

Abstract
Rationale: Despite relative antigenic stability, respiratory syncytial virus (RSV) reinfects throughout life. After more than 40 years of research, no effective human vaccine exists and correlates of protection remain poorly defined. Most current vaccine candidates seek to induce high levels of RSV-specific serum neutralizing antibodies, which are associated with reduced RSV-related hospitalization rates in observational studies but may not actually prevent infection.
Objectives: To characterize correlates of protection from infection and the generation of RSV-specific humoral memory to promote effective vaccine development.
Methods: We inoculated 61 healthy adults with live RSV and studied protection from infection by serum and mucosal antibody. We analyzed RSV-specific peripheral blood plasmablast and memory B-cell frequencies and antibody longevity.
Measurements and Main Results: Despite moderately high levels of preexisting serum antibody, 34 (56%) became infected, of whom 23 (68%) developed symptomatic colds. Prior RSV-specific nasal IgA correlated significantly more strongly with protection from polymerase chain reaction–confirmed infection than serum neutralizing antibody. Increases in virus-specific antibody titers were variable and transient in infected subjects but correlated with plasmablasts that peaked around Day 10. During convalescence, only IgG (and no IgA) RSV-specific memory B cells were detectable in peripheral blood. This contrasted with natural influenza infection, in which virus-specific IgA memory B cells were readily recovered.
Conclusions: This observed specific defect in IgA memory may partly explain the ability of RSV to cause recurrent symptomatic infections. If so, vaccines able to induce durable RSV-specific IgA responses may be more protective than those generating systemic antibody alone.
Keywords: antibody-secreting cells, mucosal immunity, immunologic memory, respiratory syncytial virus vaccines, nontherapeutic human experimentation
At a Glance Commentary
Scientific Knowledge on the Subject
A major challenge in respiratory syncytial virus (RSV) research is to understand why infection recurs throughout life despite a high degree of viral genetic stability. Passive antibody therapy protects against hospitalization of high-risk infants, but new and better correlates of protection are needed that more accurately predict candidate vaccine efficacy.
What This Study Adds to the Field
We find that virus-specific nasal IgA correlates with protection against experimental RSV challenge in adult volunteers, and replaces serum neutralizing antibody as the best predictor of protection. However, virus-specific IgA memory B-cell responses are remarkably absent after RSV infection, possibly explaining susceptibility to reinfection. Ideally, RSV vaccines should induce durable mucosal antiviral IgA and memory B cells, thereby overcoming the defective responses seen after infection with live virus.
Respiratory syncytial virus (RSV) is the leading cause of infant hospitalization worldwide, infecting around 34 million children each year (1). It is also a major underappreciated contributor to morbidity and mortality in elderly and immunosuppressed adults (2). Although RSV infection is essentially confined to the respiratory epithelium, extensive lung inflammation frequently causes life-threatening disease in these at-risk populations (3, 4). It remains one of the few common severe infections for which there is no specific intervention and an effective vaccine represents a major unmet need.
Natural infections with most viruses including influenza and rhinoviruses induce long-lasting protective immunity, with recurrent infections then occurring because of antigenic variation. In contrast, reinfection with identical strains of RSV can occur at as little as 2-month intervals even with a healthy mature immune system (5). Although reinfection with human metapneumovirus and parainfluenza viruses has been observed, only RSV has been categorically shown to induce recurrent symptomatic disease in the absence of antigenic change (6, 7). The mechanisms underlying this phenomenon remain unclear and the lack of durable immunity after natural infection has made it difficult to define precise correlates of protection. With more than 51 RSV vaccine candidates currently in development (8), the ability to predict efficacy via immune correlates is urgently required. Although the humanized monoclonal IgG palivizumab can reduce infant hospitalizations with RSV by at least 50% (9), the association between serum antibody levels and protection is imprecise (10, 11). Therefore, the value of serum antibody induction as the primary readout of potential vaccine candidates and in vaccine licensure may be limited.
Although animal models of RSV infection provide mechanistic insights, they do not recapitulate all aspects of human disease and remain imprecise guides to the clinical efficacy of vaccines (12). Investigation of hospitalized children and high-risk adults allows natural disease to be observed, but cannot correlate prior immune status with susceptibility to infection. In addition, lack of uniformity in virus inoculum, comorbidities, medical interventions, and atypical disease presentations limit the conclusions that can be reached from these observational studies. Experimental infection of volunteers therefore offers an important complementary approach, allowing intensive sampling and the opportunity to perform detailed investigations of preexisting and presymptomatic responses (13, 14).
To investigate the mechanisms underlying recurrent RSV infection, we analyzed antibody-mediated protection and B-cell responses before and after experimental human challenge. We found that serum antibody was loosely correlated with protection, but that there was a tight relationship between preexisting RSV-specific nasal antibody and resistance to infection. These data allowed us to derive a model that accurately predicts infection risk. In addition, we demonstrate a novel RSV-specific defect in IgA memory that may explain the susceptibility to reinfection with RSV and discuss its implications for RSV vaccine development. Some of the results of this study have been previously reported in the form of abstracts (15–19).
Methods
Study Design
Healthy nonsmoking adults aged 18–55 were eligible for inclusion. All were screened to exclude underlying immunodeficiencies including IgA deficiency by serum immunoglobulin class levels. Subjects were inoculated with 104 plaque-forming units of RSV A Memphis 37 (RSV M37) by intranasal drops (as defined in previous dose-ranging studies) and quarantined from 1 day before inoculation to the 10th day after (13). Nasal washes and blood were taken immediately before inoculation and regularly for the next 10 days (see Figure E1A in the online supplement). Infection was defined as RSV detectable by polymerase chain reaction (PCR) in nasal lavage on greater than or equal to 2 days between Day +2 and Day +10 to avoid false-positives from detection of the viral inoculum and to align case definitions with previous challenge studies using RSV M37 (13). Subjects completed a diary of upper respiratory tract symptoms (see online supplement) from Day −1 to Day +14. All subjects returned for further nasal and blood sampling on Day +14, Day +28, and optionally 6–12 months after inoculation (nominally Day +180). All subjects provided written informed consent and the study was approved by the UK National Research Ethics Service (study numbers 10/H0711/94 and 11/LO/1826).
