


Abstract
The c-Myc transcription factor represses the mRNA expression of the platelet-derived growth factor receptor beta gene (PDGFRB). Using chromatin immunoprecipitation, we show that c-Myc binds to the proximal promoter of the PDGFRB gene in proliferating rat fibroblasts. Interestingly, mutant c-Myc proteins that are unable to repress PDGFRB gene expression, c-MycdBR and c-Mycd106-143, are still able to bind to the promoter in vivo. Hence, promoter-binding and repression of PDGFRB by c-Myc are separable activities. We also show that Myc repression of PDGFRB is not dependent on previously described or known transactivator-binding regions, suggesting Myc may be recruited to the promoter by multiple or yet unidentified transcription factors. In the presence of intact promoter-binding by Myc, trichostatin A (TSA) can block Myc repression of PDGFRB in vivo, again demonstrating that promoter-binding and repression are separable. Taken together, we hypothesize that Myc repression of PDGFRB expression occurs by a multi-step mechanism in which repression is initiated after Myc is recruited to the promoter.
INTRODUCTION
Fibroblasts proliferate in response to platelet-derived growth factor (PDGF) during physiological circumstances such as wound healing (1). Receiving, and communicating this mitogenic signal to the proliferative machinery in the nucleus, is the PDGF receptor beta polypeptide (PDGFRB). Activation of cell proliferation by the signal transduction cascade through PDGFRB activates the immediate early genes, such as c-myc (2). c-Myc itself is a nuclear transcription factor that is a potent regulator of cell growth, proliferation and progression of the cell cycle through the G1/S phase transition (3). When deregulated, c-Myc drives neoplasia and tumourigenesis (4,5). Interestingly, at physiological levels, c-Myc has also been shown to downregulate the expression of growth promoting genes such as c-myc itself, and PDGFRB (6,7). Downregulation of these genes is believed to be part of a negative autoregulatory loop in which c-Myc controls the duration and/or extent that cells are stimulated to proliferate. Deregulation of these regulatory loops, which limit cell growth, are observed in cancers and may potentiate tumour progression (8). Thus, elucidating how c-Myc mechanistically regulates these loops and represses gene expression may provide insight into disease states, their progression and their eradication.
The c-Myc transcription factor activates and represses the expression of many genes (www.myc-cancer-gene.org) (9). To activate transcription, c-Myc heterodimerizes with Max, which facilitates direct DNA-binding to E-boxes (CACGTG) (10,11). Once bound, c-Myc recruits histone acetyltransferase (HAT) activities that modify the local histones, and/or recruits PTEFb, which likely affects RNA polymerase II (RNAPII) phosphorylation and promoter clearance (12–15). How c-Myc represses gene expression is an emerging field and several models have been proposed. Recent works have shown that c-Myc can bind to the promoters of many repressed genes in vivo (16–18). However, the precise molecular mechanism of c-Myc repression at the promoter remains unclear. While recent evidence suggests an important role for Max, previous models have focused on the idea that c-Myc itself represses genes by binding to, and therefore inhibiting, proteins important for transcriptional activation, such as Miz-1, Sp1 and NF-Y (17–20). For example, PDGFRB is transactivated by NF-Y (19). c-Myc binds to a subunit of NF-Y, NF-YC, and inhibits its transactivation in an in vitro promoter–luciferase assay (19). Moreover, a c-Myc mutant protein that is unable to bind NF-Y, is also unable to repress PDGFRB (19). Similar data exists for Miz-1 at the CDKN2B (p15ink4b) and CDKN1A (p21waf1/cip1) genes (16,21). One commonality amongst all models is that c-Myc binds indirectly to repressed promoters and occurs through gene-specific activator proteins. Hence, repression of gene expression is thought to be simply a consequence of its recruitment and binding to key proteins at the promoter (3,22). We sought to test this model by assessing if mutant c-Myc proteins, that are unable to repress PDGFRB expression, are able or unable to bind to the PDGFRB promoter. We provide evidence that promoter-binding and repression functions of c-Myc at repressed genes are separable activities. Moreover, we show that the repression mechanism can be blocked with TSA, without blocking promoter binding by c-Myc, and that it affects a post-RNAPII recruitment mechanism.
MATERIALS AND METHODS
Cell culture
The Rat-1c-myc−/− (clone HO15.19) and the parental TGR-1 cell lines were cultured in DMEM H21 (Life Technologies cat. no. 12100) + 10% CS (23). Rat-1c-myc−/− cells were infected with the replication incompetent retrovirus pBabeMN-ires-GFP carrying vector-only, human c-myc, or human c-mycd106-143; these cell lines have been previously characterized (23,24). The human c-mycdBR construct was generated by site-directed mutagenesis using the PCR primers CCGAGGAGAATGTCAACGAGCTAAAACGGAGC and GCTCCGTTTTAGCTCGTTGACATTCTCCTCGG. A purified population of Rat-1c-myc−/− cells expressing c-mycdBR was obtained by flourescence activated cell sorting (FACS) using GFP as a marker of infectivity.
