

ABSTRACT
Mono-(2-ethylhexyl) phthalate (MEHP) is the active metabolite of the most commonly used plasticizer, di-(2-ethylhexyl) phthalate, and is considered to be a reproductive toxicant. However, little is known about the effects of MEHP on ovarian antral follicles. Thus, the present study tested the hypothesis that MEHP inhibits follicle growth via oxidative stress pathways. The data indicate that MEHP increases reactive oxygen species (ROS) levels and inhibits follicle growth in antral follicles, whereas N-acetylcysteine (NAC; an antioxidant) restores ROS levels to control levels and rescues follicles from MEHP-induced inhibition of follicle growth. To further analyze the mechanism by which MEHP induces oxidative stress and inhibits follicle growth, the expression and activities of various key antioxidant enzymes (copper/zinc superoxide dismutase [SOD1], glutathione peroxidase [GPX], and catalase [CAT]) and the expression of key cell-cycle regulators (Ccnd2, Ccne1, and Cdk4) and apoptotic regulators (Bcl-2 and Bax) were compared in control and MEHP-treated follicles. The data indicate that MEHP inhibits the expression and activities of SOD1 and GPX; does not inhibit Cat expression; inhibits the expression of Ccnd2, Ccne1, Cdk4, and Bcl-2; but increases the expression of Bax compared to controls. Furthermore, NAC blocks these toxic effects of MEHP. Collectively, these data suggest that MEHP induces oxidative stress by disrupting the activities of antioxidant enzymes. This may lead to decreased expression of cell-cycle regulators and antiapoptotic regulators and increased expression of proapoptotic factors, which then may lead to inhibition of follicle growth.
Keywords: ovary, oxidative stress, toxicology
Mono (2-ethylhexyl) phthalate (MEHP) inhibits growth in cultured mouse ovarian follicles by inducing oxidative stress, inhibiting the expression of cell proliferation factors (Ccnd2, Ccne1, and Cdk4) and the antiapoptotic gene Bcl-2, and increasing expression of the proapoptotic gene Bax.
INTRODUCTION
Phthalates are synthetic plasticizers widely used in plastics and other common consumer products, such as food packaging, toys, cosmetics, clothing, and biomedical devices [1]. Di-(2-ethylhexyl) phthalate (DEHP), one of the most common phthalates, is not covalently bound to polymers in plastic. Thus, it leaches out of products and gets into the environment, making it a widespread environmental contaminant [2]. Humans are exposed to DEHP predominantly via contaminated food or beverages. Studies have shown that the exposure level of DEHP in the general population is close to the tolerable daily intake (2 mg/day); however, individuals undergoing certain medical procedures may be exposed to even higher levels (more than 100 fold) via plastic medical devices [1, 3]. Numerous human epidemiologic studies have reported an association between increased plasma levels of DEHP and various adverse reproductive outcomes in women, including endometriosis, uterine leiomyoma, increased risk of premature labor, and various pregnancy complications [4–7]. Although no clear conclusions about the effects of DEHP on human reproduction have been made due to small sample sizes and study design limitations, the published studies have raised a public health concern.
Numerous animal studies indicate that DEHP is an endocrine-disrupting chemical and reproductive toxicant. In male rats, perinatal DEHP exposure (10 mg/kg/day) disrupts the development of androgen-dependent structures, mainly by inhibiting testicular testosterone biosynthesis [6]. DEHP exposure (1 g/kg) induces germ cell apoptosis in the rat pubertal testis [8]. In female adult rats, high doses of DEHP (2 g/kg/day) result in prolonged estrous cycles, reduced serum estradiol levels, and absence of ovulation [9]. In rats, neonatal DEHP exposure (15 mg/kg/day) results in delayed onset of puberty, and higher neonatal DEHP exposure (405 mg/kg/day) increases the number of ovarian atretic tertiary follicles [10].
These previous in vivo studies provide important information, but they cannot be used to determine whether the parent compound, DEHP, or metabolites of DEHP cause toxicity. This is because once DEHP gets into the body, it is hydrolyzed into an active metabolite, mono-(2-ethylhexyl) phthalate (MEHP), by lipases and esterases in the intestine and liver [11]. Like DEHP, MEHP is thought to be an endocrine disruptor and reproductive toxicant because several in vitro studies indicate that MEHP adversely affects the structure and function of reproductive tissues. For example, in vitro studies indicate that MEHP (1 μM) reduces steroid production and increases reactive oxygen species (ROS) generation in MA-10 Leydig cells [12]. Further, studies by Muzcynski et al. [13] show that MEHP exposure (10 μM) significantly increases the rate of apoptosis in cultured human and mouse fetal testes [13]. MEHP exposure (100 μM) lowers estradiol production by reducing the expression of aromatase via peroxisome proliferator-activated receptor (PPAR)-dependent signaling pathways in granulosa cells [14]. Both DEHP and MEHP reduce estradiol production in antral follicles and inhibit follicle growth in mice [15, 16]. Thus, MEHP, like DEHP, is thought to be a reproductive toxicant.
Although previous studies suggest that DEHP and MEHP have similar activities in terms of endocrine-disrupting ability and reproductive toxicity, some studies indicate the chemicals may act differently depending on dose and tissue type. For example, studies indicate that MEHP causes toxic effects at lower doses than DEHP in Leydig cells and liver macrophages, suggesting that MEHP may be more potent than its parent compound [17–19]. Further, several in vitro studies indicate that MEHP has different effects than DEHP. For example, although MEHP inhibits rat follicle development, induces testicular cell apoptosis, and disrupts fetal testis morphology and functions, DEHP does not have these same effects [20–22]. Similarly, Thomas et al. [17] have reported that DEHP, but not MEHP, decreases rat gonadal zinc levels.
Collectively, these studies suggest that the parent compound, DEHP, and its major metabolite, MEHP, might affect biological systems via different mechanisms. However, whether DEHP and MEHP have similar or different effects on the ovary is not known. Our previous study [15] showed that DEHP induces oxidative stress in isolated antral follicles by suppressing the expression and activity of copper/zinc superoxide dismutase (SOD1) and that this inhibits follicle growth, but it is not known whether MEHP induces oxidative stress in antral follicles and, if so, whether it does this via mechanisms similar to those of DEHP. It is important to determine whether MEHP causes oxidative stress via similar mechanisms as DEHP; for animals normally exposed to the parent compound, DEHP, some of the compound may reach the ovary as DEHP, but some will likely be metabolized to MEHP. This raises the question of whether the oxidative stress and follicle growth inhibition observed with DEHP is due to DEHP or, after conversion by lipase and esterases in tissues such as the intestine and liver, to MEHP. One way to determine if MEHP is capable of damaging ovarian follicles is by directly treating follicles with MEHP. Thus, the goals of the present study were to test whether MEHP 1) inhibits follicle growth by inducing oxidative stress in mouse antral follicles, 2) alters similar enzymatic antioxidants as DEHP, and 3) is more toxic than DEHP. If studies indicate that DEHP causes toxicity, it may be important to limit exposure to the parent compound. If studies indicate that MEHP also causes damage and/or is more potent than DEHP, it may be important to develop ways to inhibit the ability of the body to convert DEHP to MEHP.