Antibody Assays
Serum neutralizing antibody titer was determined by plaque reduction neutralization titer (PRNT) in HEp-2 cells and expressed as midpoint titers (EC50). Sera from four hospitalized RSV PCR-negative infants hospitalized were included as negative control subjects and three RSV immune reference sera (Wyeth 06937, 06938, 06939; BEI Resources, Manassas, VA) as positive control subjects (20). Nasal wash IgA end point binding titer to RSV lysate and F protein was determined by ELISA as the highest titer exhibiting an optical density of greater than two times the background. End point titers were used because midpoint titers could not be calculated in view of the dilute nature of nasal lavage. Observed end point titers were corrected for dilution using the ratio of serum to nasal lavage urea before analysis as described (21).
Detection of Antibody-Secreting Cells by B-Cell Enzyme-linked Immunospot
Antibody-secreting cells (ASCs) were detected using enzyme-linked immunospot (ELISpot) as previously described (22), using whole RSV M37 lysate from HEp-2 cells and recombinant F protein based on the RSV A2 strain (see online supplement for detailed methods). For memory B cells (MBCs), additional plates were coated with 10 μg/ml measles antigen (Meridian Lifesciences, Memphis, TN), and 5 μg/ml HEp-2 antigen, 2.5 μg/ml keyhole limpet hemocyanin (Sigma, Dorset, UK), and media as negative controls. Total ASCs were expressed as spot-forming cells/106 peripheral blood mononuclear cells (PBMCs) and antigen-specific ASCs as percent of total immunoglobulin-secreting cells.
Polyclonal Activation of MBCs
PBMCs were cultured in vitro according to the method of Crotty and coworkers (23). The alternative polyclonal activation mix described by Tengvall and coworkers (24) was used in a subset of samples. ASCs were detected by B-cell ELISpot as above. Subjects exhibiting total immunoglobulin-positive cell responses below the 10th centile in either the Day 0 or Day 28 sample for either IgG or IgA were excluded (n = 9) as inadequate responders to stimulation.
Statistical Analysis
All data analyses and graphs were produced using the software R (25, 26). Results are expressed as median and interquartile range (IQR). Nonparametric data were compared using Mann-Whitney Wilcoxon tests with Holm correction for multiple comparisons. Binary response variables were related to continuous explanatory variables using logistic regression. Odds ratios (OR) and 95% confidence intervals of the OR for the explanatory variables were calculated (see online supplement). For estimation of serum neutralizing antibody titers, weighted (1/y) four-parameter logistic models were fitted to the plaque counts and the 50% neutralizing titer (EC50) was derived from the midpoint of the curve using package “drc” (27).
Results
Nasal Antibody Correlates Strongly with Protection from Infection by RSV
Sixty-one healthy nonsmoking adult volunteers were enrolled (age, 18–50; median, 22 yr) (Table 1) without preselection according to anti-RSV antibody levels. All were inoculated with RSV M37 via nasal drops and admitted to a residential quarantine facility for 10 days. Thirty-four (56%) subjects became infected as defined by PCR positivity on greater than or equal to 2 consecutive days between Days 2 and 10 postinoculation (see Figure E1B). Five individuals deemed “uninfected” had a low level of detectable viral shedding on Day 1 postinoculation with no virus detected at any point thereafter, which likely indicated the remains of the virus inoculum and not evidence of productive infection. Two “uninfected” subjects had RSV detected with a high cycle threshold (Ct) value on a single day during Days 2–10. Neither of these had any subsequent change in their RSV-specific immune responses. Median (IQR) cumulative symptom scores were 26.5 (9.6–41.9) for PCR-positive “infected” and 9.0 (0.8–20.3) for PCR-negative “uninfected” subjects (P = 0.004). Daily nasal lavage viral load measurements were made during the quarantine period and cumulative viral load for PCR-positive subjects was 4.0 (3.1–5.2) log10 copies per milliliter. No difference in the number infected was observed for differing age, sex, or ethnicity.
Table 1.
Demographic, Clinical, and Virologic Data from 61 Individuals Experimentally Inoculated with RSV A Memphis 37
| Uninfected (n = 27) | Infected (n = 34) | P Value | |
|---|---|---|---|
| Median (range) age, yr | 23 (18–39) | 21 (18–50) | ns |
| Sex, M:F | 17:10 | 18:16 | ns |
| Ethnicity | |||
| White | 21 | 28 | ns |
| Black | 4 | 1 | ns |
| Asian | 0 | 2 | ns |
| Mixed | 2 | 1 | ns |
| Other | 0 | 2 | ns |
| Median cumulative symptom score (IQR)* | 9 (0.8–20.3) | 26.5 (9.6–41.9) | 0.004 |
| Median cumulative viral load (IQR), log10 copies/ml* | 0 | 4.0 (3.1–5.2) | NA |
Definition of abbreviations: IQR = interquartile range; NA = not applicable; ns = not significant; RSV = respiratory syncytial virus.
P values for unpaired Mann-Whitney Wilcoxon U test or chi-squared test for comparison of uninfected versus infected are shown.
Trapezoidal area under the curve.
Preexisting PRNTs did not differ significantly between PCR-negative (median, 7.8 log2) and PCR-positive (median, 7.6 log2) subjects (Figure 1A). Similarly, there was no significant difference in anti-RSV or anti–F protein IgG (RSV-IgG, 7.8 vs. 7.6 log2; F-IgG, 7.9 vs. 7.2 log2) or IgA (RSV-IgA, 9.0 vs. 8.0 log2; F-IgA, 8.0 vs. 7.5 log2) in serum between PCR-negative and -positive individuals (see Figures E2A and E2B). By contrast, nasal anti-RSV and anti–F protein IgA titers in individuals who remained uninfected were significantly higher (RSV-IgA, 10.1 vs. 9.4 log2; F-IgA, 10.2 vs. 9.6 log2) (Figure 1A). However, serum IgA correlated poorly with nasal IgA (the Spearman correlation coefficient of 0.25 implying only a weak relationship between responses against RSV lysate in these compartments with a borderline P value of 0.05), suggesting that it is IgA in the airway that specifically plays a role in protection against RSV infection and that serum IgA may not derive from the same source (see Figure E2C).
Figure 1.