Chromatin immunoprecipitation
The chromatin immunoprecipitation (ChIP) procedure used has been described (18). Briefly, the chromatin from formaldehyde-fixed (1% v/v) rat fibroblasts was sonicated and immunoprecipitated using antibodies that recognize c-Myc (N-262), Max (C-124), and RNA polymerase II (N-20) (Santa Cruz Biotechnology) or acetylated histone H3 (06-599) and H4 (06-866) (Upstate). The chromatin immunoprecipitate was PCR amplified using promoter-specific primers (see below). The PCR product was resolved by agarose gel electrophoresis and visualized by UV fluorescence. ChIP–PCR primers (5′ to 3′): PDGFRB proximal (–0.4 kb): ACACGGACTCCCACACCTC and CACCACCACCACACACTTTG; PDGFRB middle (–0.9 kb): GAATACTGTTTTCACACGGGG and CAGGAAGGGAGTGGCTGAG; PDGFRB distal (–1.5 kb): GGCAAAATCCCATCCTGC and TCTTCCCAGCGTGACTGC. The primer sequences for NUC (+574), GCK and CAD have been described (14).
Immunoprecipitation and western blot analysis
Rat-1c-myc−/− cells were harvested in ice-cold F-buffer pH 7.05 (10 mM Tris pH 7.05, 50 mM NaCl, 30 mM Na4P2O7, 50 mM NaF, 5 mM ZnCl2, 10% glycerol, 1% Triton X-100), with protease inhibitors, incubated on ice for 5–10 min and centrifuged at 12 000 r.p.m. for 10 min at 4°C (Eppendorf rotor A-4-44). 500 μg of total protein lysate was immunoprecipitated using antibodies directed against c-Myc (C-33) and Max (C-124) and 20 μL of A/G-agarose beads (Santa Cruz Biotechnology). Immunoprecipitated proteins were resolved by SDS–PAGE and immunoblotted using an anti-c-Myc (9E10) and anti-actin (Sigma, A-2066) antibody.
Transient transfection and promoter luciferase assays
The 1.6 kb Sac I mouse PDGFRB promoter–luciferase construct (gift of K. Funa), containing sequences between nucleotides −1994 and −396 relative to the translation start site of the mouse PDGFRB gene, has been described previously (25). Rat-1 c-MycERTAM cells were transiently transfected with 0.9 μg of mouse PDGFRB promoter construct and 0.1 μg of pCMV-β-galactosidase using FuGene 6 (Roche). Luciferase and β-galactosidase assays were performed as previously described (7). The mutant PDGFRB promoter constructs were generated by site-directed mutagenesis (Stratagene) using the following sequences: NF-Y mutant: CCCCAAGCTTGGCTGATCAGAATCGGCCCTGC; AP2 mutant: CCCCCCACCTCCCCGCCTTCCGCTAGCTTGGCAATCAGAATCG; Sp1 mutant, AP2 disrupted: ACGCGTCCACCGTCGACGCTGAATATTTCCTAGCACCTAATGCGCATCAACAAGCTT; Sp1 mutant, AP2 intact: ACGCGTCCACCGTCGACGCTGAATATTTCCTAGCACCTAATGCGCACCCCAAGCTT. The introduced mutation(s) abolished the binding site for each element, did not create a new site(s), and maintained the natural nucleotide length of the promoter. Promoter analyses were performed using ProScan and MacVector.
Reverse transcriptase (RT)–PCR
Total RNA was isolated by Trizol extraction. Of the total RNA, 5μg was used in a 20 μL reverse transcriptase reaction and, upon completion, was diluted 10-fold. Of the RT reaction, 1 μL was PCR amplified and visualized on an ethidium-bromide-stained agarose gel by UV light. Expression between cell lines and/or conditions was quantified using ImageQuant version 5.2 (Molecular Dynamics) and then normalized using 36B4 as a loading control. Rat RT-PCR primers sequences (5′ to 3′): CDKN1A (p21cip1/waf1): GAGAACGGTGGAACTTTGACTTC and AGAAATCTGTTAGGCTGGTCTGC; GAD D45A: TGAATGTGGGTTCGTCACCAG and TTCGTGCTTTCTGTTGCGAG; PDGFRB: AACTGCCCAGACCTTGACTCG and GCTGACTTCCCCCACTCCTTAC; 36B4: AACAAACCCGCTCTGGAGAAGC and CCTCTGGAGATTTTAGTGGTGATGC.
