

Abstract
MicroRNAs are increasingly implicated in the modulation of the progression of various cancers. We previously observed that KAI1 C-terminal interacting tetraspanin (KITENIN) is highly expressed in sporadic human colorectal cancer (CRC) tissues and hence the functional KITENIN complex acts to promote progression of CRC. However, it remains unknown that microRNAs target KITENIN and whether KITENIN-targeting microRNAs modulate CRC cell motility and colorectal tumorigenesis. Here, through bioinformatic analyses and functional studies, we showed that miR-124, miR-27a, and miR-30b negatively regulate KITENIN expression and suppress the migration and invasion of several CRC cell lines via modulation of KITENIN expression. Through in vitro and in vivo induction of mature microRNAs using a tetracycline-inducible system, miR-124 was found to effectively inhibit the invasion of CT-26 colon adenocarcinoma cells and tumor growth in a syngeneic mouse xenograft model. Constitutive overexpression of precursor miR-124 in CT-26 cells suppressed in vivo tumorigenicity and resulted in decreased expression of KITENIN as well as that of MYH9 and SOX9, which are targets of miR-124. Thus, our findings identify that KITENIN-targeting miR-124, miR-27a, and miR-30b function as endogenous inhibitors of CRC cell motility and demonstrate that miR-124 among KITENIN-targeting microRNAs plays a suppressor role in colorectal tumorigenesis.
Introduction
MicroRNAs (miRNAs, miRs) are short noncoding RNAs (~22 nucleotides) that bind directly to the complementary sequences in the 3′-untranslated regions (3′UTR) of their corresponding mRNA transcripts and acts as posttranscriptional silencers of their target genes.1 miRNAs play pivotal roles in physiological and pathological processes, and the deregulation of miRNAs is associated with a wide range of diseases, including human malignancies.2 Because miRNA genes are frequently located at the chromosomal fragile sites of cancer genomes,3 miRNAs are considered a novel class of oncogenes (oncomirs) and tumor suppressors (antioncomirs). In addition, specific miRNAs can act as both oncomirs and antioncomirs depending on the cellular environment in which they are expressed.4,5 All of these previous reports highlight the important roles of miRNAs in tumor development and provide new insights into the molecular mechanisms underlying carcinogenesis; however, the roles of most of these miRNAs in physiological and pathological processes remain to be elucidated.
The molecular carcinogenesis of colorectal cancer (CRC) is complex and poorly understood. CRC development involves a multistep process including both genetic and epigenetic changes, which leads to the activation of oncogenes and inactivation of tumor-suppressor genes in cancer cells.6 The expression levels of miRNAs are reproducibly altered in CRC, and their expression patterns are associated with diagnosis, prognosis, and therapeutic outcome in CRC.7 Recently, an emerging evidence has suggested that deregulation of miRNAs in CRC can contribute to cancer development if their target mRNAs are encoded by oncogenes or tumor suppressors.8 Although recent evidence indicated that altered expression of miRNAs is causally associated with the initiation and progression of CRC, the roles and potential mechanisms of miRNAs in CRC are still largely unknown.9 Moreover, the regulation of CRC cell motility by miRNAs and the consequent modulation of CRC progression are not fully understood.