Respiratory syncytial virus (RSV)–specific nasal IgA is a superior correlate of protection to serum neutralization titer. (A) Serum neutralizing antibody (Ab) was determined by plaque reduction neutralization and nasal IgA by ELISA. Baseline serum neutralizing (top), nasal IgA against whole RSV (middle), and nasal anti-RSV fusion F protein (bottom) Ab levels in uninfected (open circles) versus infected (solid circles) subjects are shown. Serum neutralization includes four infant samples (open triangles) and three reference serum standards (solid squares; Wyeth 06937, 06938, and 06939). Horizontal bars indicate the median. (B) Logistic regression of probability of protection with baseline serum neutralizing (top), nasal whole RSV IgA (middle), and nasal RSV fusion F protein IgA (bottom) Ab levels with predicted probability (solid line) and point-wise 95% confidence band (shaded) are shown. EC50 = half-maximal effective concentration; OR = odds ratio (and 95% confidence interval). P values for unpaired Mann-Whitney Wilcoxon U test or two-tailed z test as appropriate are shown. ns = P > 0.05; *P < 0.05; **P < 0.01.
Serum antibody (PRNT) did not affect the likelihood of infection (OR, 1.6; 95% confidence interval, 0.9–2.9; P = 0.1), but virus- and F protein–specific nasal IgA were again significantly associated with protection (RSV-IgA: OR, 1.9 (1.2–3.4); P = 0.014) (F-IgA: OR, 1.7 (1.1–2.9); P = 0.016) (Figure 1B). Thus, serum antibody correlates poorly with protection, whereas a 1 log2 increase in RSV-specific nasal IgA titers renders subjects nearly twice as likely to be protected.
Increased RSV-Specific Antibody Levels after Infection Are Poorly Maintained
Following RSV challenge, RSV PCR-positive subjects showed a median 3.4-fold increase in PRNT (IQR, 2.3–4.6). Similar increments were seen in nasal anti-RSV IgA (2.6-fold; IQR, 2.0–5.4) and nasal anti–F protein IgA (3.0, 1.4–4.5). By contrast, in the group that was inoculated but remained uninfected, no significant rise in the median neutralization titer (1.4, 1.0–2.0), nasal anti-RSV IgA (0.9, 0.6–2.5), or nasal anti–F protein IgA (0.8, 0.6–1.6) (Figure 2A) was seen by Mann-Whitney test. Although two uninfected individuals displayed a fourfold rise in PRNT and eight showed approximately twofold increases in nasal anti-RSV IgA, none were PCR-positive on any day. Repetition of these assays showed no run-to-run variability, suggesting that these changes represented either normal biologic variability in serum antibody levels or responses to RSV antigen in the inoculum in the absence of infection.
Figure 2.

Serum and nasal antibody (Ab) increase after infection but are not maintained. Healthy adult volunteers were challenged intranasally with respiratory syncytial virus (RSV) A M37. Serum and nasal lavage was taken at baseline and 28 days postinoculation. A subset of subjects returned for further blood and nasal sampling 6–12 months later (nominally Day +180). Serum neutralizing Ab was determined by plaque reduction neutralization and nasal IgA by ELISA. (A) Fold change versus baseline of serum in serum plaque reduction neutralization titer (top), nasal IgA against whole RSV (middle), and nasal anti-RSV fusion F protein (bottom) Ab levels in uninfected (open circle) versus infected (solid circle) subjects. Horizontal bars indicate the median. (B) Individual plots showing trend in Ab levels at baseline and Days +28 and +180. P values for unpaired Mann-Whitney Wilcoxon U test are shown. EC50 = half-maximal effective concentration. ***P < 0.001.
Although prior nasal IgA levels correlated closely with protection, only 21% of individuals judged as infected reached postinfection nasal anti-RSV IgA titers predicted to give greater than or equal to 80% protection (see Table E1). Even fewer subjects demonstrated an equivalently protective rise in IgA specific to purified RSV F protein (9%), suggesting that a proportion of antibody boosted by natural infection might be against strain-specific variable sites on surface glycoproteins. Therefore, even the boosted levels of antibody seen shortly after natural infection are unlikely to provide complete immunity in most individuals.
Previous studies have suggested that antibodies induced by RSV infection are poorly maintained in both adults (28) and children (29). We therefore measured serum and nasal anti-RSV antibodies in infected subjects approximately 6–12 months postinoculation. Even in infected subjects showing the strongest antibody responses in serum and/or nasal secretions at 1 month, antibody levels were not maintained at later follow-up. Instead, levels returned in many to almost exactly the preinfection level for that individual (Figure 2B).
Induction of Higher Plasmablast Frequencies Correlates with Increased Antibody Levels
To better understand why RSV-specific antibodies neither reach consistently protective titers nor persist beyond 6 months, we studied B-cell responses in peripheral blood by flow cytometry. RSV-induced plasmablasts (CD3−/CD19+/CD38hi/CD27hi) peaked around 10 days postinoculation (see Figure E3A). Similarly, using B-cell ELISpots, antigen-specific ASCs against RSV were shown to arise after Day +7 and peak around Day +10 (Figure 3A). Peak virus-specific ASC frequencies ranged from 5% to 50% of total ASCs, comparable with that after influenza or yellow fever vaccination (Figure 3B) (30–33). Thus, the virus-specific acute ASC responses to RSV infection appeared quantitatively intact.
Figure 3.