RESULTS
c-Myc binds to the PDGFRB proximal promoter
PDGFRB gene expression in asynchronous, subconfluently growing Rat-1c-myc−/− cells (clone HO15.19) expressing human c-myc (Rat-1c-myc−/− c-myc), and in the parental TGR-1 cell line, is repressed in comparison to levels in Rat-1c-myc−/− vector cells (Figure 1A) (7,23). This repression can be recapitulated in a luciferase reporter assay in which a 1.6 kb Sac I mouse PDGFRB promoter construct is transiently transfected into Rat-1 cells stably expressing the c-MycERTAM fusion protein. Activation of c-MycERTAM by 4-hydroxytamoxifen (OHT) resulted in a clear and reproducible repression of the luciferase activity detected (Figure 1B) (7). While it is known that c-Myc can repress PDGFRB gene expression in vivo and in vitro, there is a lack of evidence implicating a role for c-Myc at the PDGFRB promoter. To evaluate if the repression of PDGFRB by c-Myc involves promoter binding, we performed chromatin immunoprecipitation in Rat-1c-myc−/− vector or Rat-1c-myc−/− c-myc cells. Enrichment of c-Myc chromatin immunoprecipitate (Myc IP), as compared with the no antibody control (no Ab), was detected in Rat-1c-myc−/− c-myc cells, but not in Rat-1c-myc−/− vector cells, using ChIP–PCR primers designed against the PDGFRB proximal promoter (−0.4 kb) but not ChIP–PCR primers designed against a distal upstream region (−1.5 kb), relative to the transcription start site (Figure 1C). c-Myc did not bind to glucokinase (GCK), a known negative control promoter, in any of the cell lines tested (Figure 1D) (14,18). Thus, we conclude that c-Myc is bound to the PDGFRB proximal promoter in vivo. Interestingly, Max also bound to the PDGFRB proximal promoter in Rat-1c-myc−/− vector and Rat-1c-myc−/− c-myc cells (Figure 1E). These data are consistent with the model that Max is bound to c-Myc-repressed genes before and after c-Myc protein recruitment (18). Taken together, these data support a model whereby c-Myc represses the PDGFRB gene at the proximal promoter.
Figure 1.
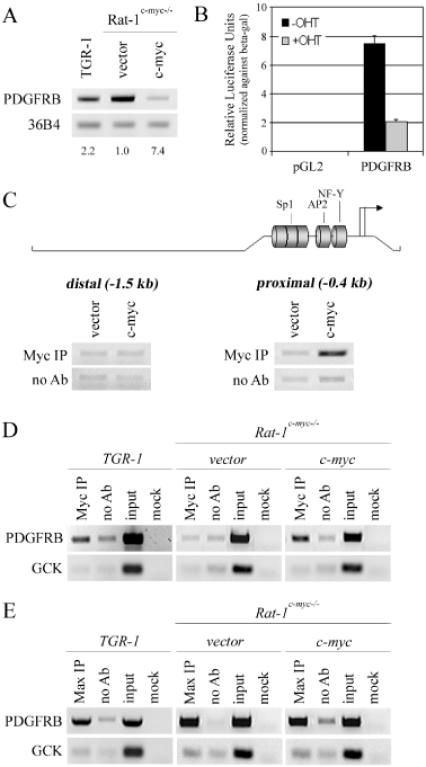
c-Myc binds to the proximal promoter of PDGFRB. (A) Expression of endogenous PDGFRB in the TGR-1 control, Rat-1c-myc−/− cells (clone HO15.19) infected with an empty retroviral vector, or retrovirus carrying human c-myc, as assessed by semi-quantitative RT–PCR. Expression of 36B4 was determined as a control for loading. Fold-repression of the normalized PDGFRB transcript level, relative to the Rat-1c-myc−/− vector control cell line, is shown. (B) A reporter luciferase assay using pGL2 vector or pGL2 vector containing 1.6 kb of the mouse PDGFRB promoter; constructs were transiently transfected into Rat-1 cells, stably expressing c-MycERTAM, and cells were then treated with or without 4-hydroxytamoxifen (OHT). (C) A chromatin immunoprecipitation (ChIP) assay using c-Myc antibody, or no antibody (no Ab) as a control, conducted in Rat-1c-myc−/− vector and Rat-1c-myc−/− c-myc cells, showing the degree of binding over the distal (−1.5 kb) and proximal PDGFRB promoter (−0.4 kb). Promoter locations are described relative to the transcriptional start region of gene promoter. A schematic of the rat PDGFRB promoter, containing putative promoter elements, is shown. (D and E) ChIP assay using c-Myc or Max specific antibody in TGR-1, Rat-1c-myc−/− vector, and Rat-1c-myc−/− c-myc cells. Binding to the PDGFRB, or control GCK, promoter is shown.
Repression and promoter-binding by c-Myc are separable functions
Since repressed promoters do not commonly contain E-boxes, c-Myc is not believed to associate with repressed promoters by contacting DNA directly; instead, binding is believed to be indirect and occurs via one or more of its many protein partners (3,22). We sought to test the mechanistic link between repression and promoter-binding at the PDGFRB promoter. As a first step, we generated Rat-1c-myc−/− cell lines that expressed human c-Myc proteins deleted for amino acids (a.a.) 106–143 (Rat-1c-myc−/− c-mycd106-143) and 355–367 (Rat-1c-myc−/− c-mycdBR) (Figure 2A). Expression of each of the two constructs in the Rat-1c-myc−/− cells was confirmed by a standard western blot analysis (Figure 2B). The 106–143 deletion includes Myc-homology box II (MbII), an evolutionarily conserved sequence motif, which has been shown to be important for binding to NF-Y as well as the repression of PDGFRB (Figure 2A) (7,19). The 355–367 deletion removes the c-Myc basic region (BR) which confers the specific in vitro recognition of chromosomal targets by c-Myc:Max heterocomplexes (Figure 2A) (26). These deletions do not alter the primary nuclear localization sequence (NLS) or the Max-binding domain (HLH-LZ) of c-Myc (Figure 2A). c-Mycd106-143 and c-MycdBR can both localize to the nucleus which is consistent with the published literature (data not shown) (27). We also show that both c-Mycd106-143 and c-MycdBR retain the capacity to bind Max in a co-immunoprecipitation assay (Figure 2C).
Figure 2.
