


Abstract
We have used DNA footprinting and fluorescence melting experiments to study the sequence-specific binding of a novel minor groove binding ligand (thiazotropsin A), containing an isopropyl substituted thiazole polyamide, to DNA. In one fragment, which contains every tetranucleotide sequence, sub-micromolar concentrations of the ligand generate a single footprint at the sequence ACTAGT. This sequence preference is confirmed in melting experiments with fluorescently labelled oligonucleotides. Experiments with DNA fragments that contain variants of this sequence suggest that the ligand also binds, with slightly lower affinity, to sequences of the type XCYRGZ, where X is any base except C, and Z is any base except G.
INTRODUCTION
A large number of ligands have been characterized, which bind in the minor groove of duplex DNA. Many of these are based on the natural products distamycin and netropsin or the synthetic bisbenzimidazole Hoechst 33258 (1–3). These compounds are generally selective for AT-rich sequences, to which they bind in a 1:1 mode (4,5). Binding to GC-containing sequences is prevented by steric clash with the 2-amino group of guanine, while the curved molecules show good contact with the narrow minor groove that is typically found in AT-rich sequences. The observation that distamycin can bind to some sequences in an antiparallel side-by-side 2:1 mode (6) has led to the development of the pyrrole–imidazole polyamides as general sequence-reading agents (7–9). These compounds recognize specific sequences by the side-by-side pairing of aromatic amino acids in the minor groove, in which pyrrole opposite imidazole (Py/Im) targets a C–G base pair, Im/Py targets G–C and the Py/Py pair binds to both A–T and T–A.
Although these molecules make specific hydrogen bond interactions with groups in the DNA minor groove, a large part of their binding energy is derived from hydrophobic interactions (10). We have therefore sought to develop novel distamycin analogues with heterocyclic rings of different shapes and sizes so as to produce molecules with improved potential for following the contours of the minor groove (11). In preliminary studies we have shown that a distamycin analogue (thiazotropsin A, Figure 1A), in which one of the N-methylpyrroles is substituted by an isopropyl-substituted thiazole, is selective for the sequence 5′-ACTAGT-3′ (11). It is thought that two molecules of this compound bind side-by-side in the minor groove but, because of the large size of the substituted ring, the molecules are staggered relative to each other and the complex therefore reads a total of 6 bp. In the present study we have determined the sequence specificity of this compound by footprinting and fluorescence melting studies.
Figure 1.
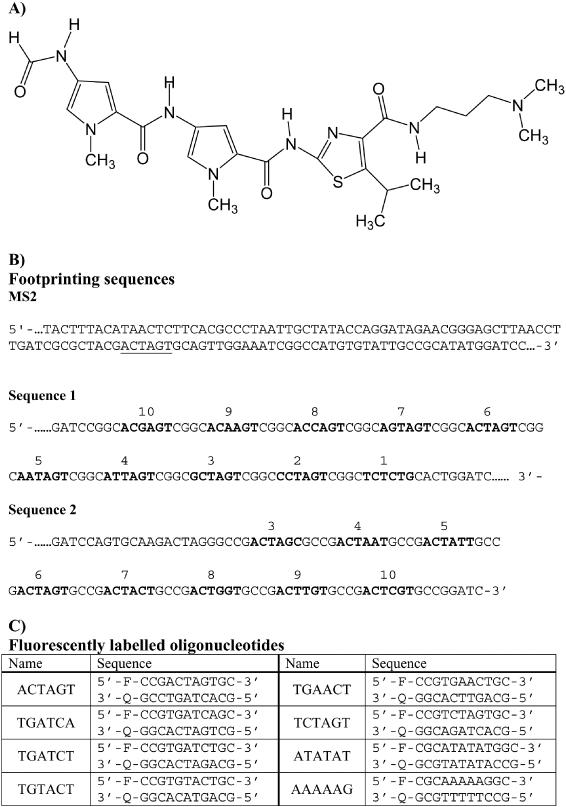
(A) Structure of thiazotropsin A. (B) Sequences of the footprinting substrates MS2, sequences 1 and 2. The fragments were each labelled at the 3′ end and only the labelled strand is shown. For sequences 1 and 2 the potential binding sites are numbered and are shown in bold. (C) Sequence of the fluorescently labelled oligonucleotides that were used for the melting studies. F indicates the fluorophore (fluorescein, dR-FAM) while Q is the quencher (methyl red, dR-MeRed).
