptors for BMP were expressed in the horse blastocyst. Importantly, a target of BMP activity in mouse, the inhibitor of differentiation (ID) gene, was up-regulated in the horse ICM (Supplemental Table S3). Similarly, other members of the TGFβ superfamily were expressed, suggesting active involvement of this pathway in early horse embryogenesis.
The TE molecular signature included genes such as transcription factors CDX2 and GATA3 and extracellular matrix protein FREM2 (Fig. 4). While CDX2 was not statistically different between samples, similar to findings in bovine embryos [3], the overall expression levels were low, and considering our RPKM thresholds for expression (RPKM > 0.4), these genes would be considered expressed in each of the three TE samples and none of the ICM samples. TEAD4 was also numerically higher only in the TE versus the ICM samples, but its downstream effector, GATA3, was significantly up-regulated in the TE. Overall, this information suggests that, like in mouse, TEAD4-stimulated GATA3 and CDX2 activity are involved in specification of the TE in the horse. Also, consistent with the nutrient uptake and secretion properties of the trophectoderm, biological processes related to transport activities, were up-regulated in the TE (Fig. 4).
Interestingly, PE markers such as GATA4, GATA6, FOXA2, HNF4A, and PDGFRA were present in both ICM and TE samples but typically at higher levels in the ICM. The presence of PE markers in transcriptome indicated that the PE lineage is already present in the Day 8 blastocyst, as previously reported [20]. Since the PE seems to develop and cover the inner surface of the blastocoele at an earlier stage in the horse embryos, it is possible that our ICM and TE isolation approaches resulted in contamination of both samples with PE cells but more significantly those of the ICM since all the PE would be included after the immunosurgery approach, while microsurgery would include only those PE cells in direct contact with the mural TE.
Lineage-specific data presented here not only validated the RNA-seq data but also provided insights into the expression patterns of pluripotent genes in different compartments of the equine blastocyst.
Expression of Epigenetic Regulators
Expression of genes regulating chromatin remodeling and DNA modification enzymes were also analyzed. DNA methyltransferases DNMT1, DNMT3A, and DNMT3B (1.84× higher in ICM) were highly expressed (RPKM > 5, average of 41) in both ICM and TE (Supplemental Table S4). Some genes known to take part in active DNA demethylation, such as TET1, TET2, and GADD45A, were also present in both the ICM and TE, whereas TET3 was detected at a much lower level (RPKM < 2; Supplemental Table S4).
The expression of histone-modifying methylases, acetylases, demethylases, and deacetylases is summarized in Supplemental Table S4. Genes encoding important histone modifiers that were expressed significantly higher in the ICM included HDAC5, SIRT1, JMJD1C, EZH2, KAT7, and KDM4A, whereas the histone modifiers expressed higher in TE were EHMT2, SMYD2, and SETBP1 (P < 0.05) [12]. Histone-modifying genes such as SET (average RPKM = 129.74), HDAC1 (average RPKM = 41.06), and NSD1 (average RPKM = 38.72) were expressed at high levels in both lineages (Supplemental Table S4), suggesting that they are the major histone modifiers at Day 8 of horse development. High expression of epigenetic modifiers may underscore the active remodeling occurring at this stage of embryo development. Expression of many histone and DNA modification enzymes suggests active DNA and chromatin modification in the developing blastocyst, processes that have been shown to be critical for blastocyst development [21].
Expression of Genes Involved in Blastocyst Growth and Expansion
Embryo development after Day 7 in the horse is characterized by a rapid increase in cell number and size due to cell proliferation and expansion of the blastocoel [13]. The expression profile of major genes associated with cell division was analyzed. Many cell cycle regulatory genes active in ES-cells [22], including AURKA, AURKB, AURKC, CCNG1, CCNF, BUB1, MAD2L1, and CDC20, were expressed in both ICM and TE (Supplemental Table S6). Plasma membrane sodium/potassium pumps (Na+, K+-ATPase) facilitate the influx of fluid inside the blastocoel [23]. Na+, K+-pump ATP1A1, ATP1B1, and ATP1B3 were the major isoforms expressed (average RPKM > 50). Transport of fluid into the blastocoel is mediated by specialized proteins present in TE cells called aquaporin (AQP). Our results showed that aquaporin genes such as AQP3 and AQP5 were expressed with AQP5 up-regulated 3.63× (P < 0.05) in the TE. Transcripts for tight junction and gap junction proteins, important for morula compaction and blastocoel formation [13], such as CLDN7, TJAP1, TJP1, TJP2, CDH1, OCLN, CGN, and GJA1 (CONNEXIN43) were also expressed. CDH1, CDH2, and CDH8 were expressed significantly higher in ICM (Supplemental Table S5). Gap junction gene GJC1 was expressed significantly higher in TE compared to ICM (Supplemental Table S5).