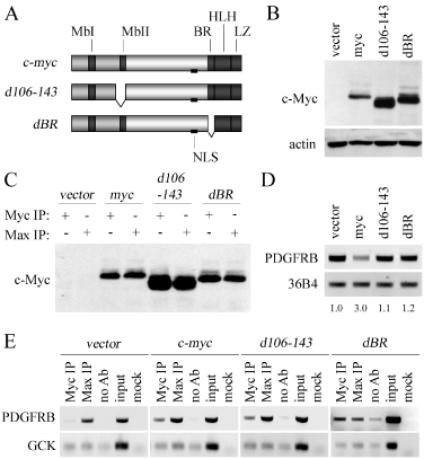
Promoter-binding and gene repression by c-Myc are separable functions. (A) Schematic of the human c-Myc, c-Mycd106-143 (d106-143), and c-MycdBR (dBR) proteins and the domains they contain Myc-homology box 1 (MbI) and 2 (MbII); nuclear localization signal (NLS); basic region (BR); helix–loop–helix (HLH); leucine zipper (LZ). (B) Immunoblot of c-Myc protein (9E10) and actin expression in Rat-1c-myc−/− cells infected with an empty retroviral vector, or retrovirus carrying c-myc, c-mycd106-143 (d106-143), or c-mycdBR (dBR). (C) Immunoblot for c-Myc protein (9E10) following immunoprecipitation (IP) using c-Myc (N-262) or Max (C-124) antibody in the Rat-1c-myc−/− cell lines indicated. (D) Expression of endogenous PDGFRB and control 36B4 in the Rat-1c-myc−/− cell lines indicated, as assessed by RT–PCR. Fold-repression of the normalized PDGFRB transcript level, relative to the Rat-1c-myc−/− vector control cell line, is shown. (E) ChIP using c-Myc or Max antibody in the Rat-1c-myc−/− cell lines indicated were conducted as described in Figure 1D and E.
Both the c-Mycd106-143 and c-MycdBR proteins were unable to repress the expression of PDGFRB, as assessed by semi-quantitative RT–PCR (Figure 2D). To assess if promoter-binding and repression of PDGFRB expression by c-Myc are the same or separable qualities, we performed ChIPs in Rat-1c-myc−/− c-mycd106-143 and Rat-1c-myc−/− c-mycdBR cells using anti-c-Myc and anti-Max antibodies. Deletion of amino acids 106–143 or the BR did not eliminate c-Myc's ability to bind to the PDGFRB promoter, as assessed by ChIP (Figure 2E). No binding by wild-type c-Myc or either of the two mutant c-Myc proteins was observed at GCK, the negative control promoter, in any of the cell lines tested (Figure 2E). Max bound to the PDGFRB promoter in all cell lines shown (Figure 2E). Hence, promoter binding by c-Myc to the PDGFRB is insufficient to affect its transcription repression function. Moreover, it implies that c-Myc's repression function may be initiated after it is bound to the promoter.
Repression does not occur solely through NF-Y, AP2 or Sp1
The observation that c-Myc binds to the PDGFRB proximal promoter in vivo is consistent with previous observations by ourselves and others. The minimal promoter region, containing the NF-Y, AP2 and Sp1 sites, is conserved between rat and mouse and is sufficient for Myc to mediate its repressive effect on transcription (Figure 3A). NF-Y, AP2 and Sp1 are transactivating proteins all capable of binding to c-Myc (19,20,28). c-Myc:NF-Y complexes have been implicated in transcriptional repression of this promoter (19,29). These data are consistent with the model that c-Myc represses PDGFRB gene expression through elements and/or proteins located within the proximal promoter region. We reasoned that if the NF-Y, AP2 or Sp1 binding site(s) were important for the repression of PDGFRB by c-Myc, then mutation of this site(s) should abrogate repression by c-Myc.
Figure 3.
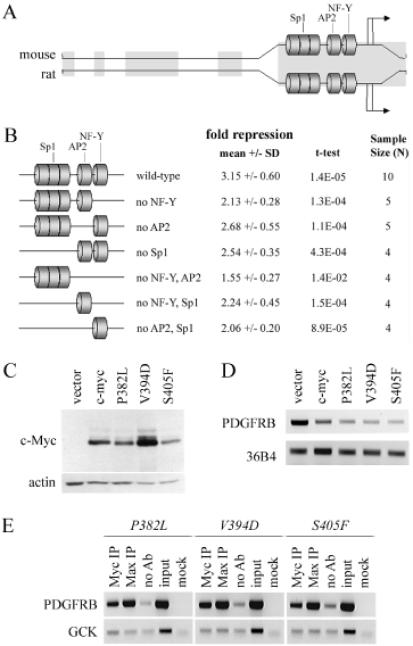
Repression does not occur solely through NF-Y, AP2, Sp1 or Miz-1. (A) The mouse (upper) and rat (lower) PDGFRB promoters are shown. Homologous regions between the two promoters are denoted by grey bars; the Sp1, AP2 and NF-Y binding sites in the proximal promoter are shown. (B) A luciferase reporter assay using a 1.6 kb fragment of the mouse PDGFRB promoter, or one of 6 binding site mutants (shown), transiently transfected into Rat-1c-MycERTAM cells. The mean fold repression +/− SD, and its associated p-value (Student's t-test), is shown. (C) Immunoblot for c-Myc (9E10) and actin expression in Rat-1c-myc−/− cells containing vector, c-myc, c-mycP382L (P382L), c-mycV394D (V394D), or c-mycS405F (S405F). (D) Expression of endogenous PDGFRB and 36B4 in the Rat-1c-myc−/− cells lines indicated. (E) ChIP using Myc or Max antibody in the Rat-1c-myc−/− cell lines indicated were conducted as described in Figure 1D and E.