We previously cloned KITENIN and identified it as a metastasis-enhancing gene.10,11 KITENIN participates in the dissemination of colorectal12 and squamous cancer cells,13 and the interaction of KITENIN with dishevelled (Dvl)/PKCδ is important in regulating CRC cell invasion via ERK/AP-1 activation.12 KITENIN is highly expressed in sporadic human CRC tissues; however, the mechanisms underlying how KITENIN expression is aberrantly regulated are not fully understood. In this study, we chose a miRNA system instead of conducting a promoter study to delineate the regulatory mechanism of KITENIN expression, which has the potential for new therapeutic intervention in CRC progression. We therefore focused on identifying miRNAs that target KITENIN and modulate its expression, as well as affect CRC cell motility. In addition, we investigated whether these identified miRNAs can be used as suppressors of colorectal tumorigenesis. We initially tried to identify KITENIN-targeting miRNAs by screening a miRNA library and by bioinformatic analyses, followed by subsequent functional studies with synthetic miRNAs and inhibitors. We next aimed to find therapeutically valuable antioncomirs that act against colorectal tumorigenesis by assessing conditional expression of mature miRNAs using a tetracycline-inducible system. Finally, we confirmed the role of antioncomirs by constitutive overexpression of candidate precursor miRNAs in a mouse xenograft model. Our results showed that KITENIN-targeting miRNAs, such as miR-27a, miR-30b, and miR-124, suppress the migration and invasion of several CRC cell lines via modulation of KITENIN expression. Among these miRNAs, miR-124 displays effective tumor-suppressor activity on colorectal tumorigenesis. Our results also suggest that these KITENIN-targeting miRNAs may play a significant role in the maintenance of an invasive phenotype in CRC cells.
Results
miR-124, miR-27a, and miR-30b negatively regulate KITENIN expression by targeting KITENIN
Previously, we observed a tumor regression effect of KITENIN siRNA when given intravenously in a mouse colon tumor model.11 We found a higher level of endogenous KITENIN in CRC tissues from advanced-stage patients and elucidated that the interaction of KITENIN with Dvl is important in regulating CRC cell invasion.12 However, transcriptional regulatory mechanisms underlying the expression of KITENIN have not yet been investigated. We therefore used a miRNA system to study the regulation of KITENIN expression. To identify miRNAs that alter expression of KITENIN in human CRC cells, we used a combination of miRNA library screening and bioinformatic analyses. HEK293 cells were cotransfected with a miRNA library that contains all the predicted miRNAs that bind to the seed sequences on KITENIN 3′UTR by bioinformatic analyses and the 3′-UTR fragment of human KITENIN cloned downstream of the luciferase reporter gene. Luciferase activity was detected in these cells. After liposomal transfection of a miRNA library of 215 pre-miRNAs into HEK293T cells, we identified several miRNAs that lowered luciferase activity (Supplementary Figure S1a). Using a threshold of 30% reduction, we selected nine of these miRNAs for further analysis. In addition, miR-27a, miR-30b, and miR-124 were chosen for further specific analyses because of their strong repressive effects on KITENIN expression levels in HEK293 cells (Supplementary Figure S1b). We initially found sequences of miR-124, miR-30b, and miR-27a that bound to the 3′UTR of KITENIN mRNA (Figure 1a); therefore, we further evaluated these three synthetic miRNA precursors using a luciferase assay. Cotransfection of synthetic miR-27a, miR-30b, or miR-124 with a reporter construct suppressed luciferase activity in HEK293 cells with similar reduction levels to those of the miRNA library screening (Figure 1b). By contrast, the repressive effects of synthetic miR-27a, miR-30b, or miR-124 on luciferase activity was abrogated by mutations in binding sites (Figure 1a) within the seed region of each miRNA (Figure 1b), confirming KITENIN as a direct target of miR-27a, miR-30b, and miR-124.
Figure 1.