Respiratory syncytial virus (RSV)–induced plasmablast expansion correlates with IgG and IgA increment. (A) Antigen-specific antibody (Ab)-secreting cells (ASCs) were enumerated using B-cell enzyme-linked immunospot with whole RSV lysate where sufficient peripheral blood mononuclear cells (PBMCs) were available. Uninfected (polymerase chain reaction–negative) subjects (n = 12) are shown in blue, infected (polymerase chain reaction–positive) subjects (n = 16) in red. (B) Peak antigen-specific ASC frequencies as percentage of total immunoglobulin-positive cells for infected subjects. Horizontal bars indicate the median. (C) Correlation of fold change in serum neutralizing Ab with peak IgG+ ASC frequency. (D) Correlation of fold change in nasal IgA against RSV with peak IgA+ ASC frequency (data from 27 subjects; nasal IgA in one subject could not be measured). (E) Correlation of peak ASC frequency with preinfection baseline serum neutralizing Ab titers. ASC frequencies are expressed as log10 per million PBMC. P values for unpaired Mann-Whitney Wilcoxon U tests and/or Spearman rank correlation coefficient (Rs) are shown as appropriate. Open circles = uninfected subjects; solid circles = infected subjects. Ab = antibody; EC50 = half-maximal effective concentration. ns = P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
By Day +14, RSV-specific ASCs had declined and by Day +28 were completely undetectable. IgM-producing ASCs were largely absent throughout (as expected of a recall response). However, IgG- and IgA-producing RSV-specific ASCs both appeared at similar frequencies, consistent with generation at a mucosal site of infection (Figure 3B). Furthermore, the frequency of RSV- and F-specific IgG+ ASCs correlated strongly with fold change in serum neutralizing antibody (Figure 3C; see Figure E3B) and virus-specific IgA+ ASCs correlated strongly with the increment in nasal IgA (Figure 3D; see Figure E3B). However, preexisting serum neutralizing antibody levels negatively correlated with peak ASC frequencies, suggesting that even though antibodies in this compartment did not provide sterilizing immunity, they might still have blunted responses against homologous antigens in the challenge agent (Figure 3E; see Figure E3C). Furthermore, cumulative viral load and symptom scores (by trapezoidal area under the curve) were not associated with peak ASC frequencies, indicating that once infection was established the frequency of antigen-specific B cells had no effect on virus control or disease severity (see Figures E4A and E4B). Therefore, the peak frequency of ASCs depended on preexisting antibodies inasmuch as antibody can prevent the establishment of infection. Once infection occurred, ASCs proliferated rapidly; this correlated with the transient production of anti-RSV IgG and IgA, but otherwise had little impact on disease course or outcome.
IgG+ Memory B-Cell Frequencies Prior to Infection Do Not Influence the Outcome of Viral Challenge
Circulating MBCs rapidly differentiate into ASCs and plasma cells on reencounter with cognate antigen and are essential for rapid humoral response to recurrent infection. To assess whether preinfection RSV-specific MBCs correlate with protection we analyzed RSV-specific MBC frequencies and compared with those against measles virus, a related pathogen to which protection following natural infection or live-attenuated vaccination is known to be long-lasting (34).
Identifying ASCs by B-cell ELISpot (23), there were no significant differences in IgG+ MBC frequencies against whole RSV, F protein, or measles virus before RSV challenge or between preinfection MBC frequencies in subsequently infected or uninfected subjects (Figure 4A). Preinfection MBC frequencies neither correlated with later symptom score nor viral load in infected subjects (see Figures E5A and E5B), nor with PRNT or nasal IgA antibody levels (see Figures E5C–E5E). This confirmed that preexisting RSV-specific MBCs per se neither influence infection rates nor contribute to control of disease or viral replication once infection is established.
Figure 4.
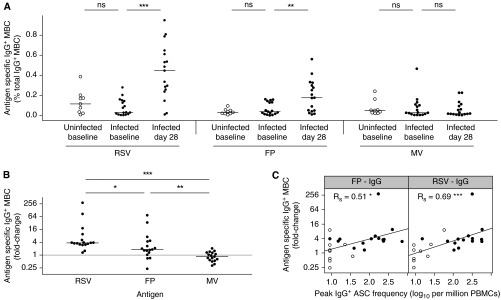
Antigen-specific IgG+ memory B cells (MBC) are induced by respiratory syncytial virus (RSV) infection. Peripheral blood mononuclear cells (PBMCs) were obtained on Days 0 and 28 postinoculation and MBCs polyclonally activated. Total and antigen-specific immunoglobulin-producing cells were then enumerated using enzyme-linked immunospot. (A) Antigen-specific IgG+ MBC frequencies for uninfected (open circles) versus infected (solid circles) subjects at baseline and Day 28 postinoculation (infected only). P > 0.05 for all comparisons except where shown. MBC frequencies are expressed as percentage of total immunoglobulin-positive cells. Horizontal bars indicate the median. (B) Fold change (log2) of IgG+ MBCs postinfection compared with baseline for infected subjects (n = 17). Horizontal bars indicate the median. (C) Correlation between peak acute antibody-secreting cell (ASC) frequency at Day 10 and fold change over baseline (log2) of RSV-specific IgG+ MBC. Spearman rank correlation coefficient (Rs) or P values for unpaired Mann-Whitney Wilcoxon U test are shown as appropriate. FP = purified RSV fusion protein; MV = measles virus; RSV = whole RSV. ns = P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Following infection, subjects demonstrated a median (IQR) increase in IgG+ MBC frequencies of 0.4% (0.2−0.5) against whole RSV and 0.1% (0−0.2) against F protein (Figure 4A). These equated to a median 3.8-fold (3.1−5.1) increase in RSV-specific MBCs and 1.8-fold (1.3−4.4) increase in F-specific MBCs compared with preinfection frequencies (Figure 4B). By contrast, measles-specific IgG+ MBC frequencies remained unchanged (Figures 4A and 4B). The fold change in both virus- and F-protein–specific IgG+ MBCs correlated with the size of ASC burst specific for the homologous antigens, suggesting that these populations were generated by associated mechanisms (Figure 4C). IgG+ MBCs therefore incremented appropriately following RSV infection but did not affect the course of acute disease.
RSV Infection Fails to Induce Virus-Specific IgA MBCs
To investigate the mechanisms underlying the decline in virus-specific nasal IgA, we studied RSV-specific IgA+ MBCs. In contrast to IgG+ MBCs, RSV- and F-specific IgA-producing MBCs (and measles-specific IgA+ MBCs) were undetectable preinfection (Figure 5A). Strikingly, anti-RSV IgA+ MBCs then demonstrated no detectable increase in frequency even shortly after RSV infection; these remained undetectable at convalescence in the same way as measles (Figure 5B).
Figure 5.