Because our data indicated that MEHP inhibits follicle growth, another goal of the present study was to expand our knowledge about the mechanisms by which phthalates such as MEHP affect follicle growth. Follicle growth depends on the proliferation of follicular cells [23]. Like other cells in the body, follicular cell proliferation is primarily regulated by different phase-specific cell-cycle regulators, cyclin:cyclin-dependent kinase (CDK) complexes. Any disruption of these regulators will cause cell-cycle arrest and induce apoptosis, therefore inhibiting follicle growth [24–26]. In the cell, cyclin D2/cyclin-dependent kinase 4 complex is activated at the early G1 phase, which can induce the expression of cyclin E1 and push the cell into the S phase [27]. Studies have shown that estrogenic endocrine-disrupting chemicals, such as methoxychlor and bisphenol A, inhibit antral follicle growth by altering the expression of G1/S-phase cell-cycle regulators [25, 26]. Studies have also shown that oxidative stress induces cell-cycle arrest at the G1/S transition in breast cancer cells and Chinese hamster ovary cells [28, 29] and that it is a common mediator of apoptosis [30]. Thus, another goal of the present study was to test whether MEHP-induced inhibition of follicle growth is due to disruption of the expression of G1/S-phase cell-cycle regulators, such as cyclin D2 (Ccnd2), cyclin E1 (Ccne1), and cyclin-dependent kinase 4 (Cdk4), and apoptotic factors, such as B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax).
MATERIALS AND METHODS
Chemicals
The MEHP was purchased from AccuStandard (New Haven, CT). Stock solutions of MEHP were prepared using dimethyl sulfoxide (DMSO; Sigma) as a solvent and in various concentrations (0.133, 1.33, 13.3, and 133 mg/ml). Stock solutions were used to make final working concentrations in culture of 0.1, 1, 10, and 100 μg/ml of MEHP, which are equivalent to approximately 0.359, 3.59, 35.9, and 359 μM. N-acetylcysteine (NAC) was purchased from Sigma. A stock solution of NAC (100 mM) was prepared using alpha-minimum essential medium (α-MEM; Invitrogen), and the final concentrations of NAC in each well of the culture were 0.5 and 1 mM.
The MEHP concentrations were chosen based on studies showing that micromolar concentrations of MEHP impair rat follicle development in vitro [20] and inhibit hormone production via nuclear PPAR pathways in mouse antral follicles as well as in rat and human granulosa cells [16, 31–33]. The selected MEHP concentrations also are environmentally relevant levels. MEHP plasma concentrations in healthy women have been reported to be approximately 0.6 μg/ml, and peritoneal fluid concentrations have been reported to be 0.4 μg/ml [34]. These concentrations are in the range of the lower doses (0.1 and 1 μg/ml) used in the current experiments. In addition, patients undergoing intensive medical care usually have markedly higher MEHP levels than healthy people [35, 36]. MEHP plasma levels in blood transfusion patients can reach as high as 50 μM, and MEHP levels of 15.1 μg/ml have been detected in infants in neonatal intensive care units [37]. These concentrations are encompassed by the two higher doses (10 and 100 μg/ml) used in current experiments.
Animals
CD-1 mice were maintained at the University of Illinois at Urbana-Champaign, Veterinary Medicine Animal Facility, under a 12L:12D photoperiod. Mice were given food and water ad libitum. Animals were euthanized at 32–35 days of age. The ovaries were removed and the antral follicles isolated as described below. All animal procedures were approved by the University of Illinois Institutional Animal Care and Use Committee.
Follicle Culture
Based on appearance and relative size (diameter, 250–400 μm), antral follicles were isolated mechanically from ovaries of cycling, young CD-1 mice, and interstitial tissue was removed using fine watchmaker forceps and individually placed in wells of 96-well culture plates [38]. At least three mice were used per experiment, providing approximately 25–40 follicles per mouse. The isolated follicles were randomly divided into different treatment groups (n = 10–32 follicles/group).
Doses of vehicle control (DMSO) or MEHP (0.1, 1, 10, and 100 μg/ml) with or without NAC (0.5 or 1 mM) were individually prepared in supplemented α-MEM as described previously [15, 33]. Supplemented α-MEM was prepared with 1% ITS (10 ng/ml of insulin, 5.5 ng/ml of transferrin, and 5.5 ng/ml of selenium), 100 mg/ml of streptomycin, 100 IU/ml of penicillin, 5% fetal calf serum (Atlanta Biologicals), and 5 IU/ml of human recombinant follicle-stimulating hormone (Dr. A. F. Parlow, National Hormone and Peptide Program, Harbor-UCLA Medical Center, Torrance, CA). An equal volume of chemical was added to each treatment group to control for the amount of vehicle in each preparation. Nontreated controls (supplemented medium only) were used in each experiment as a control for culture conditions. All follicles were cultured in 150 μl of medium for 24–96 h in an incubator at 37°C supplying 5% CO2. At the end of culture, follicles were collected, snap-frozen, and stored at −80°C for later use.
Analysis of Follicle Growth
Follicle growth was examined every 24 h by measuring follicle diameter on perpendicular axes with an inverted microscope equipped with a calibrated ocular micrometer. Follicle diameter measurements were averaged and plotted to compare the effects of chemical treatments on growth. All measurements were done without knowledge of the treatment group. Data are presented as the percentage change over time. At least three separate experiments were performed for each treatment to obtain enough power for statistical analysis.
Gene Expression Analysis
Antral follicles were collected and snap-frozen at the end of culture for quantitative real-time PCR (qPCR) analysis. Total RNA was extracted from follicles using the RNeasy Micro Kit (Qiagen, Inc.) according to the manufacturer's protocol. Total RNA (200 ng) was reverse transcribed to cDNA using an iScript RT kit (Bio-Rad) following the manufacturer's instructions. The qPCR analysis was performed using CFX96 Real-Time PCR Detection System (Bio-Rad) and accompanying software (CFX Manager Software) according to the manufacturer's instructions. Specific qPCR primers for Sod1, glutathione peroxidase (Gpx), catalase (Cat), Ccnd2, Ccne1, Cdk4, Bcl-2, Bax, and β-actin (Actb) were used in each reaction (see Table 1 for sequences). An initial incubation of 95°C for 10 min was followed by 45 cycles of denaturing at 94°C for 10 sec, annealing at 60°C for 10 sec, and extension at 72°C for 10 sec, with a final extension at 72°C for 10 min. A melting curve was generated at 55–90°C to monitor the generation of a single product. Actb was used as a reference gene for each sample because preliminary experiments indicated that expression of this gene did not change in response to DMSO and MEHP treatments (data not shown). Relative fold-changes were calculated as the ratio to the DMSO treatment group level, which was set as 1.0. All samples were measured in triplicate from at least three separate experiments.
TABLE 1.
Sequences of primer sets used for gene expression analysis.

Enzyme Activity Assays
Antral follicles were collected and snap-frozen at the end of culture for enzyme activity assays. Protein was extracted, and the activities of SOD1, GPX, and CAT were measured using specific enzyme activity kits (Cayman) according to the manufacturer's instructions. For each sample, enzyme activity was normalized to its own protein concentration (measured by BCA Protein Assay Kit; Thermo Scientific), and then the relative fold-changes were determined by setting that of DMSO (vehicle control) at 1.0. All samples were run in duplicate from at least three separate experiments.
In Vitro ROS Assays
Antral follicles were collected and snap-frozen at the end of culture and then homogenized on ice and spun at 14 000 rpm for 15 min. The supernatant was subjected to in vitro assays for measurement of the levels of ROS, predominantly superoxide (O2−) and hydrogen peroxide (H2O2), using an OxiSelect In Vitro ROS Assay Kit (Cell Biolabs, Inc.) according to the manufacturer's instructions. Data were first normalized to protein level (measured by BCA Protein Assay Kit), and then relative fold-changes were determined after setting that of DMSO (vehicle control) at 1.0. All samples were run in duplicate from at least three separate experiments.