Identification of miR-124, miR-27a, and miR-30b as KITENIN-targeting microRNAs, which modulate the expression of KITENIN. (a) Putative binding sites and sequences of miR-124, miR-30b, and miR-27a in the 3′UTR of KITENIN mRNA via bioinformatic analyses with TargetScan. Several nucleotides shown as red colors within the seed region were mutated in the 3′UTR of KITENIN (Mut-124: binding sites within the seed region of miR-124 were mutated). hsa refers to Homo sapiens. WT, wild-type. (b) A reporter assay to examine the effects of miR-27a, miR-30b, and miR-124 on the luciferase constructs containing the wild-type 3′-UTR or mutated 3′-UTR of KITENIN. A whole 3′-UTR fragment (3′-UTR WT) or mutated binding sites within the seed region of miR-124 (3′-UTR Mut-124), miR-30b (3′-UTR Mut-30b), or miR-27a (3′-UTR Mut-27a) of human KITENIN was cloned into the pSYC-31 vector downstream of the luciferase reporter. HEK293T cells were cotransfected with miR-con, miR-27a, miR-30b, or miR-124 (each 30 nmol/l) and the reporter construct. For each experiment, the data were normalized to the luciferase activity detected in cells transfected with fluorescein-labeled miRNA mimic negative control (miR-con). Luciferase activity was measured at 48 hours posttransfection and set to 1. Each bar represents mean ± SEM for triplicate samples. An asterisk indicates a significant difference between cells transfected with miR-con and miRNA (**P < 0.01, ***P < 0.001). NS, not significant. (c) Binding of miR-124 or miR-30b to its cognate seed sequence on KITENIN 3′UTR in which the binding sites for both miRNAs are very close is specific and independent. HEK293T cells were cotransfected with KITENIN 3′UTR Mut-124 and miR-con, miR-30b, or miR-30b plus increasing doses of miR-124 (left), or cotransfected with KITENIN 3′UTR Mut-30b and miR-con, miR-124, or miR-124 plus increasing doses of miR-30b (middle), or cotransfected with KITENIN 3′UTR WT and miR-con, miR-30b alone, miR-124 alone, or both of them. (d) Comparison of expression levels of miR-27a, miR-30b, or miR-124 in various CRC cells. The expression levels of miRNAs in CRC cells were analyzed by quantitative RT-PCR. The data were log10-transformed after initially being normalized to the U6 internal control and later to the HEK293T cell external control. Each bar represents the mean for triplicate samples. (e) Effects of miR-27a, miR-30b, or miR-124 on KITENIN mRNA expression levels in CRC cells. Synthetic miR-27a, miR-30b, or miR-124 (each 30 nmol/l) was transfected into Caco2 and HCT116 cells and expression levels of KITENIN mRNAs were analyzed by quantitative RT-PCR. Values represent the mean ± SEM from triple experiments. (f) Effects of miR-27a, miR-30b, or miR-124 and the corresponding miRNA inhibitor on KITENIN expression in CRC cells. Synthetic miR-27a, miR-30b, or miR-124 (each 30 nmol/l, left) and each corresponding synthetic miRNA inhibitor (each 30 nmol/l, right) were transfected into Caco2 and HCT116 cells, and the expression levels of KITENIN proteins were examined by immunoblot analyses. Actin was used as a loading control. The levels of KITENIN expression normalized to the loading control (KITENIN/actin ratio) were shown below the each blot. CRC, colorectal cancer; KITENIN, KAI1 C-terminal interacting tetraspanin; RT-PCR, reverse transcription-polymerase chain reaction.
As the binding sites for miR-124 (position 520–526 of 3′ UTR) and miR-30b (position 568–574 of 3′ UTR) are very close (apart by ~50 nucleotides), we examined whether binding of one miRNA to its cognate seed sequence could impact on the binding of the other miRNA. First, we tested whether miR-124 or miR-30b nonspecifically affects binding of miR-30b or miR-124 to its cognate seed sequence, respectively. When we cotransfected HEK293T cells with KITENIN 3′UTR-luciferase construct mutated in miR-124 binding site and miR-30b or miR-30b plus increasing doses of miR-124, roughly the same level of lowered luciferase activity was observed among them (Figure 1c, left) and vice versa (Figure 1c, middle). Next, we cotransfected HEK293T cells with KITENIN wild-type 3′UTR-luciferase construct and miR-30b alone, miR-124 alone, or both of them and tested the effect of introducing both miRNAs simultaneously. When we compared the luciferase activity among them, neither additive nor synergistic effects were detected (Figure 1c, right). As a mRNA can be simultaneously repressed by more than one miRNA species, the level of repression achieved is dependent on both the amount of mRNA and the amount of available miRNA complexes.14 These results indicated that miR-124 or miR-30b specifically binds to its cognate seed sequence and does not affect the binding of the other miRNA, although the binding sites for both miRNAs are very close.