MATERIALS AND METHODS
Oligodeoxyribonucleotides
Unmodified oligonucleotides were purchased from Oswel DNA service, (Southampton, UK) and were synthesized on an Applied Biosystems 394 DNA/RNA synthesizer. Oligonucleotides containing fluorophores and quenchers were prepared on either the 0.2 or 1 μmol scale and were purified by high-performance liquid chromatography (HPLC) as a single peak. Methyl red and fluorescein were incorporated into these oligonucleotides using MeRed-dR or Fam-dR (12). The sequences of all the fluorescently labelled oligonucleotides used in these studies are shown in Figure 1C. The Tm values of the duplexes, determined by fluorescence melting, confirmed that the sequences were indeed complementary. Oligonucleotides for preparing footprinting fragments were synthesized on the 40 nmol scale.
DNA footprinting substrates
The preparation of DNA fragment MS2, which was designed to contain all 136 tetranucleotide sequences has been previously described (13). The fragment was isolated from the parent plasmid by cutting with HindIII and SacI, and was labelled at the 3′ end of the HindIII site with [α-32P]dATP. The sequence of the labelled strand is shown in Figure 1B. DNA fragments containing variations on the putative ligand binding site (ACTAGT) were obtained by cloning appropriate synthetic oligonucleotides into the BamHI site of pUC18. The sequences of the resulting fragments, which are shown in Figure 1B, were confirmed by DNA sequencing using a T7 sequencing kit (Pharmacia). Radiolabelled DNA fragments were prepared by digesting these plasmids with HindIII and SacI, and were labelled at the 3′ end of the HindIII site with [α-32P]dATP using reverse transcriptase. Radiolabelled DNA fragments were separated from the remainder of the plasmid on 6% non-denaturing polyacrylamide gels. The bands containing the radiolabelled DNA were excised and eluted into 10 mM Tris–HCl pH 7.5 containing 0.1 mM EDTA. The DNA was then precipitated with ethanol in the presence of 0.3 M sodium acetate and dissolved in 10 mM Tris–HCl pH 7.5, containing 0.1 mM EDTA at a concentration of 10–20 c.p.s./μl, as determined on a hand-held Geiger counter.
DNA footprinting
DNase I and hydroxyl radical footprinting reactions were performed as previously described (14). Briefly, 1.5 μl of the radiolabelled DNA solution was mixed with 1.5 μl ligand, dissolved 10 mM Tris–HCl pH 7.5 containing 10 mM NaCl. This mixture was allowed to equilibrate for at least 30 min before digesting with either DNase I or a hydroxyl radical generating mixture. DNase I digestion was achieved by adding 2 μl enzyme (typically 0.01 U/ml) dissolved in 20 mM NaCl, 2 mM MgCl2, 2 mM MnCl2. The digestion was stopped after 1 min by adding 5 μl of 80% formamide containing 10 mM EDTA, 10 mM NaOH and 0.1% (w/v) bromophenol blue. Hydroxyl radical cleavage was performed by adding 6 μl of a freshly prepared mixture containing 50 μM ferrous ammonium sulphate, 100 μM EDTA, 2 mM ascorbic acid and 0.05% hydrogen peroxide. The reaction was stopped after 10 min by precipitating with ethanol. The DNA was finally redissolved in 8 μl of 80% formamide containing 10 mM EDTA, 10 mM NaOH and 0.1% (w/v) bromophenol blue.