Statistical analysis
Data are expressed as the mean ± SEM from at least three separate experiments. One-way ANOVA followed by Tukey post hoc comparisons were used to make multiple comparisons between treatment groups. Student t-tests were used to make comparisons between two groups. Statistical significance was assigned at P ≤ 0.05 for all comparison.
RESULTS
Effect of MEHP and NAC Cotreatment on ROS Levels in Antral Follicles In Vitro
Elevated ROS levels are a direct indicator of oxidative stress in biological systems [39]. To examine whether MEHP induces oxidative stress in antral follicles, we compared the levels of ROS in cultured follicles in the presence of vehicle or MEHP. Compared to DMSO controls, MEHP (1–100 μg/ml) significantly increased the level of ROS in follicles at 96 h (Fig. 1).
FIG. 1.
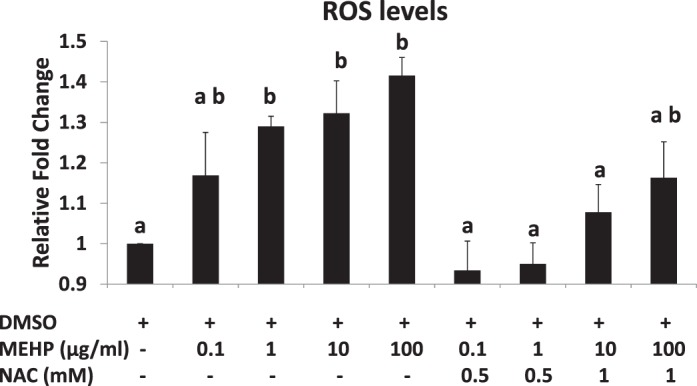
Effect of MEHP and NAC on ROS levels in antral follicles. Antral follicles were exposed to DMSO or MEHP (0.1–100 μg/ml) with or without NAC (0.5–1 mM) for 96 h in vitro and subjected to in vitro ROS assays to measure ROS levels. The levels of ROS were normalized to protein level in each sample and reported as relative fold-change compared to DMSO controls. All data represent the mean ± SEM from three independent experiments (n = 35 follicles/treatment/experiment). Bars with different letters are significantly different from each other (P ≤ 0.05).
Next, we determined if antioxidant NAC (0.5–1 mM) cotreatment protects follicles against MEHP-induced ROS production. The concentrations of NAC were based on the results of previous studies showing that they inhibit DEHP-induced oxidative stress [15]. NAC (0.5–1 mM) cotreatment with MEHP (0.1, 1, and 10 μg/ml) completely reduced the levels of ROS to control levels, whereas NAC (1 mM) cotreatment with MEHP (100 μg/ml) partially restored the levels of ROS to control levels (Fig. 1).
Effect of MEHP Treatment on Follicle Growth
To determine whether MEHP affects antral follicle growth, we treated antral follicles with medium (nontreated controls), DMSO (vehicle controls), or MEHP (0.1–100 μg/ml) for 96 h. The growth of DMSO-treated follicles was similar to that of nontreated controls (data not shown). By 72 h, the three highest doses of MEHP (1, 10, and 100 μg/ml) significantly decreased antral follicle growth compared to that of DMSO controls. This MEHP-inhibited follicle growth remained throughout the 96-h culture. By 96 h, the lowest dose of MEHP (0.1 μg/ml) also inhibited growth compared to that of DMSO controls (Fig. 2).
FIG. 2.
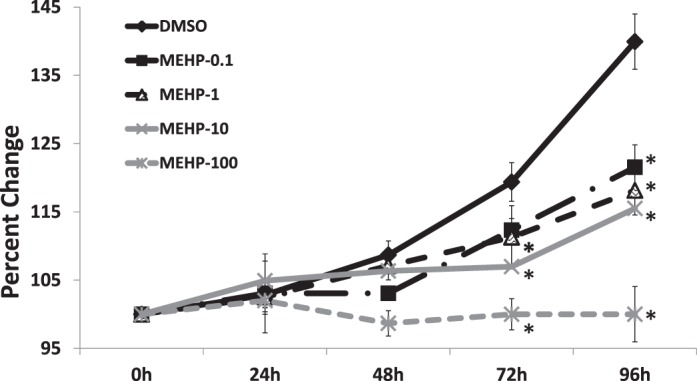
Effect of MEHP exposure on antral follicle growth. Antral follicles were cultured in the presence of DMSO or MEHP (0.1–100 μg/ml) for 96 h. Growth of follicles was monitored during culture and reported as percentage change over time. The graph represents the mean ± SEM from at least three separate experiments. Lines with an asterisk (*) are significantly different from DMSO controls at selected time points (n = 10–16 follicles/treatment/experiment; P ≤ 0.05).
Effect of MEHP on Expression of Cell-Cycle Regulators and Apoptotic Genes
Studies indicate that cell-cycle regulators control follicular cell proliferation and, therefore, control follicle growth [26, 40]. Thus, we compared the expression profiles of several cell-cycle regulators (Ccnd2, Ccne1, and Cdk4) in DMSO- and MEHP-treated follicles. Compared to DMSO controls, MEHP (1 μg/ml, which is the lowest dose of MEHP that inhibited follicle growth by 72 h) significantly inhibited the expression of cell-cycle genes. Specifically, MEHP significantly decreased the expression of Ccnd2 at 48 h, decreased the expression of Ccne1 at 72 and 96 h, and decreased the expression of Cdk4 starting at 48 h and continuing through 96 h (Fig. 3A).
FIG. 3.

Effect of MEHP exposure on the expression of cell-cycle regulators and apoptotic genes. After exposure of antral follicles to DMSO controls or MEHP (1 μg/ml) for 24–96 h in vitro, the follicles were collected and subjected to qPCR analysis for the expression profiles of Ccnd2, Ccne1, and Cdk4 (A) and the expression profiles of Bcl-2 and Bax (B). All values were normalized to Actb as loading control and reported as relative fold-change compared to DMSO levels. The graph represents the mean ± SEM from at least three separate experiments. Lines with an asterisk (*) are significantly different from DMSO controls at selected time points (n = 10–16 follicles/treatment/experiment; P ≤ 0.05).
Another possible reason for follicle growth inhibition could be apoptosis of follicular cells. Thus, we determined if MEHP alters the expression of selected apoptotic regulators (Bcl-2 and Bax). Exposure to MEHP (1 μg/ml) significantly reduced the expression of the antiapoptotic factor, Bcl-2, starting at 24 h and continuing through 72 h. MEHP significantly enhanced the expression of the proapoptotic factor, Bax, starting at 48 h and continuing through 96 h (Fig. 3B).