We next examined the expression levels of endogenous miR-27a, miR-30b, and miR-124 in various CRC cells and found high levels of these miRNAs in Caco2 and HCT116 CRC cells (Figure 1d). We chose Caco2 and HCT116 cells for analysis of KITENIN expression by miRNAs. Transfection of synthetic miR-27a, miR-30b, or miR-124 into Caco2 and HCT116 cells lowered KITENIN mRNA (Figure 1e) and KITENIN protein (Figure 1f, left panel) levels compared to those of cells transfected with fluorescein-labeled miRNA mimic negative control (miR-con), which is not targeting KITENIN mRNA. Importantly, transfection of a synthetic miRNA inhibitor to miR-27a, miR-30b, or miR-124 into Caco2 and HCT116 cells increased KITENIN expression levels (KITENIN/actin ratio: 1.23–1.44, Figure 1f, right panel). Additionally, synthetic miR-27a, miR-30b, or miR-124 and their synthetic miRNA inhibitors were transfected into SW480, SW620, and DLD1 CRC cells (Supplementary Figure S2), which express lower levels of endogenous miR-27a and miR-124 than Caco2 and HCT116 cells (Figure 1d). Compared with repressed KITENIN level by three miRNAs in Caco2 (KITENIN/actin ratio: 0.55–0.73) and HCT116 cells (KITENIN/actin ratio: 0.52–0.82), the magnitude of repressed KITENIN level by these miRNAs were relatively small in SW480 (Supplementary Figure S2a, KITENIN/actin ratio: 0.83–0.92), SW620 (Supplementary Figure S2b, KITENIN/actin ratio: 0.87–0.99), and DLD1 CRC cells (Supplementary Figure S2c, KITENIN/actin ratio: 0.80–0.91). Although the magnitude of repression is small, these results indicated that miR-27a, miR-30b, or miR-124 functions as a KITENIN-targeting miRNA in SW480, SW620, and DLD1 cells like in Caco2 and HCT116 cells. Overall, these validation experiments confirmed the initial screening results that miR-27a, miR-30b, and miR-124 are KITENIN-targeting miRNAs, and indicated that these three miRNAs negatively modulate KITENIN expression in CRC cells, which express measurable levels of these endogenous KITENIN-targeting miRNAs. Among the examined CRC cells, the maximum repression of KITENIN by miR-30b or miR-27a was observed in Caco2 cells, whereas the peak effect by miR-124 was observed in HCT116 cells (Figure 1f and Supplementary Figure S2).
Inhibition of cell motility and proliferation of CRC cells by KITENIN-targeting miRNAs
We next addressed whether KITENIN-targeting miRNAs were involved in the regulation of cell motility and proliferation in CRC cells via modulation of KITENIN expression. We again chose Caco2 and HCT116 cells for further functional analysis. Compared to miR-con-transfected Caco2 and HCT116 cells, cell invasion was markedly suppressed in miR-27a-, miR-30b-, or miR-124-transfected Caco2 and HCT116 cells (Figure 2a). In contrast, transfection of a synthetic miRNA inhibitor to miR-27a, miR-30b, or miR-124 significantly increased cell invasion of Caco2 and HCT116 cells (Figure 2b). Moreover, in HCT116 cells, transfection of miR-27a, miR-30b, or miR-124 significantly attenuated cell migration as assessed by a wound-healing model (Figure 2c), as well as significantly decreased cell proliferation (Figure 2d). These results indicate that these three KITENIN-targeting miRNAs play a role in the maintenance of cell motility in CRC cells, which express high levels of KITENIN-targeting miRNAs.
Figure 2.