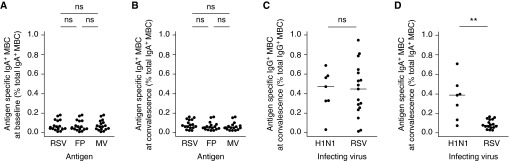
Respiratory syncytial virus (RSV) infection fails to induce IgA+ memory B cells (MBCs). Peripheral blood mononuclear cells were obtained on Days 0 and 28 postinoculation and MBCs activated polyclonally in vitro. Total and antigen-specific immunoglobulin-producing cells were then enumerated using B-cell enzyme-linked immunospot as before. Data for infected subjects exhibiting adequate peripheral blood mononuclear cell responses to in vitro stimulation only (n = 17) are shown. (A) Frequency of IgA+ MBCs at baseline (preinfection) and (B) convalescence (Day +28). Subjects experimentally challenged with RSV A M37 were compared with patients naturally infected with pandemic influenza A/H1N1/09 (seven exhibited adequate response to stimulation and were analyzed). (C) IgG+ MBC frequencies and (D) IgA+ MBC frequencies at Day 28 postinoculation (RSV) and Day 28–42 posthospitalization (H1N1) are shown. Horizontal bars indicate the median. P values for unpaired Mann-Whitney Wilcoxon U test are shown as appropriate. FP = purified RSV fusion protein; MV = measles virus; RSV = whole RSV. ns = P > 0.05; **P < 0.01.
IgA+ and IgG+ MBCs differ in their requirements for optimal in vitro activation (24). To ensure that the apparent absence of RSV-specific IgA+ MBCs was not simply caused by inadequate activation, we repeated the in vitro activation of PBMC from six selected subjects who had previously demonstrated a robust RSV-specific IgG+ MBC response with an alternative protocol using CpG, B-cell activation factor (BAFF), and IL-15 to enhance IgA+ MBC stimulation (24). However, this did not improve the polyclonal stimulation of IgA+ MBCs, which were induced at similar frequencies to the original method, whereas total IgG+ MBCs were less well induced (see Figures E6A and E6B). Again, RSV-specific IgA+ MBCs were undetectable both at baseline and convalescence using either stimulation method (see Figures E6C and E6D).
To determine whether poor IgA+ MBC generation was RSV-specific, we compared these responses with those after natural influenza A(H1N1)pdm09 infection. Convalescent PBMCs were obtained as part of the Mechanisms of Severe Acute Influenza Consortium study from eight patients hospitalized with virologically confirmed pdmH1N1 infection during the 2009–2010 and 2010–2011 influenza seasons. All were previously healthy with no known immunosuppressive disease. Antigen-specific MBCs were polyclonally stimulated under identical conditions and identified by B-cell ELISpot. As expected, the virus-specific IgG+ MBCs induced following both pdmH1N1 and RSV were found at similar frequencies (Figure 5C). However, PBMCs from patients naturally infected with influenza showed no defect in IgA+ MBC response, with significantly higher numbers of virus-specific IgA+ MBCs than in those infected with RSV (Figure 5D). In influenza, virus-specific IgA+ and IgG+ MBC numbers were therefore comparable and IgA+ MBCs were generally abundant in those who had recovered from infection, contrasting with the paucity of IgA+ MBCs after RSV infection.
Discussion
In this study, we show that mucosal IgA titers are predictive of susceptibility to RSV infection (in contrast to serum antibody levels, which are not), and identify a previously unrecognized defect in IgA MBC responses to RSV. The few previous studies of mucosal IgA in RSV disease have been restricted in their applicability by study design and other technical limitations (35–37). Our investigations now show the power of the experimental human infection model, which for the first time has allowed development of a tightly predictive model of RSV infection risk in seropositive individuals.
A previous study examining the protective role of RSV-specific antibodies during experimental human RSV challenge (38) showed that serum neutralization and IgG titers against F protein were both significantly higher in those remaining uninfected, whereas no difference was seen in nasal IgA titers. The reasons for the discrepancy between those results and our present findings may be methodologic: in the earlier study, a different method of nasal lavage was used, resulting in a more dilute sample and average twofold to threefold lower nasal IgA titer. The normalization according to total protein content may also have led to underestimation of true IgA level. In addition, the laboratory-adapted RSV A2 strain used in the previous study induced little symptomatic disease in infected subjects and may have had different host-virus dynamics compared with RSV M37, a more recent, nonattenuated, clinically derived strain. Finally, the previous study included fewer subjects (a total of 28 inoculated individuals), some of whom were prestratified on the basis of serum antibody levels, which may have affected the analyses.
Interestingly, consistently protective levels of anti-RSV IgA were only seen in a small minority of individuals, even during early convalescence. Furthermore, although antibodies were temporarily boosted by infection, titers waned to preinfection steady-state levels within 6–12 months. This contrasts sharply with known responses to viruses, such as influenza or rhinovirus, in which strain-specific serum IgG can persist life-long and nasal IgA remain significantly raised for a year or more (39, 40). Antibody responses to RSV infection are therefore remarkably transient, accounting for the frequency of adult reinfection. This study now sets a new goal for vaccine-induced immunity: mucosal IgA responses in the respiratory mucosa beyond those induced naturally. Indeed, if mucosal IgA could be induced in the major at-risk population of young children, there would be advantages not only in reducing the incidence of RSV infection but also by limiting the extent of virus transmission, although extrapolation of these data to children should be undertaken with caution.
Importantly, we found that short-lived RSV-specific IgG+ and IgA+ B cells were generated robustly at comparable frequencies with those observed following other challenges including influenza infection or vaccination (30, 32). With acute production of short-lived ASCs apparently intact, a vaccine capable of inducing higher frequencies of these cells should also induce greater antibody responses and therefore enhanced protection. Previous studies in natural RSV infection of adults indicated that this might be further modulated by duration of antigenic stimulation, with prolonged viral shedding driving ASC responses (41). However, anti-RSV antibody titers were poorly maintained, implying a defect in long-lived plasma cells (LLPCs) that arise from the ASC population and which normally migrate to survival niches including bone marrow and respiratory mucosa (42, 43). Future studies to address this contention will require direct sampling of these compartments, which may be difficult.