Effect of NAC Cotreatment on MEHP-Induced Follicle Growth Inhibition
Because our data indicated that MEHP causes oxidative stress and NAC reduces oxidative stress in follicles (Fig. 1), and that MEHP inhibits follicle growth (Fig. 2), we determined if NAC treatment protects against MEHP-induced inhibition of follicle growth. At 96 h, MEHP (0.1–100 μg/ml) inhibited follicle growth compared to that of DMSO controls (Fig. 4). NAC (0.5–1 mM) cotreatment, however, blocked the ability of MEHP to inhibit follicle growth. Specifically, compared to MEHP alone (0.1–10 μg/ml), NAC (0.5–1 mM) cotreatment with MEHP (0.1–10 μg/ml) significantly increased follicle growth compared to that observed in DMSO controls (Fig. 4). The highest dose of MEHP (100 μg/ml) further inhibited follicle growth compared to lower doses of MEHP (0.1–10 μg/ml), and NAC (1 mM) cotreatment partially blocked the ability of MEHP (100 μg/ml) to inhibit follicle growth (Fig. 4).
FIG. 4.
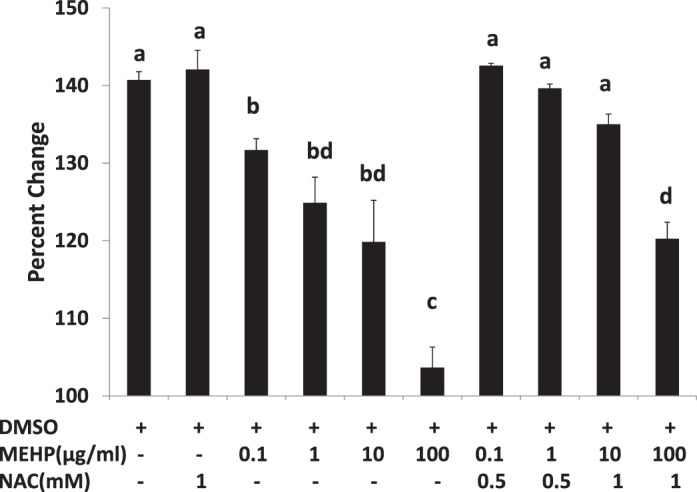
Effect of MEHP and NAC cotreatment on antral follicle growth. Antral follicles were cultured in the presence of DMSO or MEHP (0.1–100 μg/ml) with or without NAC (0.5–1 mM) for 96 h. Growth of follicles was reported as percentage change at 96 h. Bars with different letters are significantly different from each other (n = 10–16 follicles/treatment/experiment; P ≤ 0.05). Data on the graph represent the mean ± SEM from at least three separate experiments.
Effect of MEHP and NAC Cotreatment on Cell-Cycle Regulators and Apoptotic Genes in Antral Follicles
Because NAC rescues follicle growth from MEHP treatment (Fig. 4), we examined the effects of NAC cotreatment on the expression of selected cell-cycle and apoptotic regulators. At 96 h, MEHP (10 and 100 μg/ml) significantly reduced the expression of Ccnd2 and Ccne1, and MEHP (100 μg/ml) significantly decreased Cdk4 levels (Fig. 5A). Further, NAC cotreatment restored the expression of these genes to control levels (Fig. 5A). Similarly, exposure to MEHP (10 and 100 μg/ml) significantly reduced the expression of Bcl-2 but enhanced the expression of Bax (Fig. 5B). NAC cotreatment (1 mM), however, counteracted the effects of MEHP on the expression of these genes and restored the levels of Bcl-2 and Bax to control levels (Fig. 5B).
FIG. 5.

Effect of MEHP and NAC cotreatment on cell-cycle regulators and apoptotic genes. Antral follicles were exposed to DMSO or MEHP (10 and 100 μg/ml) with or without NAC (1 mM) for 96 h and then subjected to qPCR analysis for Ccnd2, Ccne1, and Cdk4 (A) or Bcl-2 and Bax (B) mRNA expression levels. All values were normalized to Actb as loading control and reported as relative fold-change compared to DMSO levels. Data on the graph represent the mean ± SEM from at least three separate experiments. Bars with an asterisk (*) are significantly different from DMSO controls (n = 10–16 follicles/treatment/experiment; P ≤ 0.05).
Effect of MEHP Treatment on Gene Expression of Antioxidant Enzymes
Because MEHP induced oxidative stress in cultured antral follicles (Fig. 1), experiments were conducted to determine if it did so by altering the expression of antioxidant enzymes required to detoxify the ROS in the system. Specifically, the expression levels of the endogenous antioxidant enzymes Sod1, Gpx, and Cat were compared in DMSO- and MEHP-treated follicles. At 96 h, only MEHP (100 μg/ml) significantly decreased the expression of Sod1 and Gpx compared to DMSO controls. Other MEHP doses (0.1–10 μg/ml) did not significantly affect Sod1 and Gpx expression compared to controls (Fig. 6, A and B). MEHP did not significantly affect Cat expression compared to DMSO controls at any of the tested doses (Fig. 6C). NAC cotreatment significantly increased the expression of Sod1 and Gpx compared to DMSO controls and MEHP alone (Fig. 6D).
FIG. 6.

Effect of MEHP and NAC cotreatment on Sod1, Gpx, and Cat mRNA expression levels. After exposure of antral follicles to DMSO or MEHP (0.1–100 μg/ml) for 96 h in vitro, the follicles were collected and subjected to qPCR analysis for Sod1 (A), Gpx (B), and Cat (C) mRNA expression levels. In addition, antral follicles were exposed to DMSO or MEHP (100 μg/ml) with or without NAC (1 mM) for 96 h and subjected to qPCR to measure the mRNA expression levels of Sod1 and Gpx (D). All values were normalized to Actb as a loading control and reported as relative fold-change compared to DMSO controls. Data on the graph represent the mean ± SEM from at least three separate experiments. An asterisk (*) indicates a significant difference from the DMSO controls (n = 10–16 follicles/treatment/experiment; P ≤ 0.05).
Effect of MEHP Treatment on Activities of Antioxidant Enzymes
To assess the effects of MEHP on the activities of antioxidant enzymes, we measured the activities of SOD1, GPX, and CAT at 72 and 96 h, which are the time points when we observed MEHP-induced growth inhibition. At 72 h, MEHP (10 and 100 μg/ml) significantly inhibited GPX activity, whereas MEHP (10 μg/ml) significantly increased SOD1 activity compared to DMSO (Fig. 7A). By 96 h, all doses of MEHP significantly inhibited the activity of GPX (Fig. 7B). MEHP (10 μg/ml) increased SOD1 activity, whereas 100 μg/ml MEHP inhibited SOD1 activity. Only certain doses of MEHP (0.1 and 10 μg/ml) significantly increased CAT activity compared to DMSO (Fig. 7B).
FIG. 7.

Effect of MEHP and NAC on SOD1, GPX, and CAT activities in antral follicles. A) Antral follicles were exposed in vitro to DMSO or MEHP (0.1–100 μg/ml) for 72 or 96 h and then subjected to specific assays to measure the enzyme activities of SOD1, GPX, and CAT. All values were normalized to protein level as a loading control and reported as relative fold-change compared to DMSO controls. Data on the graph represent the mean ± SEM from at least three separate experiments. An asterisk (*) indicates a significant difference from DMSO controls (n = 24 follicles/treatment/experiment; P ≤ 0.05). B and C) Antral follicles were cultured in the presence of DMSO or MEHP (10 and 100 μg/ml) with or without NAC (0.5–1 mM) for 96 h. After culture, the follicles were collected and subjected to specific activity assays for SOD1 activity (B) and GPX activity (C). The activities of SOD1 and GPX were normalized to protein level as a loading control. All values are reported as relative fold-change compared to DMSO controls. Data on the graph represent the mean ± SEM from at least three separate experiments. Bars with different letters are significantly different from each other (n = 16–24 follicles/treatment/experiment; P ≤ 0.05).