Inhibition of cell motility and proliferation by KITENIN-targeting micro RNAs in CRC cells. (a) Effects of miR-27a, miR-30b, or miR-124 on cell invasiveness of Caco2 and HCT116 cells. Caco2 (upper) and HCT116 (lower) cells were transiently transfected with miR-con, miR-27a, miR-30b, or miR-124 (each 30 nmol/l) and subjected to an invasion assay using the Transwell migration apparatus and fibronectin as a chemotactic factor. The pictures shown represent three independent experiments. The histogram represents invading cells, which were counted at five chosen areas and represented by bar graphs (mean ± SEM, n = 3). An asterisk indicates a significant difference between the miR-con and each miR group (***P < 0.001). (b) Invasion assay in Caco2 and HCT116 cells after transfection of miRNA inhibitor. Caco2 (upper) and HCT116 (lower) cells were transiently transfected with miR-con or a synthetic miRNA inhibitor (30 nmol/l) to miR-27a, miR-30b, or miR-124 and subjected to an invasion assay as in a. An asterisk indicates a significant difference between the miR-con and each miR inhibitor group (mean ± SEM, n = 3; *P < 0.05, **P < 0.01, ***P < 0.001). (c) Cell migration assay in HCT116 cells after treatment with KITENIN-targeting miRNAs. HCT116 cells after transfection of KITENIN-targeting miRNAs were subjected to a wound-healing assay (left). The pictures shown represent those from three independent experiments. The effects of each miR (30 nmol/l) on cell migration are displayed as relative healing distances (right). Values are mean SEM for three independent experiments (n = 3). (d) Cell growth assays in HCT116 cells after treatment with KITENIN-targeting miRNAs. The effects of miRNA on cell proliferation were examined by a MTT assay and an asterisk indicates a significant difference in cell proliferation between the miR-con and each miR group. Values represent the mean ± SEM from triple experiments. (e) The relative expression levels of HOXA13 mRNA in CRC cells after treatment with KITENIN-targeting miRNAs. Synthetic miR-27a, miR-30b, or miR-124 (each 30 nmol/l) was transfected into HEK293T, Caco2, and HCT116 cells, and the expression levels of HOXA13 mRNAs were analyzed by quantitative RT-PCR. Values represent the mean ± SEM from triple experiments. An asterisk indicates a significant difference between the miR-con and each miR group. CRC, colorectal cancer; KITENIN, KAI1 C-terminal interacting tetraspanin.
Having found that miR-124, miR-27a, and miR-30b, which share KITENIN as a common target, acts as inhibitory modulators of the invasiveness of CRC cells, we next investigated whether other oncogenes exist as common targets of these three miRNAs and contribute to modulation of CRC cell motility. Through bioinformatic analyses, we found that HOXA13 was also predicted to be a common target of miR-124, miR-27a, and miR-30b. HOXA13 is reported to act as an oncogene in leukemia,15 hepatoma,16 esophageal cancer,17 and gastric18 cancer. Using expression analyses, we examined whether HOXA13 is the real target of the three miRNAs in CRC cells. However, expression of HOXA13 was not consistently modulated by the three miRNAs in HEK293T, Caco2, and HCT116 cells (Figure 2e); In HEK293T cells, only miR-27a repressed expression of HOXA13, whereas only miR-124 repressed expression of HOXA13 in HCT116 cells, but no repression was observed in Caco2 cells. It is known that the cellular context-dependent functions of miRNAs in the various functional processes could be explained in part by the variations in transcriptome composition in diverse cell types in different species and the ratio of copy numbers between a specific miRNA and its target may also influence its functions.19 However, the precise understanding of the cellular context-dependent functions of miRNAs in cancer cells is unknown.20 Thus, due to vastly different effects of three KITENIN-targeting miRNAs on HOXA13 expression in three cell lines resulting from cell-type specific functions of miRNA, we ruled out the possibility that HOXA13 works as a common target of three KITENIN-targeting miRNAs, such as miR-124, miR-27a, and miR-30b. Therefore, our results suggest that miR-124, miR-27a, and miR-30b, which share KITENIN as a common target, play a role in the maintenance of invasiveness of CRC cells via modulation of KITENIN expression.
Induced expression of ectopic mature miR-27a, miR-30b, and miR-124 via a tetracycline-inducible system suppresses KITENIN expression and the invasiveness of CT-26 cells.