In animal models, IgA-producing B cells are abundant at mucosal sites (44) and appear rapidly in mucosal tissues under the influence of mediators, such as BAFF and APRIL (a proliferation-inducing ligand) (45–47). However, to generate high affinity antibodies and long-term memory responses, antigen-presenting cells, B cells, and T follicular helper cells must interact in germinal centers (48, 49). T-cell help allows not only class-switching to IgA but also the promotion of ASC and MBC formation (50, 51). MBCs require many of the same differentiation signals as LLPCs and we therefore hypothesized that MBC responses would also be suboptimal. Our analysis of IgG+ MBCs preinfection and postinfection found no evident defect, but remarkably, we were unable to detect any RSV-specific IgA+ MBCs before infection and no increment in IgA+ MBC frequency afterward. Because the acute ASC response was dominated by RSV-specific IgA+ ASCs, which are likely to have arisen from preexisting IgA+ MBCs, the finding that RSV-specific IgA+ MBCs were so infrequent both preinfection and postinfection was highly unexpected. Despite this, moderate levels of serum and nasal IgA against RSV could be detected in all volunteers. Thus, although incremental boosting of IgA+ MBCs might be poor, over multiple reinfections LLPCs in bone marrow or respiratory tract are likely to have developed that are capable of producing RSV-specific IgA long-term but whose population size or antibody-producing capabilities are minimally augmented following rechallenge.
RSV is known to express several immunomodulatory proteins and we therefore suggest that this defect in IgA memory represents a viral immune evasion strategy. However, some alternative explanations should be considered. It is possible that IgA+ MBCs following respiratory infections in general are so infrequent that these assays cannot detect them. Alternatively, IgA+ MBCs might be preferentially retained in mucosal sites and therefore not detectable in the circulation. However, if either of these were the case, we would also not have expected to find influenza-specific IgA+ MBCs in convalescent PBMCs. Furthermore, studies of oral cholera vaccination show that vaccine antigen-specific IgA+ MBCs do appear in the circulation of vaccinees (52). It could be argued that the more vigorous inflammatory response to influenza is responsible for induction of a greater overall humoral response, thus explaining the longer persistence of mucosal IgA and higher frequencies of influenza-specific MBCs. However, there was no difference in the frequency of IgG+ MBCs between the two infections, suggesting that generalized deficiency of inflammatory signaling was unlikely to have been the cause of reduced anti-RSV IgA+ MBCs. By contrast, intranasal infection with attenuated influenza strains and rhinoviruses is still capable of inducing persistent humoral responses despite causing less inflammation than RSV (40, 53).
We therefore propose the hypothesis that RSV infection is unusual in limiting the induction of B-cell memory responses in the respiratory tract. This would explain why the defect is specifically in IgA+ MBCs (which are preferentially generated at mucosal sites) and does not affect IgG+ MBCs. Recent studies have suggested that although BAFF is elevated in bronchoalveolar lavage and bronchial brushings from children with RSV bronchiolitis, APRIL is not (54). In addition, defects in RSV antigen presentation and T-cell memory generation have been observed in several animal models, which may impact on T follicular helper cell activity (55). Thus, both T cell–independent and –dependent mechanisms may be influenced by RSV to specifically impair local immunity. If few IgA+ MBCs are induced following infection, this may explain why antibody-mediated immunity plateaus despite recurrent virus exposure. Further mechanistic investigation to support this interpretation will require invasive respiratory tract sampling of infected individuals, because no animal model experiences recurrent symptomatic RSV infections. It also remains to be seen whether similar mechanisms exist in human metapneumovirus or other respiratory viruses where recurrent infections occur.
The ability to predict infection risk using nasal IgA titers and discovery of a specific defect in IgA memory have important implications for RSV vaccine development. Although findings from experimental infection of young adults may not be directly applied to infants and elderly individuals, they can be extrapolated to women of child-bearing age in whom mucosal vaccination may reduce RSV transmission to their children and confer a “cocooning” effect. Community cohort studies will support the applicability of these data in high-risk populations, but our results suggest that vaccines inducing mucosal RSV-specific IgA are likely to be more effective than those stimulating systemic antibodies alone. For vaccines that induce local virus-specific IgA, we can also now accurately predict their protective efficacy before commitment to phase II/III clinical trials and for licensing purposes. However, the unique absence of IgA+ MBCs following RSV infection will necessitate reconsideration of which types of vaccine to pursue. Live-attenuated RSV vaccines, for example, may also be subject to this same defect and be incapable of inducing sufficient long-lived IgA to provide solid protection (56). Even novel vaccine constructs that might bypass this defect will encounter this limitation, with the paucity of preexisting IgA+ MBCs necessitating multiple-boost regimens.
In conclusion, we demonstrate that nasal antibody strongly predicts protection from RSV infection in adults. However, antibody levels wane much more rapidly than reported in other respiratory viral infections and are almost always below those needed for protection within months. Unexpectedly, we found that RSV-specific IgA+ MBCs were not significantly induced by infection, implying that effective RSV vaccines may need to overcome this defect to provide durable immunity.
Acknowledgments
Acknowledgment
The authors thank Gaynor Campbell, Natasha Gunawardana, Laura Medrano González, and Jonathan Spencer for their technical assistance, and the study volunteers for their generosity and tolerance. Purified respiratory syncytial virus fusion protein was a gift from Mucosis B.V., the Netherlands.
Footnotes
Supported by The Medical Research Council UK (G0902266, C.C.), Wellcome Trust (087805/Z/08/Z, P.J.M.O.), the National Institute for Health Research Imperial Biomedical Research Centre, a National Institute for Health Research Senior Investigator award (P.J.M.O.), and the Wellcome Trust/Medical Research Council–cofunded Mechanisms of Severe Acute Influenza Consortium Program (090382/Z/09/Z).