Effect of NAC Cotreatment on Activities of SOD1 and GPX
Because addition of NAC protects follicle growth from MEHP-induced inhibition by reducing the oxidative stress in the system (Figs. 1 and 4), we conducted experiments to investigate whether NAC cotreatment protects the follicle from MEHP-induced oxidative stress by restoring the activity of SOD1 and GPX to control levels. Compared to DMSO controls, MEHP (10 μg/ml) significantly increased SOD1 activity, whereas MEHP (100 μg/ml) inhibited SOD1 activity at 96 h (Fig. 7B). Addition of NAC (1 mM) did not change the effect of MEHP on SOD1 activity at 10 μg/ml but restored SOD1 activity to control levels at 100 μg/ml (Fig. 7C). MEHP (10 and 100 μg/ml) significantly inhibited GPX activity, and NAC (1 mM) cotreatment prevented the inhibitory effects of MEHP on GPX activity (Fig. 7C).
DISCUSSION
Numerous studies have shown that endocrine-disrupting chemicals induce oxidative stress in reproductive tissues by disrupting internal antioxidant protective mechanisms [18, 38, 41–43]. Our previous study [15] indicated that DEHP inhibits growth of mouse antral follicles, increases ROS levels, and decreases the expression and activity of SOD1 in vitro. However, studies have suggested that MEHP is more potent than DEHP and that it causes different effects than DEHP on rat follicle development, testicular cell survival, and fetal testis morphology and function [18–22]. Thus, the present study was designed to test whether MEHP, the active metabolite of DEHP, also induces oxidative stress in antral follicles and, if so, to elucidate the mechanisms by which it does.
In the present study, we found that MEHP induces ROS levels, disrupts the expression and activities of SOD1 and GPX, and inhibits follicle growth. We also found that MEHP reduces the expression of cell-cycle regulators (Ccnd2, Ccne1 and Cdk4) and the antiapoptotic factor (Bcl-2) but increases the expression of the proapoptotic factor (Bax). In addition, our data indicate that cotreatment with NAC, a known antioxidant [44], rescues the toxic effects of MEHP in antral follicles by restoring the expression and activity of antioxidant enzymes and by restoring the expression of proliferation and apoptotic factors to control levels.
Oxidative stress is caused by an imbalance of pro- and antioxidants in the system, which raises the physiological level of ROS, including free oxygen species and peroxides, and leads to oxidative DNA, lipid, and protein damage [45]. Antioxidant enzymes, such as SOD1, GPX, and CAT, compose the most important intracellular antioxidant defense system to prevent cellular damage caused oxidative stress. SODs (SOD1 and SOD2) are responsible for dismutation of O2− to H2O2 and oxygen. Further, CAT and GPX, coupled with the glutathione cycle, reduce H2O2 to water and oxygen. Any disruption of this defense system will cause accumulation of ROS and lead to oxidative damage [46]. ROS and antioxidants have been implicated in the regulation of follicle development, oocyte maturation, fertilization, implantation, embryo development, and pregnancy in human and animal models [47–51]. Several studies have shown that extra accumulation of ROS is directly associated with the age-related decline in follicle quality [52], endometriosis [48], unexplained infertility, and low success rates in assisted reproductive techniques [46, 53] and that it may play an important role in the initiation of apoptosis in antral follicles [54]. Oxidative stress is also suggested as a common mechanism in endocrine disruptor-mediated dysfunction in reproduction, and it is thought that ROS may serve as an early marker for toxicity evaluation [38, 55–57].
Our data suggest that the ability of MEHP to inhibit GPX activity is one of the main mechanisms by which it exerts its toxic effects. Our findings are consistent with other studies of MEHP, which show that MEHP induces oxidative stress by decreasing the expression and activity of GPX in kidney and prostate cancer cells [58, 59], and studies of GPX, which show that dysregulation of GPX leads to apoptosis in endothelial cells and luteal cells in the ovary [60, 61].
Inhibition of GPX induced by MEHP can lead to accumulation of H2O2 in antral follicles, which then could activate other antioxidant enzymes to try to protect follicles from oxidative damage. This may explain why MEHP (10 μg/ml) increased SOD1 activity and MEHP (0.1 and 10 μg/ml) increased CAT activity (Fig. 7B). However, the highest dose of MEHP (100 μg/ml) likely overwhelmed the antioxidant system and inhibited the activity of SOD1 by 96 h, which then led to a further increase ROS levels and resulted in blockage of follicle growth. These data are consistent with previous studies showing that H2O2 induces oxidative stress and serves as a signal to elevate the activity of antioxidant enzymes to help eliminate the toxic effects of H2O2 in testicular germ cells [62, 63].
One of the important findings of present study is that MEHP induces oxidative stress via different mechanisms than its parent compound, DEHP. Specifically, MEHP affects different antioxidant enzymes compared to DEHP. In turn, this may cause different ROS accumulation in MEHP- and DEHP-treated antral follicles. Our previous study [15] showed that DEHP predominantly affects the expression and activity of SOD1 but not GPX and CAT. The present data indicate that MEHP induces oxidative stress mainly by suppressing the expression and activity of GPX. We speculate that DEHP decreases in SOD1 activity would lead to accumulation of superoxide (O2−) and then cause damage in antral follicles. MEHP-induced decreases in both SOD1 and GPX activity would lead to accumulation of H2O2 and cause more damage in antral follicles than observed with DEHP. This could be why lower doses of MEHP (0.1 μg/ml) cause damage to follicles compared to DEHP (1 μg/ml) [15]. The different antioxidant enzymes affected by DEHP and MEHP might cause different ROS accumulation in follicles. Further, it is possible that follicles are more sensitive to H2O2 than O2− and that this leads to MEHP being more toxic than DEHP in antral follicles.
The mechanisms by which MEHP affects antioxidant enzyme expression and activity remain unclear. It is known, however, that two classes of transcription factors, nuclear factor κB (NF-κB) and activator protein 1 (AP-1), are involved in the oxidative stress response in mammalian systems [64]. Studies have shown that antioxidant response elements (AREs) and motifs for NF-κB and AP-1 are present in the promoter regions of most of the antioxidant enzymes, including SOD1, SOD2, GPX, and CAT [64, 65]. Elevated ROS levels regulate the expression and activity of these antioxidant enzymes by activating different signaling pathways in different biological systems, which include extracellular signal-regulated kinases (ERKs) and p38 mitogen-activated protein kinases (MAPKs) [66], tyrosine kinase [67], phosphotidylinositol-3-kinase (PI3K)/Akt [68], and protein kinase C [69]. DEHP induces oxidative stress and causes apoptosis in hepatocytes by activating ERK/MAPK and p38/MAPK [70, 71]. MEHP activates PI3K/Akt and NF-κB signaling in the testis and induces oxidative stress and germ cell apoptosis [72]. We speculate that the structural differences between DEHP and MEHP lead to their distinct effects on antioxidant enzymes in antral follicles. DEHP has two 2-ethylhexanol branched chains, which cause DEHP to be lipophilic and allow it to easily cross the lipid membrane to activate intracellular signal cascades. MEHP, with only one 2-ethylhexanol branched chain, is less lipophilic than DEHP. This could cause MEHP to activate or inhibit the signaling molecules located on membrane instead of activating intracellular molecules. Thus, it is possible that DEHP and MEHP affect different signaling pathways and transcription factors in antral follicles and that this leads to distinct effects on the expressions and activities of SOD1 and GPX.