Denaturing gel electrophoresis
Products of DNase I or hydroxyl radical digestion were separated on 8% polyacrylamide gels containing 8 M urea. DNA samples were boiled for 3 min immediately before loading onto the gels. Polyacrylamide gels (40 cm long) were run at 1500 V for 2 h. These were then fixed in 10% acetic acid, transferred to Whatmann 3MM paper, dried under vacuum at 80°C and exposed to a phosphorimager screen (Kodak) overnight. This was scanned using a Molecular Dynamics Storm 860 phosphorimager. The products of digestion were assigned by comparison with Maxam–Gilbert marker lanes specific for guanine and adenine.
Fluorescence melting
Fluorescence melting curves were determined in a Roche LightCycler, as previously described (12) in a total reaction volume of 20 μl. For each reaction the oligonucleotides (Figure 1C) were diluted to a final concentration of 0.25 μM in 10 mM sodium phosphate buffer pH 7.4 containing 20 mM NaCl. In a typical experiment the oligonucleotides were first denatured by heating to 95°C for 5 min. They were then annealed by cooling to 30°C at a rate of 0.1°C/s (the slowest heating and cooling rate for the LightCycler). The samples were maintained at 30°C for 5 min before slowly heating to 95°C (0.1°C/s). Recordings were taken during both the annealing and melting steps. The LightCycler has one excitation source (488 nm) and the changes in fluorescence were measured at 520 nm. In these experiments we found no significant differences between the heating and melting curves. Melting temperatures (Tm) were determined from the maximum in the first derivatives of the melting profiles using the Roche LightCycler programme.
RESULTS
Footprinting
We have previously suggested that the polyamide thiazotropsin A has very different sequence binding properties to the natural minor groove binding agent distamycin (11). In contrast to the AT selectivity displayed by the natural compounds, this agent appeared to bind selectively to the sequence ACTAGT. Figure 2 shows DNase I and hydroxyl radical digestion patterns for this compound on the MS2 DNA fragment, which possesses all possible arrangements of tetranucleotide sequences. It can be seen that concentrations between 0.3 and 10 μM produce a single DNase I footprint which is located around the symmetrical sequence ACTAGT. Other regions are also protected at higher ligand concentrations, which are located close to the sequences TATT, TTAA, TATA, TAATT, ATAA and TTTA and are typical of the patterns expected with AT-selective minor groove binding ligands. This selectivity is confirmed by the hydroxyl radical footprinting patterns which also reveal a single clear region of protection in the sequence ACTAGT with 0.3 μM ligand. Other regions of attenuated cleavage are also visible at higher ligand concentrations, especially around the sequence AATT. It therefore appears that, amongst the sequences that are present in this fragment, the ligand is selective for ACTAGT, or a portion of this (such as ACTA or CTAG) that is not found elsewhere in the fragment.
Figure 2.
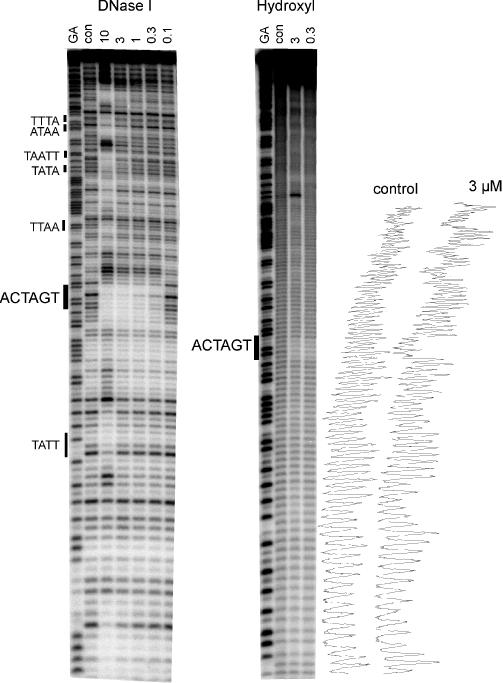
DNase I and hydroxyl radical footprinting of thizaotropsin A on the MS2 DNA fragment. The ligand concentration (μM) is shown at the top of each lane. Digestion of the DNA in the absence of added ligand is indicated by ‘con’. Tracks labelled ‘GA’ are Maxam–Gilbert sequence markers specific for purines. Densitometer traces of the control and 3 μM lanes of the hydroxyl radical cleavage are shown on the right.