Author Contributions: C.C., P.J.M.O., M.S.H., J.P.D., and J.W. designed and conceived the study. M.S.H., C.C., and A.P. performed the experimental infection study. M.S.H., A.J., C.C., S.M., and C.A.M.d.H. performed the laboratory experiments. J.D. and Mechanisms of Severe Acute Influenza Consortium Investigators performed the collection and processing of influenza samples. M.S.H., C.C., and P.J.M.O. wrote the manuscript. M.S.H., A.J., S.M., J.D., A.P., J.P.D., C.A.M.d.H., J.W., P.J.M.O., and C.C. reviewed the manuscript prior to submission.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201412-2256OC on March 2, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O’Brien KL, Roca A, Wright PF, Bruce N, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol. 2002;76:5654–5666. doi: 10.1128/JVI.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner PS, McQuillin J, Court SDM. Speculation on pathogenesis in death from respiratory syncytial virus infection. BMJ. 1970;1:327–330. doi: 10.1136/bmj.1.5692.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall CB, Walsh EE, Long CE, Schnabel KC. Immunity to and frequency of reinfection with respiratory syncytial virus. J Infect Dis. 1991;163:693–698. doi: 10.1093/infdis/163.4.693. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto M, Sugawara K, Takashita E, Muraki Y, Hongo S, Nishimura H, Matsuzaki Y. Longitudinal course of human metapneumovirus antibody titers and reinfection in healthy adults. J Med Virol. 2010;82:2092–2096. doi: 10.1002/jmv.21920. [DOI] [PubMed] [Google Scholar]
- 7.Glezen WP, Frank AL, Taber LH, Kasel JA. Parainfluenza virus type 3: seasonality and risk of infection and reinfection in young children. J Infect Dis. 1984;150:851–857. doi: 10.1093/infdis/150.6.851. [DOI] [PubMed] [Google Scholar]
- 8.PATH 2014. Respiratory syncytial virus: vaccine development against a major cause of childhood respiratory illness [accessed 2014 Dec 16]. Available fromhttp://sites.path.org/vaccinedevelopment/respiratory-syncytial-virus-rsv/
- 9.The IMpact-RSV Study Group. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics. 1998;102:531–537. [PubMed] [Google Scholar]
- 10.Falsey AR, Walsh EE. Relationship of serum antibody to risk of respiratory syncytial virus infection in elderly adults. J Infect Dis. 1998;177:463–466. doi: 10.1086/517376. [DOI] [PubMed] [Google Scholar]
- 11.Piedra PA, Jewell AM, Cron SG, Atmar RL, Glezen WP. Correlates of immunity to respiratory syncytial virus (RSV) associated-hospitalization: establishment of minimum protective threshold levels of serum neutralizing antibodies. Vaccine. 2003;21:3479–3482. doi: 10.1016/s0264-410x(03)00355-4. [DOI] [PubMed] [Google Scholar]
- 12.Bem RA, Domachowske JB, Rosenberg HF. Animal models of human respiratory syncytial virus disease. Am J Physiol Lung Cell Mol Physiol. 2011;301:L148–L156. doi: 10.1152/ajplung.00065.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeVincenzo JP, Wilkinson T, Vaishnaw A, Cehelsky J, Meyers R, Nochur S, Harrison L, Meeking P, Mann A, Moane E, et al. Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am J Respir Crit Care Med. 2010;182:1305–1314. doi: 10.1164/rccm.201002-0221OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollard AJ, Savulescu J, Oxford J, Hill AV, Levine MM, Lewis DJ, Read RC, Graham DY, Sun W, Openshaw P, et al. Human microbial challenge: the ultimate animal model. Lancet Infect Dis. 2012;12:903–905. doi: 10.1016/S1473-3099(12)70292-X. [DOI] [PubMed] [Google Scholar]
- 15.Chiu C, Habibi MS, Jozwik A, Paras A, Makris S, Openshaw PJM.B cell responses following experimental RSV challenge in adult volunteers. Presented at the RSV Vaccines for the World conference. October 14–16, 2013Porto, Portugal [Google Scholar]
- 16.Habibi MS, Jozwik A, Makris S, Paras A, Openshaw P, Chiu C.Human B cell responses following experimental respiratory syncytial virus challenge. Presented at the Controlled Human Infection Studies in the Development of Vaccines and Therapeutics conference. January 9–11, 2013Cambridge, UK [Google Scholar]
- 17.Habibi MS, Guvenel AK, Jozwik A, Paras A, Chiu C, Openshaw PJM.Immunity to respiratory syncytial virus (RSV) infection. Presented at the Keystone Symposia Meeting on Pathogenesis of Respiratory Viruses (J5), January 19–24, 2014Keystone, CO [Google Scholar]
- 18.Habibi MS, Jozwik A, Makris S, Dunning J, Paras A, Wrammert J, Openshaw PJM, Chiu C.Defective IgA B cell memory generation following experimental human respiratory syncytial virus infection impairs protective immunity. Presented at the 9th International Respiratory Syncytial Virus Symposium. Stellenbosch, South Africa, November 9–13, 2014 [Google Scholar]
- 19.Medrano González L, Habibi MS, Jozwik A, Openshaw PJM. Chiu C. Quality of antigen-specific B cell responses as a correlate of protection against RSV [abstract] Immunology. 2014;143:75. [Google Scholar]
- 20.Yang DP, Zielinska E, Quiroz J, Madore D, Rappaport R. Preparation of a respiratory syncytial virus human reference serum for use in the quantitation of neutralization antibody. Biologicals. 2007;35:183–187. doi: 10.1016/j.biologicals.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Rennard SI, Basset G, Lecossier D, O’Donnell KM, Pinkston P, Martin PG, Crystal RG. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol (1985) 1986;60:532–538. doi: 10.1152/jappl.1986.60.2.532. [DOI] [PubMed] [Google Scholar]
- 22.Saletti G, Çuburu N, Yang JS, Dey A, Czerkinsky C. Enzyme-linked immunospot assays for direct ex vivo measurement of vaccine-induced human humoral immune responses in blood. Nat Protoc. 2013;8:1073–1087. doi: 10.1038/nprot.2013.058. [DOI] [PubMed] [Google Scholar]
- 23.Crotty S, Aubert RD, Glidewell J, Ahmed R. Tracking human antigen-specific memory B cells: a sensitive and generalized ELISPOT system. J Immunol Methods. 2004;286:111–122. doi: 10.1016/j.jim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 24.Tengvall S, Lundgren A, Quiding-Järbrink M, Svennerholm A-M. BAFF, stimulatory DNA and IL-15 stimulates IgA(+) memory B cells and provides a novel approach for analysis of memory responses to mucosal vaccines. Vaccine. 2010;28:5445–5450. doi: 10.1016/j.vaccine.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 25.R Core TeamR: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. Available from: http://www.R-project.org/