RNA-seq results showed that various mechanisms responsible for blastocyst formation and growth are active in Day 8 equine embryos.
Expression of Genes Related to Capsule Formation
When the horse embryo enters the uterus, late Day 5 after ovulation, a distinct acellular membrane is synthesized by the TE and deposited between the embryo and the zona pellucida, providing protection to the developing embryo [24, 25]. Capsule formation is a vital step in equine embryo development, and therefore analyzing its origin at the molecular level is critical to our understanding of this unique aspect of horse development. We explored our transcriptome data for components of pathways that may be responsible for synthesis of constituents of the capsule, as reported earlier [26] (Supplemental Table S6). Biochemically, the equine capsule is composed of mucin-like glycoproteins, mostly sialic acid- Neu5Ac (more than 60%), and sugars, such as N-acetyl-galactosamine (GalNAc), galactose (Gal), N-acetyl-glucosamine (GlcNAc), and sulfated polysaccharides [26]. Mucin secreted from the trophectoderm and maternal endometrium has been shown to be responsible for blastocyst capsule synthesis [27]. Our RNA-seq data revealed that mucin-coding genes, such as MUC1 and MUC20, were expressed in blastocysts at average RPKM > 1, suggesting a possible source for capsule mucoproteins. Other major genes involved in synthesis of proteoglycans, such as GALNT1, GALNT4, and GALNT12, were significantly higher in the TE compared to the ICM, whereas GALNT2, GALNT3, GALNT6, GALNT7, and GALNT10 were expressed in both tissues [28]. Genes coding for key enzymes catalyzing sialic acid biosynthesis, such as NAGK, NANS, CMAS, and GNE, were expressed in blastocysts, but GNE was significantly higher (2.42×) in the TE. Sialyltransferases (SIAE, ST6GAL1, and ST8SIA2) [29, 30] were also expressed in blastocysts. A previous study identified sialidase NEU2 as one of the sialic acid-metabolizing enzymes secreted by the Day 8 horse blastocyst [31]. Based on the RNA-seq data, NEU1 seems to be the major sialidase expressed in the Day 8 blastocyst, whereas NEU2 and NEU3 were expressed at lower levels. Genes encoding enzymes responsible for GalNac metabolism, such as galactosyltransferase, acetylglucosaminyltransferase (B3GNT2), N-acetylgalactosaminyltransferase (B3GALNT1), sialyltransferase (ST3GAL4), and glycosyltransferase (GLT8D1) [29], were expressed in the blastocyst, with sialyltransferase ST3GAL6 and ST3GAL1 expressed significantly higher in ICM and TE, respectively. Previously reported in horse blastocyst [5], CMP-sialic acid transporter (SLC35A1) and sialic acid transporter (SLC17A5) were expressed significantly higher in the TE. UDP-galactose transporters (SLC35A2 and SLC35A3 and SLC35A4) were expressed in both blastocyst compartments. Other transporters that play important roles in glycoprotein biosynthesis, such as SLC35B4 and SLC35B2, were also expressed in blastocyst but slightly higher in TE (1.33× and 2.04× higher, respectively). We also explored the RNA-seq data for genes encoding enzymes involved in biosynthesis of sulfated sugars, which is another important building block of the equine capsule [26]. Many genes encoding sulfotransferases were expressed in the blastocyst, such as carbohydrate sulfotransferases CHST8 and CHST10, whereas CHST4 was expressed significantly higher in the TE (3.15×; P < 0.0001). Many heparan sulfotransferase genes, such as HS2ST1. were also expressed in the blastocyst. Transglutaminases (TGM3) have been proposed to play important role in rabbit blastocyst coat formation and reinforcement, which is biochemically similar to the horse capsule [32]. In Day 8 horse blastocysts, high levels of TGM3 are observed mainly in the TE (1.75×; P < 0.05 higher than in the ICM). Our finding confirms the previous report of expression of capsule-related genes in the horse blastocyst [5] and also introduces many new genes. Several of the putative genes involved in capsule biosynthesis are selectively up-regulated in TE, suggesting an active role for the TE in capsule biosynthesis.