Although fragment MS2 contains every possible tetranucleotide sequence, we cannot exclude the possibility that the ligand can also bind to other hexanucleotides that are not represented. It is clearly not possible to generate a single fragment that contains every one of the 2080 possible hexanucleotide sequences. We therefore prepared two fragments which contain several variations around the sequence ACTAGT in which each potential site is separated by a common tetranucleotide (CGGC/GCCG) to which we would not expect the ligand to bind. The results of DNase I and hydroxyl radical footprints with these fragments are shown in Figure 3. These DNase I patterns show clear footprints on both fragments at sites 8 (ACCAGT), 6 (ACTAGT) and 3 (GCTAGT). The original sequence identified with MS2 (ACTAGT), site 6, can be seen to produce the best footprint, and the bands within this site are still attenuated at the lowest ligand concentration. Sites 1 (TCTCTG), 2 (CCTAGT), 5, (AATAGT), 9 (ACAAGT) and 10 (ACGAGT) are hardly affected, even at the highest concentration (10 μM), while sites 4 (ATTAGT) and 7 (AGTAGT) show attenuated cleavage at 3 and 5 μM. These results, which are summarized in Table 1, demonstrate that the central tetranucleotide CTAG is neither necessary nor sufficient to produce a ligand binding site. Site 8 does not possess this sequence, yet produces a clear footprint, while site 2 contains a central CTAG and does not yield a footprint. In contrast, the hydroxyl radical cleavage patterns shows attenuated cleavage at most of the potential target sites, though this is again most pronounced at site 6.
Figure 3.

DNase I and hydroxyl radical footprinting of thizaotropsin A on sequences 1 and 2. The ligand concentration (μM) is shown at the top of each lane. Digestion of the DNA in the absence of added ligand is indicated by ‘Con’. Tracks labelled ‘GA’ are Maxam–Gilbert sequence markers specific for purines. The location of the 10 potential binding sites is shown.
Table 1. Interaction of the thiazaole polyamide with the potential binding sites in sequences 1 and 2.
| Site No. | Sequence | Footprint (5 μM) |
|---|---|---|
| 1 | TCTCTG/CAGAGA | No |
| 2 | CCTAGT/ACTAGG | No |
| 3 | GCTAGT/ACTAGC | Yes |
| 4 | ATTAGT/ACTAAT | No |
| 5 | AATAGT/ACTATT | No |
| 6 | ACTAGT/ACTAGT | Yes |
| 7 | AGTAGT/ACTACT | No |
| 8 | ACCAGT/ACTGGT | Yes |
| 9 | ACAAGT/ACTTGT | No |
| 10 | ACGAGT/ACTCGT | No |
The final column indicates the presence or absence of a footprint with 5 μM ligand.
Fluorescence melting experiments
We further examined the sequence selectivity of this ligand by measuring its effect on the melting of several short DNA duplexes. For these experiments we used the fluorescence melting technique that we have previously described (12). This uses synthetic duplexes in which one strand is labelled with fluorescein at the 5′ end and the other strand is labelled with methyl red at the 3′ end (Figure 1C). These groups are in close proximity when the oligonucleotides associate to form a duplex, and the fluorescence is quenched. When the duplex melts, the fluorophore and quencher are separated and there is a large increase in the fluorescence signal. For these experiments we used 11mer duplexes which contain the potential hexanucleotide binding site at the centre. These sites are flanked by GC pairs, to which we would not expect the ligand to bind, and which were arranged so as to prevent misannealing or hairpin formation. Figure 4 presents the fluorescence melting curves for four of these duplexes in the presence of varying concentrations of the ligand. Plots showing the effect of ligand concentration on the changes in the melting temperatures of all eight duplexes are shown in Figure 5, and these results are summarized in Table 2, along with the results of similar experiments with distamycin.