To test this, we performed luciferase reporter assays using the 1.6 kb mouse PDGFRB promoter, or constructs containing NF-Y, AP2 and/or Sp1 site(s) mutations, transiently transfected into Rat-1 c-MycERTAM cells (Figure 3B). All constructs, assayed in at least four independent experiments, had statistically significant levels of repressed luciferase activity upon c-Myc activation by OHT treatment (Student's t-test, p < 0.014). This includes a 1.6 kb PDGFRB promoter construct that harboured a mutated NF-Y site (from CCAAT to CTGAT). This is in contrast to the findings of Izumi et al. (19) in which a minimal PDGFRB promoter construct, containing a mutated NF-Y site (CTGAT), was not repressed by c-Myc. It remains unclear why the results differ but may be due to the different length of PDGFRB promoter construct, luciferase vector and the experimental cell system used. Therefore, the deletion of the NF-Y, AP2 and/or Sp1 binding sites are insufficient to abrogate the c-Myc-mediated repression of the 1.6 kb Sac I mouse PDGFRB promoter.
Repression of PDGFRB by c-Myc does not require c-Myc:Miz-1 complex formation
The c-Myc-interacting zinc finger protein 1 (Miz-1) is known to be involved in the repression of several known c-Myc-repressed genes such as CDKN2B (p15ink4b) and CDKN1A (p21waf1/cip1) (16,21). To assess if Miz-1 was involved in the repression mechanism of PDGFRB, we generated three Rat-1c-myc−/− cell lines that express the c-Myc mutant proteins, c-MycP382L, c-MycV394D and c-MycS405F, that are impaired in their ability to bind Miz-1 (21). Expression of these mutant c-Myc proteins was confirmed by standard western blot analysis (Figure 3C). The relative mRNA expression of PDGFRB, assessed by RT–PCR, was lower in each of the asynchronous, subconfluently growing Rat-1c-myc−/− c-myc, Rat-1c-myc−/− c-mycP382L, Rat-1c-myc−/− c-mycV394D and Rat-1c-myc−/− c-mycS405F cell lines relative to the Rat-1c-myc−/− vector cell line (Figure 3D). These data show that each of the mutant proteins, like wild-type c-Myc, was able to repress PDGFRB. Moreover, we found that the three mutant proteins were able to bind to the PDGFRB promoter as assess by ChIP (Figure 3E). We conclude that c-Myc promoter-binding and repression of PDGFRB does not require c-Myc:Miz-1 complex formation.
TSA blocks c-Myc-induced repression but not promoter-binding by c-Myc
Histone deacetylation is one mechanism by which a promoter's activity can be downregulated. Thus, as an alternative approach, we sought to assess the effect of trichostatin A (TSA), a deacetylase inhibitor (DACi), on the c-Myc-mediated repression of PDGFRB. To test this, asynchronous, subconfluently growing Rat-1c-myc−/− cells, infected with vector or c-myc, were treated with increasing amounts of TSA (0, 300 or 600 nM) for 16 h. Cells were harvested for total RNA and then assessed for gene expression by RT–PCR. The expression of CDKN1A, a known TSA-inducible gene, is upregulated in response to TSA in this cell system (Figure 4A). We also show that TSA reduces the basal mRNA expression of PDGFRB and GADD45A through an unknown mechanism (Figure 4A). Interestingly, ectopic Myc expression repressed CDKN1A (p21waf1/cip1) and GADD45A expression in the presence of TSA (Figure 4A) (20), whereas PDGFRB was not repressed by Myc in Rat-1c-myc−/− c-myc cells treated with either 300 nM or 600 nM TSA (Figure 4A). We conclude that TSA blocks the c-Myc-mediated repression of PDGFRB expression in rat fibroblasts.
Figure 4.
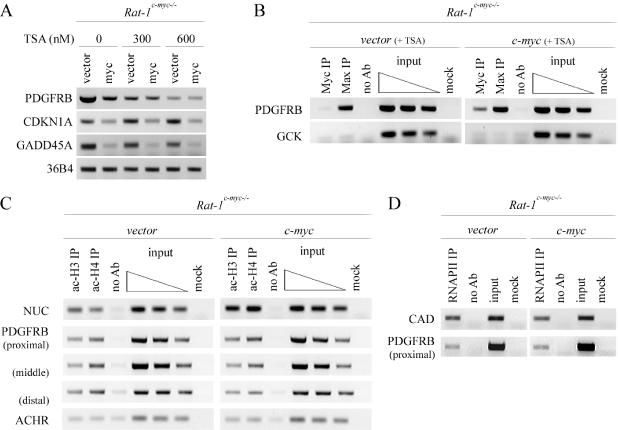
c-Myc represses a post-RNAPII recruitment step that is TSA sensitive. (A) Expression of PDGFRB, CDKN1A, GADD45 and 36B4 in Rat-1c-myc−/− vector and Rat-1c-myc−/− c-myc cells treated with 0, 300 or 600 nM of trichostatin A for 16 h, as assessed by RT–PCR. (B) ChIP using c-Myc or Max antibody in Rat-1c-myc−/− vector and Rat-1c-myc−/− c-myc cells treated with 300 nM TSA for 16 h. Binding to the PDGFRB, or control GCK, promoter is shown. (C and D) ChIP using acetylated histone H3 (ac-H3), ac-H4, or RNA polymerase II (RNAPII) antibody in Rat-1c-myc−/− vector and Rat-1c-myc−/− c-myc cells. PCR-amplified regions include NUC, ACHR, CAD as well as the PDGFRB proximal (−0.4 kb), middle (−0.9 kb), and distal promoter (−1.5 kb).