Taken together, our data provide comprehensive insight into the gene expression program during the first differentiation event in horse embryonic development. A comparative global expression profiling approach between ICM and TE revealed a subset of genes that were differentially expressed. ICM transcriptome analysis provides clues as to the establishment and maintenance of horse pluripotent cells and could help in the derivation of horse ESCs and improve our understanding of the early horse developmental program. The identification of gene expression programs involved in ICM and TE will further our understanding of the complex molecular and cellular events in early developmental processes and provide a foundation for future studies of metabolism, growth, and differentiation in mammalian embryos.
Supplementary Material
Footnotes
Supported by the Center for Equine Health UC Davis with funds provided by the State of California pari-mutuel fund and contributions by private donors to P.J.R. and J.F.R. and by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) by R01HD070044 to P.J.R. Sequencing data were deposited in the Gene Expression Omnibus (GEO) repository under accession number GSE51431. Presented in part at the 39th Annual Conference of the International Embryo Transfer Society, January 19–22, 2013, Hannover, Germany.
REFERENCES
- Giritharan G, Delle Piane L, Donjacour A, Esteban FJ, Horcajadas JA, Maltepe E, Rinaudo P. In vitro culture of mouse embryos reduces differential gene expression between inner cell mass and trophectoderm. Reprod Sci. 2012;19:243–252. doi: 10.1177/1933719111428522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatomo H, Kagawa S, Kishi Y, Takuma T, Sada A, Yamanaka K, Abe Y, Wada Y, Takahashi M, Kono T, Kawahara M. Transcriptional wiring for establishing cell lineage specification at the blastocyst stage in cattle. Biol Reprod. 2013;88:158. doi: 10.1095/biolreprod.113.108993. [DOI] [PubMed] [Google Scholar]
- Ozawa M, Sakatani M, Yao J, Shanker S, Yu F, Yamashita R, Wakabayashi S, Nakai K, Dobbs KB, Sudano MJ, Farmerie WG, Hansen PJ. Global gene expression of the inner cell mass and trophectoderm of the bovine blastocyst. BMC Dev Biol. 2012;12:33. doi: 10.1186/1471-213X-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betteridge KJ. Equine embryology: an inventory of unanswered questions Theriogenology 2007. 68 (suppl 1): S9 S21 [DOI] [PubMed] [Google Scholar]
- Klein C, Troedsson MH. Transcriptional profiling of equine conceptuses reveals new aspects of embryo-maternal communication in the horse. Biol Reprod. 2011;84:872–885. doi: 10.1095/biolreprod.110.088732. [DOI] [PubMed] [Google Scholar]
- Chitwood JL, Rincon G, Kaiser GG, Medrano JF, Ross PJ. RNA-seq analysis of single bovine blastocysts. BMC Genomics. 2013;14:350. doi: 10.1186/1471-2164-14-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers-Brown G, Bidstrup LA, Famula TR, Colgin M, Roser JF. Treatment with recombinant equine follicle stimulating hormone (reFSH) followed by recombinant equine luteinizing hormone (reLH) increases embryo recovery in superovulated mares. Anim Reprod Sci. 2011;128:52–59. doi: 10.1016/j.anireprosci.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Bermejo-Alvarez P, Rizos D, Rath D, Lonergan P, Gutierrez-Adan A. Sex determines the expression level of one third of the actively expressed genes in bovine blastocysts. Proc Natl Acad Sci U S A. 2010;107:3394–3399. doi: 10.1073/pnas.0913843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang L, Li X, Li K, Fang J, Yao Y. Ovarian stimulation retards postimplantation development and alters global gene expression profile of blastocysts in mouse. Fertil Steril. 2010;93:2770–2773. doi: 10.1016/j.fertnstert.2010.03.018. [DOI] [PubMed] [Google Scholar]