Figure 4.
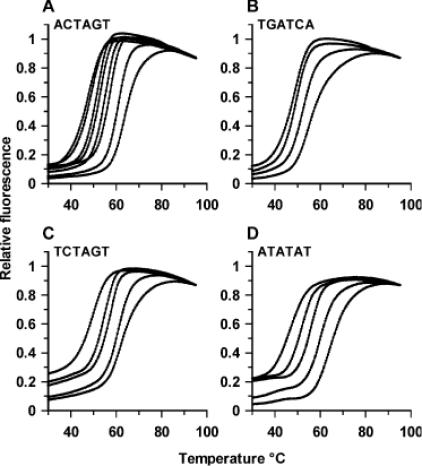
Fluorescence melting curves in the presence of thiazotropsin A. (A) ACTAGT, in the presence of 0, 1, 2, 5, 10, 20, 60 and 100 μM ligand. (B) TGATCA, in the presence of 0, 20, 60 and 100 μM ligand. (C) TCTAGT, in the presence of 0, 10, 20, 60 and 100 μM ligand and (D), ATATAT in the presence of 0, 5, 20, 60 and 100 μM ligand. In each case the fluorescence values have been normalized to the same final value.
Figure 5.
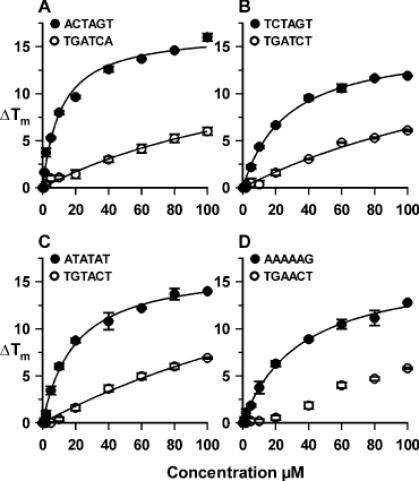
Plots showing the variation of ΔTm with ligand concentration for the eight fluorescently labelled duplexes. The curves were fitted to the data using the equation ΔTm = ΔTm,max × L/(L+C50) and are described by the parameters shown in Table 2. The C50 values have no direct physical meaning, but provide a convenient means for comparing the relative effects of the ligand on the different sequences.
Table 2. Effects of ligands on the melting temperature of the fluorescently labelled oligonucleotides.
| Sequence | Thiazotropsin A | Distamycin | ||
|---|---|---|---|---|
| ΔTm with 10 μM | C50 (μM) | ΔTm with 10 μM | C50 (μM) | |
| ACTAGT | 8.0 | 11.2 ± 1.6 | 5.0 | 22.8 ± 2.1 |
| TGATCA | 1.1 | 175 ± 60 | 5.3 | 23.0 ± 2.7 |
| TGATCT | 0.4 | 210 ± 90 | 2.6 | * |
| TGTACT | 0.4 | 290 ± 150 | 2.1 | * |
| TGAACT | 0.2 | >300 | 3.0 | * |
| TCTAGT | 4.4 | 27 ± 3 | 1.5 | * |
| ATATAT | 6.0 | 20 ± 2 | 13.9 | 3.0 ± 0.4 |
| AAAAAG | 3.8 | 35 ± 3 | 15.0 | 0.9 ± 0.2 |
The second and fourth columns show the increase in melting temperature (ΔTm, °C) produced by addition of 10 μM of the ligands. C50 is the ligand concentration (μM) that produces half the maximal change in melting temperature and was obtained by fitting the data to the equation ΔTm = ΔTm,max × L/(L+C50), where ΔTm,max is the maximum change in melting temperature and L is the ligand concentration. These C50 values are only used to provide a convenient means for comparing the relative effects of the ligand on the different sequences, and do not have any direct physical meaning. Asterisks indicates that this equation did not fit the data, which showed a steady increase in melting temperature with increasing ligand concentration.