- 26.Sarkar D.Lattice: multivariate data visualization with R. New York: Springer; 2008. Available from: http://lmdvr.r-forge.r-project.org
- 27.Ritz C, Streibig JC. Bioassay analysis using R. J Stat Softw. 2005;12:1–22. [Google Scholar]
- 28.Falsey AR, Singh HK, Walsh EE. Serum antibody decay in adults following natural respiratory syncytial virus infection. J Med Virol. 2006;78:1493–1497. doi: 10.1002/jmv.20724. [DOI] [PubMed] [Google Scholar]
- 29.Sande CJ, Mutunga MN, Okiro EA, Medley GF, Cane PA, Nokes DJ. Kinetics of the neutralizing antibody response to respiratory syncytial virus infections in a birth cohort. J Med Virol. 2013;85:2020–2025. doi: 10.1002/jmv.23696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng N-Y, Mays I, Garman L, Helms C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wrammert J, Koutsonanos D, Li G-M, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med. 2011;208:181–193. doi: 10.1084/jem.20101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li G-M, Chiu C, Wrammert J, McCausland M, Andrews SF, Zheng N-Y, Lee J-H, Huang M, Qu X, Edupuganti S, et al. Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc Natl Acad Sci USA. 2012;109:9047–9052. doi: 10.1073/pnas.1118979109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wrammert J, Onlamoon N, Akondy RS, Perng GC, Polsrila K, Chandele A, Kwissa M, Pulendran B, Wilson PC, Wittawatmongkol O, et al. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J Virol. 2012;86:2911–2918. doi: 10.1128/JVI.06075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357:1903–1915. doi: 10.1056/NEJMoa066092. [DOI] [PubMed] [Google Scholar]
- 35.Mills J, V, Van Kirk JE, Wright PF, Chanock RM, Fishburne IE. Experimental respiratory syncytial virus infection of adults: possible mechanisms of resistance to infection and illness. J Immunol. 1971;107:123–130. [PubMed] [Google Scholar]
- 36.Watt PJ, Robinson BS, Pringle CR, Tyrrell DA. Determinants of susceptibility to challenge and the antibody response of adult volunteers given experimental respiratory syncytial virus vaccines. Vaccine. 1990;8:231–236. doi: 10.1016/0264-410x(90)90051-m. [DOI] [PubMed] [Google Scholar]
- 37.Walsh EE, Falsey AR. Humoral and mucosal immunity in protection from natural respiratory syncytial virus infection in adults. J Infect Dis. 2004;190:373–378. doi: 10.1086/421524. [DOI] [PubMed] [Google Scholar]
- 38.Lee FE-H, Walsh EE, Falsey AR, Betts RF, Treanor JJ. Experimental infection of humans with A2 respiratory syncytial virus. Antiviral Res. 2004;63:191–196. doi: 10.1016/j.antiviral.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Fujimoto C, Takeda N, Matsunaga A, Sawada A, Tanaka T, Kimoto T, Shinahara W, Sawabuchi T, Yamaguchi M, Hayama M, et al. Induction and maintenance of anti-influenza antigen-specific nasal secretory IgA levels and serum IgG levels after influenza infection in adults. Influenza Other Respi Viruses. 2012;6:396–403. doi: 10.1111/j.1750-2659.2011.00330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barclay WS, al-Nakib W, Higgins PG, Tyrrell DA. The time course of the humoral immune response to rhinovirus infection. Epidemiol Infect. 1989;103:659–669. doi: 10.1017/s095026880003106x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee FE-H, Falsey AR, Halliley JL, Sanz I, Walsh EE. Circulating antibody-secreting cells during acute respiratory syncytial virus infection in adults. J Infect Dis. 2010;202:1659–1666. doi: 10.1086/657158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 43.Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol. 2005;23:367–386. doi: 10.1146/annurev.immunol.23.021704.115723. [DOI] [PubMed] [Google Scholar]
- 44.Mora JR, von Andrian UH. Differentiation and homing of IgA-secreting cells. Mucosal Immunol. 2008;1:96–109. doi: 10.1038/mi.2007.14. [DOI] [PubMed] [Google Scholar]
- 45.Mora JR, Iwata M, Eksteen B, Song S-Y, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 46.He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, Shan M, Chadburn A, Villanacci V, Plebani A, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26:812–826. doi: 10.1016/j.immuni.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 47.Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interface. Annu Rev Immunol. 2011;29:273–293. doi: 10.1146/annurev-immunol-031210-101317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brandtzaeg P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine. 2007;25:5467–5484. doi: 10.1016/j.vaccine.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 49.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 50.Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. 2000;13:443–451. doi: 10.1016/s1074-7613(00)00044-3. [DOI] [PubMed] [Google Scholar]
- 51.Borsutzky S, Cazac BB, Roes J, Guzmán CA. TGF-beta receptor signaling is critical for mucosal IgA responses. J Immunol. 2004;173:3305–3309. doi: 10.4049/jimmunol.173.5.3305. [DOI] [PubMed] [Google Scholar]
- 52.Harris AM, Bhuiyan MS, Chowdhury F, Khan AI, Hossain A, Kendall EA, Rahman A, LaRocque RC, Wrammert J, Ryan ET, et al. Antigen-specific memory B-cell responses to Vibrio cholerae O1 infection in Bangladesh. Infect Immun. 2009;77:3850–3856. doi: 10.1128/IAI.00369-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clements ML, Murphy BR. Development and persistence of local and systemic antibody responses in adults given live attenuated or inactivated influenza A virus vaccine. J Clin Microbiol. 1986;23:66–72. doi: 10.1128/jcm.23.1.66-72.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McNamara PS, Fonceca AM, Howarth D, Correia JB, Slupsky JR, Trinick RE, Al Turaiki W, Smyth RL, Flanagan BF. Respiratory syncytial virus infection of airway epithelial cells, in vivo and in vitro, supports pulmonary antibody responses by inducing expression of the B cell differentiation factor BAFF. Thorax. 2013;68:76–81. doi: 10.1136/thoraxjnl-2012-202288. [DOI] [PubMed] [Google Scholar]
- 55.Openshaw PJ, Chiu C. Protective and dysregulated T cell immunity in RSV infection. Curr Opin Virol. 2013;3:468–474. doi: 10.1016/j.coviro.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karron RA, Buchholz UJ, Collins PL. Live-attenuated respiratory syncytial virus vaccines. Curr Top Microbiol Immunol. 2013;372:259–284. doi: 10.1007/978-3-642-38919-1_13. [DOI] [PMC free article] [PubMed] [Google Scholar]