Studies have shown that oxidative stress is one of the risk factors for disruption of normal cell proliferation and apoptosis [73, 74]. Follicle growth depends largely on follicular cell proliferation and health. Thus, we tested the possibility that MEHP-induced follicle growth inhibition is due to its effects on cell-cycle regulators and apoptotic factors via an oxidative stress pathway. We found that MEHP exposure suppresses the expression of key cell-cycle regulators at the G1/S transition and increases the expression of proapoptotic factor, Bax, but decreases the expression of the antiapoptotic factor, Bcl-2. Further, our data show that NAC cotreatment can rescue the effects of MEHP on the expression of G1/S-phase cell-cycle regulators, apoptotic factors, and follicle growth, indicating that these toxic effects are due to MEHP-induced oxidative stress. These findings are consistent with studies showing that oxidative stress interrupts cell proliferation and induces apoptosis in different biological systems [62, 75, 76] and that NAC acts as a free radical scavenger and glutathione precursor in reproductive tissues and protects them from oxidative stress-induced apoptosis [38, 76–78]. Interestingly, studies have shown that overexpressing GPX1 in endothelial cells reduces the Bax:Bcl-2 ratio and protects cells against oxidative stress-induced apoptosis [60] and that down-regulation of GPX leads to bovine luteal cell apoptosis [61]. These studies further support our hypothesis that the MEHP-induced disruption of cell-cycle regulators and apoptotic factors is due to MEHP-induced oxidative stress caused by inhibition of GPX.
In conclusion, the present study shows that MEHP exposure induces oxidative stress and inhibits growth in antral follicles. It also raises the question of why MEHP and DEHP induce oxidative stress via different mechanisms. Further studies should focus on distinguishing the effects and mechanisms by which MEHP and DEHP affect ovarian functions by comparing MEHP and DEHP treatment in knockout mouse models with deletions in antioxidant enzymes or by using models in which antioxidant enzymes are overexpressed. A comparison of the effects of MEHP and DEHP in vivo would also further expand our understanding on how phthalates affect female reproduction.
ACKNOWLEDGMENT
The authors thank Liying Gao for her outstanding technical help and also appreciate Bethany Karman and Ayelet Ziv-Gal for their help with follicle isolation and other technical assistance.
Footnotes
Supported by NIH R01ES019178 (J.A.F.); a Billie Field Fellowship in Reproductive Biology (W.W. and Z.R.C.); an NIEHS Endocrine, Development, and Reproductive Toxicology Training Fellowship (W.W.); an NIH Pathway to Independence Award (Z.R.C.); and an Environmental Toxicology Scholarship (M.S.B.).
REFERENCES
- Heudorf U, Mersch-Sundermann V, Angerer J. Phthalates: toxicology and exposure. Int J Hyg Environ Health 2007; 210: 623 634. [DOI] [PubMed] [Google Scholar]
- Halden RU. Plastics and health risks. Annu Rev Public Health 2010; 31: 179 194. [DOI] [PubMed] [Google Scholar]
- Lyche JL, Gutleb AC, Bergman A, Eriksen GS, Murk AJ, Ropstad E, Saunders M, Skaare JU. Reproductive and developmental toxicity of phthalates. J Toxicol Environ Health B Crit Rev 2009; 12: 225 249. [DOI] [PubMed] [Google Scholar]
- Weuve J, Hauser R, Calafat AM, Missmer SA, Wise LA. Association of exposure to phthalates with endometriosis and uterine leiomyomata: findings from NHANES, 1999–2004. Environ Health Perspect 2010; 118: 825 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyatt RM, Adibi JJ, Calafat AM, Camann DE, Rauh V, Bhat HK, Perera FP, Andrews H, Just AC, Hoepner L, Tang D, Hauser R. Prenatal di(2-ethylhexyl)phthalate exposure and length of gestation among an inner-city cohort. Pediatrics 2009; 124: e1213 e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino-Andrade AJ, Chahoud I. Reproductive toxicity of phthalate esters. Mol Nutr Food Res 2009; 54: 148 157. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chun S, Jang JY, Chae HD, Kim CH, Kang BM. Increased plasma levels of phthalate esters in women with advanced-stage endometriosis: a prospective case-control study. Fertil Steril 2010; 95: 357 359. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Zeybek ND, Giray B, Asan E, Hincal F. The effects of di(2-ethylhexyl)phthalate exposure and selenium nutrition on Sertoli cell vimentin structure and germ-cell apoptosis in rat testis. Arch Environ Contam Toxicol 2012; 62: 539 547. [DOI] [PubMed] [Google Scholar]
- Davis BJ, Maronpot RR, Heindel JJ. Di-(2-ethylhexyl) phthalate suppresses estradiol and ovulation in cycling rats. Toxicol Appl Pharmacol 1994; 128: 216 223. [DOI] [PubMed] [Google Scholar]
- Grande SW, Andrade AJ, Talsness CE, Grote K, Golombiewski A, Sterner-Kock A, Chahoud I. A dose-response study following in utero and lactational exposure to di-(2-ethylhexyl) phthalate (DEHP): reproductive effects on adult female offspring rats. Toxicology 2007; 229: 114 122. [DOI] [PubMed] [Google Scholar]
- Frederiksen H, Skakkebaek NE, Andersson AM. Metabolism of phthalates in humans. Mol Nutr Food Res 2007; 51: 899 911. [DOI] [PubMed] [Google Scholar]
- Fan J, Traore K, Li W, Amri H, Huang H, Wu C, Chen H, Zirkin B, Papadopoulos V. Molecular mechanisms mediating the effect of mono-(2-ethylhexyl) phthalate on hormone-stimulated steroidogenesis in MA-10 mouse tumor Leydig cells. Endocrinology 2010; 151: 3348 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muczynski V, Cravedi JP, Lehraiki A, Levacher C, Moison D, Lecureuil C, Messiaen S, Perdu E, Frydman R, Habert R, Rouiller-Fabre V. Effect of mono-(2-ethylhexyl) phthalate on human and mouse fetal testis: in vitro and in vivo approaches. Toxicol Appl Pharmacol 2012; 261: 97 104. [DOI] [PubMed] [Google Scholar]
- Lovekamp-Swan T, Davis BJ. Mechanisms of phthalate ester toxicity in the female reproductive system. Environ Health Perspect 2003; 111: 139 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Craig ZR, Basavarajappa MS, Gupta RK, Flaws JA. Di (2-ethylhexyl) phthalate inhibits growth of mouse ovarian antral follicles through an oxidative stress pathway. Toxicol Appl Pharmacol 2012; 258: 288 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Singh JM, Leslie TC, Meachum S, Flaws JA, Yao HH. Di-(2-ethylhexyl) phthalate and mono-(2-ethylhexyl) phthalate inhibit growth and reduce estradiol levels of antral follicles in vitro. Toxicol Appl Pharmacol 2010; 242: 224 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Curto KA, Thomas MJ. MEHP/DEHP: gonadal toxicity and effects on rodent accessory sex organs. Environ Health Perspect 1982; 45: 85 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkekoglu P, Rachidi W, Yuzugullu OG, Giray B, Favier A, Ozturk M, Hincal F. Evaluation of cytotoxicity and oxidative DNA damaging effects of di(2-ethylhexyl)-phthalate (DEHP) and mono(2-ethylhexyl)-phthalate (MEHP) on MA-10 Leydig cells and protection by selenium. Toxicol Appl Pharmacol 2010; 248: 52 62. [DOI] [PubMed] [Google Scholar]