- Cockburn K, Rossant J. Making the blastocyst: lessons from the mouse. J Clin Invest. 2010;120:995–1003. doi: 10.1172/JCI41229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, Robson P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell. 2010;18:675–685. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Choi YH, Harding HD, Hartman DL, Obermiller AD, Kurosaka S, McLaughlin KJ, Hinrichs K. The uterine environment modulates trophectodermal POU5F1 levels in equine blastocysts. Reproduction. 2009;138:589–599. doi: 10.1530/REP-08-0394. [DOI] [PubMed] [Google Scholar]
- Yamanaka Y, Lanner F, Rossant J. FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development. 2010;137:715–724. doi: 10.1242/dev.043471. [DOI] [PubMed] [Google Scholar]
- Keefer CL, Pant D, Blomberg L, Talbot NC. Challenges and prospects for the establishment of embryonic stem cell lines of domesticated ungulates. Anim Reprod Sci. 2007;98:147–168. doi: 10.1016/j.anireprosci.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Breton A, Sharma R, Diaz AC, Parham AG, Graham A, Neil C, Whitelaw CB, Milne E, Donadeu FX. Derivation and characterization of induced pluripotent stem cells from equine fibroblasts. Stem Cells Dev. 2013;22:611–621. doi: 10.1089/scd.2012.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy K, Sung HK, Zhang P, Laflamme S, Vincent P, Agha-Mohammadi S, Woltjen K, Monetti C, Michael IP, Smith LC, Nagy A. Induced pluripotent stem cell lines derived from equine fibroblasts. Stem Cell Rev. 2011;7:693–702. doi: 10.1007/s12015-011-9239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enders AC, Schlafke S, Lantz KC, Liu IKM. Endoderm cells of the equine yolk sac from Day 7 until formation of the definitive yolk sac placenta. Equine Vet J. 1993;25:3–9. [Google Scholar]
- Fulka H, Mrazek M, Tepla O, Fulka J., Jr. DNA methylation pattern in human zygotes and developing embryos. Reproduction. 2004;128:703–708. doi: 10.1530/rep.1.00217. [DOI] [PubMed] [Google Scholar]
- Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPhee DJ, Jones DH, Barr KJ, Betts DH, Watson AJ, Kidder GM. Differential involvement of Na(+),K(+)-ATPase isozymes in preimplantation development of the mouse. Dev Biol. 2000;222:486–498. doi: 10.1006/dbio.2000.9708. [DOI] [PubMed] [Google Scholar]
- Flood PF, Betteridge KJ, Diocee MS. Transmission electron microscopy of horse embryos 3–16 days after ovulation. J Reprod Fertil Suppl. 1982;32:319–327. [PubMed] [Google Scholar]
- Freeman DA, Butler JE, Weber JA, Geary RT, Woods GL. Co-culture of day-5 to day-7 equine embryos in medium with oviductal tissue. Theriogenology. 1991;36:815–822. doi: 10.1016/0093-691x(91)90347-g. [DOI] [PubMed] [Google Scholar]
- Oriol JG, Betteridge KJ, Clarke AJ, Sharom FJ. Mucin-like glycoproteins in the equine embryonic capsule. Mol Reprod Dev. 1993;34:255–265. doi: 10.1002/mrd.1080340305. [DOI] [PubMed] [Google Scholar]
- Gillies LK, Waelchli RO, Ruddock WDJ, Betteridge KJ, LaMarre J. Patterns of MUC1 expression in the equine endometrium and trophoblast during early pregnancy. Theriogenology. 1999;51:225–225. [Google Scholar]
- Arar S, Chan KH, Quinn BA, Waelchli RO, Hayes MA, Betteridge KJ, Monteiro MA. Desialylation of core type 1 O-glycan in the equine embryonic capsule coincides with immobilization of the conceptus in the uterus. Carbohydr Res. 2007;342:1110–1115. doi: 10.1016/j.carres.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhausen I, Schachter H, Stanley P. O-GalNAc glycans In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, GW Hart, Etzler ME. (eds.), Essentials of Glycobiology, 2nd ed Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2009. 115 128 [PubMed] [Google Scholar]
- Klein C, Troedsson M. Equine pre-implantation conceptuses express neuraminidase 2–a potential mechanism for desialylation of the equine capsule. Reprod Domest Anim. 2012;47:449–454. doi: 10.1111/j.1439-0531.2011.01901.x. [DOI] [PubMed] [Google Scholar]
- Denker HW. Structural dynamics and function of early embryonic coats. Cells Tissues Organs. 2000;166:180–207. doi: 10.1159/000016732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.