Figure 4A shows the results with the duplex that contains the hexanucleotide sequence identified from the footprinting experiments (ACTAGT). Under these conditions (0.25 μM duplex, 200 mM NaCl) this duplex produces a simple monophasic melting profile with a melting temperature (Tm) of 49°C. Addition of 1 μM ligand increases the Tm by 1.5°C, while further increases in concentration shift the curve to higher temperatures. At all concentrations the melting profiles show simple monophasic transitions, indicating that the bound and free ligand are in rapid exchange. This is similar to the effect of distamycin on AT-rich sequences, but contrasts with published melting profiles for hairpin polyamides which show biphasic melting curves in which the two transitions correspond to those of the free and bound DNA (15,16). We do not observe any differences between the melting and annealing profiles. Figure 5A shows the changes in melting temperature (ΔTm) as a function of the ligand concentration, from which it can be seen that it produces a maximum ΔTm of ∼16°C. A simple rectangular hyperbola fitted to these data reveals that the half-maximal change in melting temperature is produced with 11.2 ± 1.6 μM ligand. Although this value (C50) is related to the dissociation constant of the ligand it has no direct physical meaning, but is a convenient means for comparing the relative effects of the ligand on the different sequences. Distamycin also stabilizes this duplex (see Table 2), though it is less effective than the thiazole polyamide and, as expected, it is more potent at the AT-rich sequences.
Figure 4B shows the results of similar melting experiments with the reverse sequence (TGATCA). The ligand has a much smaller effect on this sequence, and a concentration of 20 μM increases the melting temperature by only 2°C. The effect of ligand concentration on ΔTm is shown in Figure 5A, from which we estimate a C50 value of 175 μM. Distamycin has a greater effect than the thiazole polyamide on this sequence, and stabilizes it by the same amount as the forward sequence ACTAGT. Figure 4C shows melting curves with the sequence TCTAGT, which differs from the original hexanucleotide sequence at only the first base. This is stabilized by the ligand, though it requires about twice the concentration to produce a given effect; e.g. a ΔTm of 10°C is produced by 20 μM ligand with ACTAGT, but 40 μM ligand with TCTAGT. The effect of ligand concentration on ΔTm is shown in Figure 5B, from which we estimate a C50 value of 27 μM. Distamycin has a much weaker effect on this sequence and we could not estimate a C50 value from the observed changes in melting temperature.
The sequences ATATAT and AAAAAG contain well-characterized binding sites for the AT-selective minor groove binder distamycin, to which it is expected to bind in the 2:1 and 1:1 mode, respectively. The melting profiles for the interaction of thiazotropsin A with ATATAT are shown in Figure 4D, while the ΔTm values produced by different concentrations of this ligand are shown in Figure 5C and D for both sequences. It can be seen that the ligand stabilizes both sequences, though to a lesser extent than ACTAGT, and that it binds better to ATATAT than AAAAAG. Distamycin affects the melting of both these sequences at low concentrations and, in contrast to thiazotropsin A, has a greater effect at AAAAAG than ATATAT. Both ligands have very little effect on the melting of TGATCT, TGTACT and TGAACT (see Figure 5 and Table 2).