Since c-Mycd106-143 can bind to the PDGFRB promoter without repressing its activity, we hypothesized that c-Myc may be initiating a multi-step repression mechanism once bound to the promoter. By this model then, we should ideally be able to isolate and visualize c-Myc at the promoter while inhibiting its repressive function by a genetic or pharmacological approach. To test this model, we performed ChIP in Rat-1c-myc−/− vector and Rat-1c-myc−/− c-myc cells treated with 300 nM TSA for 16 h. We report that c-Myc and Max are both bound to the PDGFRB promoter in Rat-1c-myc−/− c-myc cells treated with 300 nM TSA (Figure 4B). Neither did c-Myc bind to PDGFRB in Rat-1c-myc−/− vector cells treated with 300 nM TSA, nor did c-Myc or Max bind to the negative control promoter, GCK. Thus, we conclude that TSA blocks the c-Myc-mediated activity repressing PDGFRB without inhibiting c-Myc's recruitment and promoter binding.
c-Myc inhibits a post-RNAP II recruitment step
Since TSA is a DACi which also targets histone deacetylases (HDACs), we sought to assess if c-Myc recruits a HDAC activity to the PDGFRB promoter. To test this, we performed ChIPs in Rat-1c-myc−/− vector and Rat-1c-myc−/− c-myc cells using antibodies directed against acetylated histones H3 and H4. Histone H3 and H4 are acetylated at the nucleolin (NUC) promoter in response to c-Myc, whereas the acetylcholine receptor (ACHR) promoter has been previously reported to possess a low basal level of acetylated H3 or H4 regardless of the c-myc status (Figure 4C) (14). Interestingly, we did not detect any significant difference in the acetylation state of histone H3 or H4, at three locations over the PDGFRB promoter (i.e. distal, middle or proximal), in Rat-1c-myc−/− c-myc versus Rat-1c-myc−/− vector cells (Figure 4D). This data is consistent with the observation that NaBu, another HDAC inhibitor, did not block the c-Myc repression of PDGFRB (data not shown).
This data is also consistent with the observation that there are no significant differences in the amount of RNA polymerase II (RNAPII) bound to the proximal promoter region in Rat-1c-myc−/− vector versus Rat-1c-myc−/− c-myc cells (Figure 4D). Hence, c-Myc does not repress PDGFRB gene expression by preventing RNAPII loading to the proximal promoter, a key step in transcriptional initiation. RNAPII levels do not change at the CAD promoter in response to c-Myc expression and is included as a control (Figure 4D) (30). Thus, we conclude that c-Myc does not significantly alter the abundance of acetylated histone H3, acetylated histone H4, or RNAPII at the PDGFRB promoter. Taken together, these data are consistent with a model whereby c-Myc represses PDGFRB gene expression by recruiting a TSA-sensitive activity that inhibits a post-RNAPII recruitment mechanism.
DISCUSSION
Elevated c-Myc protein levels repress PDGFRB gene expression; the repression is c-Myc dependent since PDGFRB is repressed in TGR-1 cells, but not Rat-1c-myc−/− cells, which have been serum starved and then serum stimulated (7,31). We show here that c-Myc also binds to the proximal promoter region of the PDGFRB gene in Rat-1c-myc−/− c-myc cells, but not Rat-1c-myc−/− vector cells; this data is consistent with promoter–luciferase assays showing that the minimal or proximal promoter is sufficient for c-Myc to repress PDGFRB (19). Taken together, we argue that the c-Myc protein represses the mRNA expression of the PDGFRB gene through the proximal promoter.
These data are consistent with other c-Myc-repressed genes that, in general, appear to be regulated at or near their transcriptional start sites (3,22). The mechanism(s) by which c-Myc represses transcription is an area of active research. The most accepted model in the literature is the co-activator displacement model whereby c-Myc interacts with a transcriptional activator (e.g. Miz-1, Sp1, NF-Y) at the promoter and, by virtue of the interaction, displaces a necessary co-activator (e.g. p300) thereby leading to gene repression. An interesting pervading theme is that the c-Myc:protein complexes that are important for binding to these promoters are also those needed for repression. We demonstrate several clear examples in which this model is not consistent with the observed data. For example, c-Myc, in Rat-1c-myc−/− c-myc cells treated with 300 nM TSA, is able to bind to the promoter yet is unable to repress PDGFRB expression. Also, c-MycdBR and c-Mycd106-143, which are able to bind to the proximal promoter of PDGFRB, are unable to repress PDGFRB. Thus, we clearly show that promoter binding by c-Myc, while essential, is not sufficient to affect the repression of PDGFRB. Instead, we argue that c-Myc promoter-binding and repression are separate steps in a multi-step repression mechanism that c-Myc initiates at the PDGFRB promoter.