- Rose ML, Rivera CA, Bradford BU, Graves LM, Cattley RC, Schoonhoven R, Swenberg JA, Thurman RG. Kupffer cell oxidant production is central to the mechanism of peroxisome proliferators. Carcinogenesis 1999; 20: 27 33. [DOI] [PubMed] [Google Scholar]
- Wan X, Zhu Y, Ma X, Zhu J, Zheng Y, Hou J, Wang F, Liu Z, Zhang T. Effect of DEHP and its metabolite MEHP on in vitro rat follicular development [in Chinese]. Wei Sheng Yan Jiu 2010; 39: 268 270, 274. [PubMed] [Google Scholar]
- Sjoberg P, Bondesson U, Gray TJ, Ploen L. Effects of di-(2-ethylhexyl) phthalate and five of its metabolites on rat testis in vivo and in in vitro. Acta Pharmacol Toxicol (Copenh) 1986; 58: 225 233. [DOI] [PubMed] [Google Scholar]
- Chauvigne F, Menuet A, Lesne L, Chagnon MC, Chevrier C, Regnier JF, Angerer J, Jegou B. Time- and dose-related effects of di-(2-ethylhexyl) phthalate and its main metabolites on the function of the rat fetal testis in vitro. Environ Health Perspect 2009; 117: 515 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MC, Midgley AR, Jr,, Richards JS. Hormonal regulation of ovarian cellular proliferation. Cell 1978; 14: 71 78. [DOI] [PubMed] [Google Scholar]
- Quirk SM, Cowan RG, Harman RM, Hu CL, Porter DA. Ovarian follicular growth and atresia: the relationship between cell proliferation and survival. J Anim Sci 2004; 82 (suppl): E40 E52. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Meachum S, Hernandez-Ochoa I, Peretz J, Yao HH, Flaws JA. Methoxychlor inhibits growth of antral follicles by altering cell cycle regulators. Toxicol Appl Pharmacol 2009; 240: 1 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz J, Craig ZR, Flaws JA. Bisphenol A inhibits follicle growth and induces atresia in cultured mouse antral follicles independently of the genomic estrogenic pathway. Biol Reprod 2012; 87 3: 63, 1 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays 1995; 17: 471 480. [DOI] [PubMed] [Google Scholar]
- Chua PJ, Yip GW, Bay BH. Cell cycle arrest induced by hydrogen peroxide is associated with modulation of oxidative stress related genes in breast cancer cells. Exp Biol Med (Maywood) 2009; 234: 1086 1094. [DOI] [PubMed] [Google Scholar]
- Clopton DA, Saltman P. Low-level oxidative stress causes cell-cycle specific arrest in cultured cells. Biochem Biophys Res Commun 1995; 210: 189 196. [DOI] [PubMed] [Google Scholar]
- Buttke TM, Sandstrom PA. Oxidative stress as a mediator of apoptosis. Immunol Today 1994; 15: 7 10. [DOI] [PubMed] [Google Scholar]
- Lovekamp TN, Davis BJ. Mono-(2-ethylhexyl) phthalate suppresses aromatase transcript levels and estradiol production in cultured rat granulosa cells. Toxicol Appl Pharmacol 2001; 172: 217 224. [DOI] [PubMed] [Google Scholar]
- Reinsberg J, Wegener-Toper P, van der Ven K, van der Ven H, Klingmueller D. Effect of mono-(2-ethylhexyl) phthalate on steroid production of human granulosa cells. Toxicol Appl Pharmacol 2009; 239: 116 123. [DOI] [PubMed] [Google Scholar]
- Lenie S, Smitz J. Steroidogenesis-disrupting compounds can be effectively studied for major fertility-related endpoints using in vitro cultured mouse follicles. Toxicol Lett 2009; 185: 143 152. [DOI] [PubMed] [Google Scholar]
- Cobellis L, Latini G, De Felice C, Razzi S, Paris I, Ruggieri F, Mazzeo P, Petraglia F. High plasma concentrations of di-(2-ethylhexyl)-phthalate in women with endometriosis. Hum Reprod 2003; 18: 1512 1515. [DOI] [PubMed] [Google Scholar]
- Pak VM, Nailon RE, McCauley LA. Controversy: neonatal exposure to plasticizers in the NICU. MCN Am J Matern Child Nurs 2007; 32: 244 249. [DOI] [PubMed] [Google Scholar]
- Kamrin MA. Phthalate risks, phthalate regulation, and public health: a review. J Toxicol Environ Health B Crit Rev 2009; 12: 157 174. [DOI] [PubMed] [Google Scholar]
- Sjoberg PO, Bondesson UG, Sedin EG, Gustafsson JP. Exposure of newborn infants to plasticizers. Plasma levels of di-(2-ethylhexyl) phthalate and mono-(2-ethylhexyl) phthalate during exchange transfusion. Transfusion 1985; 25: 424 428. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Schuh RA, Fiskum G, Flaws JA. Methoxychlor causes mitochondrial dysfunction and oxidative damage in the mouse ovary. Toxicol Appl Pharmacol 2006; 216: 436 445. [DOI] [PubMed] [Google Scholar]
- Lim J, Luderer U. Oxidative damage increases and antioxidant gene expression decreases with aging in the mouse ovary. Biol Reprod 2010; 84: 775 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestell RG, Albanese C, Reutens AT, Segall JE, Lee RJ, Arnold A. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr Rev 1999; 20: 501 534. [DOI] [PubMed] [Google Scholar]
- Aly HA, Khafagy RM. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced cytotoxicity accompanied by oxidative stress in rat Sertoli cells: possible role of mitochondrial fractions of Sertoli cells. Toxicol Appl Pharmacol 2011; 252: 273 280. [DOI] [PubMed] [Google Scholar]
- Jin MH, Hong CH, Lee HY, Kang HJ, Han SW. Toxic effects of lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on development of male reproductive system: involvement of antioxidants, oxidants, and p53 protein. Environ Toxicol 2010; 25: 1 8. [DOI] [PubMed] [Google Scholar]
- Botelho GG, Bufalo AC, Boareto AC, Muller JC, Morais RN, Martino-Andrade AJ, Lemos KR, Dalsenter PR. Vitamin C and resveratrol supplementation to rat dams treated with di(2-ethylhexyl)phthalate: impact on reproductive and oxidative stress end points in male offspring. Arch Environ Contam Toxicol 2009; 57: 785 793. [DOI] [PubMed] [Google Scholar]
- Parasassi T, Brunelli R, Costa G, De Spirito M, Krasnowska E, Lundeberg T, Pittaluga E, Ursini F. Thiol redox transitions in cell signaling: a lesson from N-acetylcysteine. TheScientificWorldJOURNAL 2010; 10: 1192 1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007; 39: 44 84. [DOI] [PubMed] [Google Scholar]
- Agarwal A, Gupta S, Sekhon L, Shah R. Redox considerations in female reproductive function and assisted reproduction: from molecular mechanisms to health implications. Antioxid Redox Signal 2008; 10: 1375 1403. [DOI] [PubMed] [Google Scholar]