Next, we investigated whether KITENIN-targeting miRNAs effectively inhibit the invasion of CT-26 murine colon adenocarcinoma cells by in vitro induction of mature miRNA using a tetracycline-inducible system. We generated three stable CT-26 cell lines for conditional expression of miR-27a, miR-30b, or miR-124. To determine whether expression of ectopic miRNA was induced in a time-dependent manner following the addition of doxycycline (DOX, 5 μg/ml), cells were analyzed by quantitative reverse transcription-polymerase chain reaction (RT-PCR). At 48 hours following DOX addition, expression of miR-27a (Figure 3a) and miR-30b (Figure 3b) was induced, and by 96 hours the induced expression levels had increased further; however, induction of miR-124 was delayed in comparison to the other miRNAs (Figure 3c). When we compared the repression of KITENIN expression in CT-26 cells by induced ectopic mature miR-27a, miR-30b, and miR-124 via a tetracycline-inducible system, miR-30b showed greater effects than remaining two miRNAs. The level of repression achieved is dependent on both the amount of mRNA and the amount of available miRNA complexes,14 however, the current understanding of the meaning of these cell-type specific functions of miRNAs is still limited. In line with this observation, a time-course analysis showed that the level of murine KITENIN protein began to decrease in each stably miR-transfected CT-26 cell line 48 hours after the addition of DOX and stabilized by 96 hours (Figure 3d). The plateau repression of KITENIN expression by miR-27a or miR-124 was observed after 72 hours of induction, whereas miR-30b represented the peak repression on 4 days after DOX. In addition, cell invasion was suppressed in each stably miR-transfected CT-26 cell line 72 hours after DOX (Figure 3e). These results showed that the induced expression of ectopic mature miRNAs by a tetracycline-inducible system is effective in CT-26 cells and confirmed that these three KITENIN-targeting miRNAs regulate cell invasiveness of CRC cells via modulation of KITENIN expression. Moreover, it indicates that conditional expression of mature miRNAs by a tetracycline-inducible system can be useful for studying the effects of miRNAs on in vitro functional cellular phenotypes.
Figure 3.
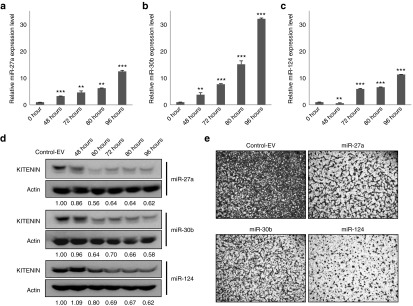
Induced KITENIN-targeting miRNAs in CT-26 cells decrease KITENIN expression and cell invasion. (a–c) A time-course expression of miR-27a (a), miR-30b (b), or miR-124 (c) in stably miR-transfected CT-26 cells after treatment with doxycycline (DOX). CT-26 cells were stably transfected with an H1-miRNA plasmid encoding mature miR-27a (a), miR-30b (b), or miR-124 (c). The induced expression level of mature miR-27a, miR-30b, and miR-124 in CT-26 cells were determined by quantitative RT-PCR following addition of doxycycline (5 μg/ml) for 96 hours. (d) Induced miR-27a, miR-30b, and miR-124 in CT-26 cells inhibit KITENIN expression in a time-dependent manner. KITENIN expression was examined in stably the control empty vector (control-EV)-transfected or miR-transfected CT-26 cells following the addition of doxycycline from 48 to 96 hours. The levels of inhibition of KITENIN expression in each time point by miRNA normalized to the loading control (KITENIN/actin ratio) were shown below the each blot. (e) Induction of miR-27a, miR-30b, and miR-124 in CT-26 cells suppresses its invasiveness to fibronectin. In vitro cell invasion was examined in stably the control-EV-transfected or miR-transfected CT-26 cells following the addition of doxycycline for 72 hours (n = 3). KITENIN, KAI1 C-terminal interacting tetraspanin.