Interestingly, although TSA, a potent DACi and HDAC inhibitor, blocked the repression of PDGFRB by c-Myc, we did not find any evidence that c-Myc effected the levels of histone H3/H4 acetylation or RNAPII loading on the promoter. These data are similar to other examples in the literature; TSA, e.g., inhibits MMTV promoter activity but does not effect the level of histone H4 acetylation (32). Taken together, since TSA does not block c-Myc from binding to the PDGFRB promoter, we propose that c-Myc utilizes additional proteins, once bound to the promoter, to repress. For example, these repression mechanisms may be the sum of larger protein complexes formed at repressed promoters of which, at least at PDGFRB, is sensitive to TSA. Supporting this notion is a growing body of literature demonstrating an essential role for Max, c-Myc's protein partner, in c-Myc-mediated transcriptional repression. For example, Max is bound to many c-Myc-repressed genes both before and after c-Myc's recruitment to these promoters (16,18,33,34); we observe the same phenomenon at the PDGFRB. Moreover, the Myc:Max interaction is essential for repression. Thus, Max may be used to recruit c-Myc to repressed, as well as activated, gene promoters (18). It will be interesting to profile the c-Myc-binding proteins that co-exist, with c-Myc, at each of its activated and repressed promoters. In doing so, the mechanistic difference(s) between activated and repressed genes, and which also likely provide c-Myc:Max complexes with the mechanistic information they require to activate or repress, may be elucidated.
Taken together, these data lead us to conclude that c-Myc represses the expression of PDGFRB in rat fibroblasts through the proximal promoter. We argue that promoter-binding and repression are separable activities of the c-Myc protein, and demonstrate three examples where c-Myc can bind to the PDGFRB promoter, but not repress its expression. We have also demonstrated that the c-Myc repression activity at PDGFRB is TSA sensitive. To fully understand the c-Myc repression mechanism, and how to precisely inhibit it, the identity of these additional protein contacts/players will need to be elucidated.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jenny Ho and the Penn Lab for critical reading of this manuscript. We also thank the Farnham Lab for technical advice. D.Y.L.M. is a recipient of a post-graduate scholarship from the Natural Sciences and Engineering Research Council (NSERC PGS-B); D.B.-L. holds a post-doctoral fellowship from the Canadian Institute of Health Research (CIHR). This work was supported by a grant from the National Cancer Institute of Canada with funds from the Canadian Cancer Society (L.Z.P.).
REFERENCES
- 1.Takehara K. (2000) Growth regulation of skin fibroblasts. J. Dermatol. Sci., 24 (Suppl 1), S70–S77. [DOI] [PubMed] [Google Scholar]
- 2.Kelly K., Cochran,B.H., Stiles,C.D. and Leder,P. (1983) Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell, 35, 603–610. [DOI] [PubMed] [Google Scholar]
- 3.Oster S.K., Ho,C.S., Soucie,E.L. and Penn,L.Z. (2002) The myc oncogene: MarvelouslY Complex. Adv. Cancer Res., 84, 81–154. [DOI] [PubMed] [Google Scholar]
- 4.Felsher D.W. and Bishop,J.M. (1999) Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell, 4, 199–207. [DOI] [PubMed] [Google Scholar]
- 5.Pelengaris S., Khan,M. and Evan,G.I. (2002) Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell, 109, 321–334. [DOI] [PubMed] [Google Scholar]
- 6.Penn L.J., Brooks,M.W., Laufer,E.M. and Land,H. (1990) Negative autoregulation of c-myc transcription. EMBO J., 9, 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oster S.K., Marhin,W.W., Asker,C., Facchini,L.M., Dion,P.A., Funa,K., Post,M., Sedivy,J.M. and Penn,L.Z. (2000) Myc is an essential negative regulator of platelet-derived growth factor beta receptor expression. Mol. Cell. Biol., 20, 6768–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C.R., Kang,Y. and Massague,J. (2001) Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor beta growth arrest program. Proc. Natl Acad. Sci. USA, 98, 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeller K.I., Jegga,A.G., Aronow,B.J., O'Donnell,K.A. and Dang,C.V. (2003) An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol., 4, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackwell T.K., Kretzner,L., Blackwood,E.M., Eisenman,R.N. and Weintraub,H. (1990) Sequence-specific DNA binding by the c-Myc protein. Science, 250, 1149–1151. [DOI] [PubMed] [Google Scholar]
- 11.Blackwood E.M. and Eisenman,R.N. (1991) Max: a helix–loop–helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science, 251, 1211–1217. [DOI] [PubMed] [Google Scholar]