DISCUSSION
We have previously suggested that the thiazole polyamide thiazotropsin A binds to the sequence ACTAGT, and produces a clear DNase I footprint around a region that contains this sequence in the MS2 fragment (11). We have shown that the ligand produces a single hydroxyl radical footprint on this sequence, covering 4 bp, which is also centred around the sequence ACTAGT. This hydroxyl radical footprint is larger than that typically observed with distamycin (2–3 bases) (17), and is therefore consistent with our suggestion (11) that this ligand binds as a dimer of overlapping subunits. However, although the MS2 fragment contains every possible tetranucleotide, it is not possible to assess whether ACTAGT is the only hexanucleotide to which the ligand binds, or whether it can also bind to other sequences that contain the tetranucleotide CTAG. We have addressed this problem by performing footprinting experiments on DNA fragments that contain variations on the sequence ACTAGT. From these results it is clear that CTAG alone is not sufficient to produce a good binding site as a footprint is produced at site 6 (ACTAGT) but not site 2 (ACTAGG). These data can be used to probe the binding requirements by examining the effect of changing each base of this symmetrical sequence in turn. Looking first at the third/fourth position (ACTXGT) footprints are seen with X = A (site 6) and G (site 8) but not C (site 10) or T (site 9). Similarly in the second/fifth position (ACTAXT) footprints are produced with X = G (site 6) and A (site 4), but not T (site 5) and C (site 7). At the first/sixth position (ACTAGX) footprints are observed with X = T (site 6) and C (site 3), but not G (site 2), though we have no information on A at this position. These results suggest that, assuming that the ligand binds as a dimer to a symmetrical sequence, the preferred binding site is RCYRGY. Examination of the MS2 fragment confirms that this sequence only occurs once (ACTAGT). The RCYRGY sequence is also found once in the tyrT fragment, which we have previously shown produces a single DNase I footprint around the sequence ACTGGT (11).
The fluorescence melting experiments confirm that the ligand has the greatest effect on ACTAGT. Weaker but significant interaction was also observed with TCTAGT (differing in the first base), while the ligand bound much more weakly to the reverse sequence TGATCA. The interaction with TCTAGT (ACTAGA) complements the footprinting data for which we had no information on this hexanucleotide, and suggests that the first base can also be T. This therefore suggests that the preferred binding site for the thiazole polyamide is XCYRGZ, where X is any base except C and Z is any base except G. The melting experiments also show that this ligand binds to the AT-rich sequences AAAAA and ATATAT, which are good target sites for AT-selective minor groove binding ligands. In contrast, the footprinting experiments only showed binding to these sites at much higher concentrations. The reasons for the different results from the two techniques with these sequences are not clear, but may be related to the higher temperatures employed in the melting experiments. The precise changes in melting temperature are a complex function of ligand binding affinity and binding enthalpy, as well as the enthalpy of oligonucleotide melting. A given shift in Tm is proportional to binding affinity for two ligands only if their binding enthalpy and stoichiometry are identical. For these reasons the melting data alone do not conclusively demonstrate differences in binding affinity to the different sequences, but they complement the footprinting data in identifying the best binding sites. It should also be noted that although distamycin binds better to AAAAA than ATATAT, the order is reversed for thiazotropsin A.
These molecules provide an additional means of recognizing GC base pairs in the minor groove, which complement and extend that achieved with the hairpin polyamides (2,3,7,8). Although recognition of 6 bp alone is unlikely to produce biological specificity it has been demonstrated that some short sequences may represent unique (specific) binding sites in some promoters (18,19). Indeed, a search of 1.2 Mbp in the human promoter database (http://zlab.bu.edu/~mfrith/HPD.html) revealed that the sequence ACTAGT may be underrepresented compared with other arrangements of the same base pairs, as this occurred with ∼40% of the frequency as sites such as GAATTC, TCTAGA, AGATCT and AAGCTT and has the same abundance as related hexanucleotides that contain a CpG step, such as TTCGAA and AACGTT.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a grant from the European Union.