- Ruder EH, Hartman TJ, Goldman MB. Impact of oxidative stress on female fertility. Curr Opin Obstet Gynecol 2009; 21: 219 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A, Gupta S, Sharma RK. Role of oxidative stress in female reproduction. Reprod Biol Endocrinol 2005; 3: 28 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gubory KH, Fowler PA, Garrel C. The roles of cellular reactive oxygen species, oxidative stress and antioxidants in pregnancy outcomes. Int J Biochem Cell Biol 2010; 42: 1634 1650. [DOI] [PubMed] [Google Scholar]
- Dennery PA. Oxidative stress in development: nature or nurture? Free Radic Biol Med 2010; 49: 1147 1151. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu Y, An Z, Huang D, Qi Y, Zhang Y. Mediating effect of ROS on mtDNA damage and low ATP content induced by arsenic trioxide in mouse oocytes. Toxicol in Vitro 2011; 25: 979 984. [DOI] [PubMed] [Google Scholar]
- Tatone C, Amicarelli F, Carbone MC, Monteleone P, Caserta D, Marci R, Artini PG, Piomboni P, Focarelli R. Cellular and molecular aspects of ovarian follicle ageing. Hum Reprod Update 2008; 14: 131 142. [DOI] [PubMed] [Google Scholar]
- Matos L, Stevenson D, Gomes F, Silva-Carvalho JL, Almeida H. Superoxide dismutase expression in human cumulus oophorus cells. Mol Hum Reprod 2009; 15: 411 419. [DOI] [PubMed] [Google Scholar]
- Devine PJ, Perreault SD, Luderer U. Roles of reactive oxygen species and antioxidants in ovarian toxicity. Biol Reprod 2012; 86: 27, 1 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Miller KP, Babus JK, Flaws JA. Methoxychlor inhibits growth and induces atresia of antral follicles through an oxidative stress pathway. Toxicol Sci 2006; 93: 382 389. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Mathur PP. Induction of oxidative stress in the rat testis after short-term exposure to the organochlorine pesticide methoxychlor. Arch Toxicol 2002; 76: 692 698. [DOI] [PubMed] [Google Scholar]
- Minamiyama Y, Ichikawa H, Takemura S, Kusunoki H, Naito Y, Yoshikawa T. Generation of reactive oxygen species in sperms of rats as an earlier marker for evaluating the toxicity of endocrine-disrupting chemicals. Free Radic Res 2010; 44: 1398 1406. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Giray BK, Kizilgun M, Rachidi W, Hininger-Favier I, Roussel AM, Favier A, Hincal F. Di(2-ethylhexyl)phthalate-induced renal oxidative stress in rats and protective effect of selenium. Toxicol Mech Methods 2012; 22: 415 423. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Rachidi W, De Rosa V, Giray B, Favier A, Hincal F. Protective effect of selenium supplementation on the genotoxicity of di(2-ethylhexyl)phthalate and mono(2-ethylhexyl)phthalate treatment in LNCaP cells. Free Radic Biol Med 2010; 49: 559 566. [DOI] [PubMed] [Google Scholar]
- Faucher K, Rabinovitch-Chable H, Cook-Moreau J, Barriere G, Sturtz F, Rigaud M. Overexpression of human GPX1 modifies Bax to Bcl-2 apoptotic ratio in human endothelial cells. Mol Cell Biochem 2005; 277: 81 87. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ishigami T, Makino N, Sakamoto K. The down-regulation of glutathione peroxidase causes bovine luteal cell apoptosis during structural luteolysis. J Biochem 2001; 129: 937 942. [DOI] [PubMed] [Google Scholar]
- Maheshwari A, Misro MM, Aggarwal A, Sharma RK, Nandan D. N-acetyl-l-cysteine counteracts oxidative stress and prevents H2O2 induced germ cell apoptosis through down-regulation of caspase-9 and JNK/c-Jun. Mol Reprod Dev 2011; 78: 69 79. [DOI] [PubMed] [Google Scholar]
- Bose Girigoswami K, Bhaumik G, Ghosh R. Induced resistance in cells exposed to repeated low doses of H2O2 involves enhanced activity of antioxidant enzymes. Cell Biol Int 2005; 29: 761 767. [DOI] [PubMed] [Google Scholar]
- Scandalios JG. Oxidative stress: molecular perception and transduction of signals triggering antioxidant gene defenses. Braz J Med Biol Res 2005; 38: 995 1014. [DOI] [PubMed] [Google Scholar]
- Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohe R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol 2005; 25: 4914 4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knock GA, Ward JP. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal 2011; 15: 1531 1547. [DOI] [PubMed] [Google Scholar]
- El-Deeb IM, Yoo KH, Lee SH. ROS receptor tyrosine kinase: a new potential target for anticancer drugs. Med Res Rev 2010; 31: 794 818. [DOI] [PubMed] [Google Scholar]
- Rojo AI, Salinas M, Martin D, Perona R, Cuadrado A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J Neurosci 2004; 24: 7324 7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara N, Oguro T, Sakabe T, Matsushima M, Takikawa O, Isobe K, Nagase F. Hemoglobin induces the expression of indoleamine 2,3-dioxygenase in dendritic cells through the activation of PI3K, PKC, and NF-kappaB and the generation of reactive oxygen species. J Cell Biochem 2009; 108: 716 725. [DOI] [PubMed] [Google Scholar]
- Lee J, Lim KT. Plant-originated glycoprotein (24 kDa) has an inhibitory effect on proliferation of BNL CL.2 cells in response to di(2-ethylhexyl)phthalate. Cell Biochem Funct 2011; 29: 496 505. [DOI] [PubMed] [Google Scholar]
- Ghosh J, Das J, Manna P, Sil PC. Hepatotoxicity of di-(2-ethylhexyl)phthalate is attributed to calcium aggravation, ROS-mediated mitochondrial depolarization, and ERK/NF-kappaB pathway activation. Free Radic Biol Med 2010; 49: 1779 1791. [DOI] [PubMed] [Google Scholar]
- Rogers R, Ouellet G, Brown C, Moyer B, Rasoulpour T, Hixon M. Cross-talk between the Akt and NF-kappaB signaling pathways inhibits MEHP-induced germ cell apoptosis. Toxicol Sci 2008; 106: 497 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci 2007; 96: 2181 2196. [DOI] [PubMed] [Google Scholar]
- Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol 2012; 22: 592 601. [DOI] [PubMed] [Google Scholar]
- Xue T, Luo P, Zhu H, Zhao Y, Wu H, Gai R, Wu Y, Yang B, Yang X, He Q. Oxidative stress is involved in Dasatinib-induced apoptosis in rat primary hepatocytes. Toxicol Appl Pharmacol 2012; 261: 280 291. [DOI] [PubMed] [Google Scholar]
- Song Y, Shi Y, Yu H, Hu Y, Wang Y. Yang K. p,p′-Dichlorodiphenoxydichloroethylene induced apoptosis of Sertoli cells through oxidative stress-mediated p38 MAPK and mitochondrial pathway. Toxicol Lett 2011; 202: 55 60. [DOI] [PubMed] [Google Scholar]
- Aggarwal A, Misro MM, Maheshwari A, Sehgal N. Differential modulation of apoptotic gene expression by N-acetyl-l-cysteine in Leydig cells stimulated persistently with hCG in vivo. Mol Cell Endocrinol 2012; 348: 155 164. [DOI] [PubMed] [Google Scholar]
- Otala M, Erkkila K, Tuuri T, Sjoberg J, Suomalainen L, Suikkari AM, Pentikainen V, Dunkel L. Cell death and its suppression in human ovarian tissue culture. Mol Hum Reprod 2002; 8: 228 236. [DOI] [PubMed] [Google Scholar]