- 12.Bouchard C., Dittrich,O., Kiermaier,A., Dohmann,K., Menkel,A., Eilers,M. and Luscher,B. (2001) Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev., 15, 2042–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon S.B., Wood,M.A. and Cole,M.D. (2000) The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol. Cell. Biol., 20, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank S.R., Schroeder,M., Fernandez,P., Taubert,S. and Amati,B. (2001) Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev., 15, 2069–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eberhardy S.R. and Farnham,P.J. (2002) Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J. Biol. Chem., 277, 40156–40162. [DOI] [PubMed] [Google Scholar]
- 16.Staller P., Peukert,K., Kiermaier,A., Seoane,J., Lukas,J., Karsunky,H., Moroy,T., Bartek,J., Massague,J., Hanel,F. and Eilers,M. (2001) Repression of p15INK4b expression by Myc through association with Miz-1. Nature Cell Biol., 3, 392–399. [DOI] [PubMed] [Google Scholar]
- 17.Seoane J., Le,H.V. and Massague,J. (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature, 419, 729–734. [DOI] [PubMed] [Google Scholar]
- 18.Mao D.Y.L., Watson,J.D., Yan,P.S., Barsyte-Lovejoy,D., Khosravi,F., Wong,W.W., Farnham,P.J., Huang,T.H. and Penn,L.Z. (2003) Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol., 13, 882–886. [DOI] [PubMed] [Google Scholar]
- 19.Izumi H., Molander,C., Penn,L.Z., Ishisaki,A., Kohno,K. and Funa,K. (2001) Mechanism for the transcriptional repression by c-Myc on PDGF beta-receptor. J. Cell Sci., 114, 1533–1544. [DOI] [PubMed] [Google Scholar]
- 20.Gartel A.L., Ye,X., Goufman,E., Shianov,P., Hay,N., Najmabadi,F. and Tyner,A.L. (2001) Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl Acad. Sci. USA, 98, 4510–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herold S., Wanzel,M., Beuger,V., Frohme,C., Beul,D., Hillukkala,T., Syvaoja,J., Saluz,H.P., Haenel,F. and Eilers,M. (2002) Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol. Cell, 10, 509–521. [DOI] [PubMed] [Google Scholar]
- 22.Wanzel M., Herold,S. and Eilers,M. (2003) Transcriptional repression by Myc. Trends Cell Biol., 13, 146–150. [DOI] [PubMed] [Google Scholar]
- 23.Mateyak M.K., Obaya,A.J., Adachi,S. and Sedivy,J.M. (1997) Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ., 8, 1039–1048. [PubMed] [Google Scholar]
- 24.Oster S.K., Mao,D.Y.L., Kennedy,J. and Penn,L.Z. (2003) Functional analysis of the N-terminal domain of the Myc oncoprotein. Oncogene, 22, 1998–2010. [DOI] [PubMed] [Google Scholar]
- 25.Ballagi A.E., Ishizaki,A., Nehlin,J.O. and Funa,K. (1995) Isolation and characterization of the mouse PDGF beta-receptor promoter. Biochem. Biophys. Res. Commun., 210, 165–173. [DOI] [PubMed] [Google Scholar]
- 26.Ferre-D'Amare A.R., Prendergast,G.C., Ziff,E.B. and Burley,S.K. (1993) Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature, 363, 38–45. [DOI] [PubMed] [Google Scholar]
- 27.Stone J., de Lange,T., Ramsay,G., Jakobovits,E., Bishop,J.M., Varmus,H. and Lee,W. (1987) Definition of regions in human c-myc that are involved in transformation and nuclear localization. Mol. Cell. Biol., 7, 1697–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaubatz S., Imhof,A., Dosch,R., Werner,O., Mitchell,P., Buettner,R. and Eilers,M. (1995) Transcriptional activation by Myc is under negative control by the transcription factor AP-2. EMBO J., 14, 1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taira T., Sawai,M., Ikeda,M., Tamai,K., Iguchi-Ariga,S.M. and Ariga,H. (1999) Cell cycle-dependent switch of up-and down-regulation of human hsp70 gene expression by interaction between c-Myc and CBF/NF-Y. J. Biol. Chem., 274, 24270–24279. [DOI] [PubMed] [Google Scholar]
- 30.Eberhardy S.R. and Farnham,P.J. (2001) c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem., 276, 48562–48571. [DOI] [PubMed] [Google Scholar]
- 31.Watson J.D., Oster,S.K., Shago,M., Khosravi,F. and Penn,L.Z. (2002) Identifying genes regulated in a Myc-dependent manner. J. Biol. Chem., 277, 36921–36930. [DOI] [PubMed] [Google Scholar]
- 32.Wilson M.A., Ricci,A.R., Deroo,B.J. and Archer,T.K. (2002) The histone deacetylase inhibitor trichostatin A blocks progesterone receptor-mediated transactivation of the mouse mammary tumor virus promoter in vivo. J. Biol. Chem., 277, 15171–15181. [DOI] [PubMed] [Google Scholar]
- 33.Wu S., Cetinkaya,C., Munoz-Alonso,M.J., von der Lehr,N., Bahram,F., Beuger,V., Eilers,M., Leon,J. and Larsson,L.G. (2003) Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene, 22, 351–360. [DOI] [PubMed] [Google Scholar]
- 34.Barsyte-Lovejoy D., Mao,D.Y.L. and Penn,L.Z. (2004) c-Myc represses the proximal promoters of GADD45a and GADD153 by a post-RNA polymerase II recruitment mechanism. Oncogene, 23, 3481–3486. [DOI] [PubMed] [Google Scholar]