REFERENCES
- 1.Neidle S. (2001) DNA minor-groove recognition by small molecules. Nat. Prod. Rev., 18, 291–309. [DOI] [PubMed] [Google Scholar]
- 2.Dervan P.B. (2001) Molecular Recognition of DNA by small molecules. Bioorg. Med. Chem., 9, 2215–2235. [DOI] [PubMed] [Google Scholar]
- 3.Wemmer D.E. (2001) Ligands recognizing the minor groove of DNA: development and applications. Biopolymers, 52, 197–211. [DOI] [PubMed] [Google Scholar]
- 4.Kopka M.L., Yoon,C., Goodsell,D., Pjura,P. and Dickerson,R.E. (1985) The molecular-origin of DNA drug specificity in netropsin and distamycin. Proc. Natl Acad. Sci. USA, 82, 1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopka M.L., Yoon,D., Goodsell,D., Pjura,P. and Dickerson,R.E. (1985) Binding of an antitumor drug to DNA netropsin and C-G-C-G-A-A-T-T-BrC-G-C-G. J. Mol. Biol., 183, 553–563. [DOI] [PubMed] [Google Scholar]
- 6.Pelton J.G. and Wemmer,D.E (1989) Structural characterization of a 2-1 distamycin A.d(CGCAAATTGGC) complex by two-dimensional NMR. Proc. Natl Acad. Sci. USA, 86, 5723–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wemmer D.E. (2000) Designed sequence-specific minor groove ligands. Annu. Rev. Biophys. Biomol. Struct., 29, 439–461. [DOI] [PubMed] [Google Scholar]
- 8.Dervan P.B. and Burli,R.W. (1999) Sequence-specific DNA recognition by polyamides. Curr. Opin. Chem. Biol., 3, 688–693. [DOI] [PubMed] [Google Scholar]
- 9.Wemmer D.E. and Dervan,P.B. (1997) Targeting the minor groove of DNA. Curr. Opin. Struct. Biol., 7, 355–361. [DOI] [PubMed] [Google Scholar]
- 10.Haq I., Ladbury,J.E., Chowdhry,B.Z., Jenkins,T.C. and Chaires,J.B. (1997) Specific binding of Hoechst 33258 to the d(CGCAAATTTGCG)(2) duplex: Calorimetric and spectroscopic studies. J. Mol. Biol., 271, 244–257. [DOI] [PubMed] [Google Scholar]
- 11.Anthony N.G., Fox,K.R., Johnston,B..F., Khalaf,A.I., Mackay,S.P., McGroarty,I.S.Parkinson,J.A., Skellern,G.G., Suckling,C.J. and Waigh,R.D. (2004) DNA binding of a short lexitropsin. Bioorg. Med. Chem. Lett., 14, 1353–1356. [DOI] [PubMed] [Google Scholar]
- 12.Darby R.A.J., Sollogoub,M., McKeen,C. Brown,L., Risitano,A., Brown,N., Barton,C., Brown,T. and Fox,K.R. (2002). High throughput measurement of duplex, triplex and quadruplex melting curves using molecular beacons and a LightCycler. Nucleic Acids Res., 30, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavesa M. and Fox,K.R. (2001) Preferred binding sites for [N-MeCys3,N-MeCys7]TANDEM determined using a universal footprinting substrate. Anal. Biochem., 293, 246–250. [DOI] [PubMed] [Google Scholar]
- 14.Fox K.R. and Waring,M.J. (2001) High Resolution footprinting studies of drug-DNA complexes using chemical and enzymic probes. Meth. Enzymol., 340, 412–430. [DOI] [PubMed] [Google Scholar]
- 15.Pilch D.S., Poklar,N., Gelfand,A.A., Law,S.M., Breslauer,K.J., Baird,E.E. and Dervan,P.B. (1996) Binding of a hairpin polyamide in the minor groove of DNA: Sequence-specific enthalpic discrimination. Proc. Natl Acad. Sci. USA, 93, 8306–8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilch D.S., Poklar,N., Baird,E.E., Dervan,P.B., Breslauer,K.J. (1999) The thermodynamics of polyamide-DNA recognition: Hairpin polyamide binding in the minor groove of duplex DNA. Biochemistry, 38, 2143–2151. [DOI] [PubMed] [Google Scholar]
- 17.Abu-Daya A. and Fox,K.R (1997) Interaction of minor groove binding ligands with long AT tracts. Nucleic Acids Res., 25, 4962–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neidle S., Puvvada,M.S. and Thurston,D.E. (1994) The relevance of drug DNA sequence specificity to anti-tumour activity. Eur. J. Cancer, 30, 567–568. [DOI] [PubMed] [Google Scholar]
- 19.Gottesfeld, Turner,J.M. and Dervan,P.B. (2000) Chemical approaches to control gene expression. Gene Expr, 9, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]