In vivo induction of ectopic KITENIN-targeting miRNAs in CT-26 cells inhibits tumor growth in a syngeneic mouse xenograft model
Because conditional expression of mature miRNAs in CT-26 cells by a tetracycline-inducible system works was effective (Figures 3a–c), we examined which miRNAs among KITENIN-targeting miRNAs effectively inhibit the in vivo tumor growth of CT-26 cells in a syngeneic mouse xenograft model. Each stably miR-transfected CT-26 cell line, such as CT-26/miR-27a, CT-26/miR-30b, and CT-26/miR-124 cells, were injected subcutaneously into a syngeneic host (Balb/c mice). The mice inoculated with CT-26/miR cells were divided into two groups: the control group was given phosphate buffered saline (PBS) while the experimental group was given DOX (100 μg) semiweekly by intraperitonial injection. Additionally, water containing DOX (2 mg/ml) was provided daily to mice in the experimental group to support the induction and maintenance of miRNA expression throughout the experimental period. Mice in the control group formed tumors around 1 week after injection. Tumor volumes were measured every other day after injection until necrosis was observed in the primary tumors (second week). Mice were sacrificed at 14 days after cell injection to obtain fresh tumor tissues, which were isolated and weighed. The contents of each miRNA and KITENIN mRNA were examined in the tumor tissues of each group. Ectopic miRNA expression levels increased by 1.5- to twofold (Figure 4a) and expression levels of KITENIN decreased in the tumor tissues of each group after in vivo induction by DOX (Figure 4b). Although in vivo induction of the miRNAs resulted in tendency of decreasing tumor weight in each group, however, miR-124 was found to significantly inhibit tumor growth (Figure 4c), suggesting miR-124 as a candidate for further analysis on the inhibition of in vivo tumor growth. The differential effects of miR-30b and miR-27a compared to miR-124 on the inhibition of in vivo tumor growth may be ascribed either to different cell systems or to different cancer cell types,19,20 and the miRNAs were reported to exert confounding effects on cell growth and proliferation within the primary tumor.21 Thus, it is crucial to confirm the functional significance of miRNAs in specific conditions. The results show that in vivo conditional expression of each mature miRNA by a tetracycline-inducible system is useful for studying the effects of candidate miRNAs on the progression of tumorigenesis.
Figure 4.
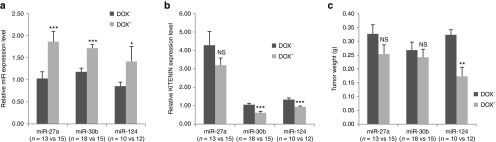
In vivo induction of KITENIN-targeting miRNAs in CT-26 cells suppresses KITENIN expression in tumor tissues and affects tumor growth in a syngeneic mouse tumor model. (a,b) Induced expression of miR-27a, miR-30b, and miR-124 with doxycycline (DOX) (a) and their effects on KITENIN expression (b) in resected tumor tissues. Expression levels of mature miR-27a, miR-30b, and miR-124 (a) and KITENIN mRNA (b) were determined by quantitative RT-PCR in isolated tumor tissues of each group obtained 14 days after CT-26 cell injection with or without in vivo DOX treatment. An asterisk indicates a significant difference between presence (+) and absence (−) of DOX treatment. NS, not significant. (c) Effects of in vivo induction of KITENIN-targeting miRNAs on tumor growth in a syngeneic mouse tumor model. Tumor weight was measured in isolated tumor tissues from each group obtained at 14 days. During in vivo mature miRNA induction with DOX, miR-124 among the KITENIN-targeting miRNAs significantly delayed the tumor growth of CT-26 cells. KITENIN, KAI1 C-terminal interacting tetraspanin.
Constitutive overexpression of pre-miR-124 in CT-26 cells suppresses tumor growth in a syngeneic mouse model
miR-124 was found to effectively inhibit tumor growth and was identified as a candidate for further analysis through a pilot study of the in vivo miRNA induction in a mouse tumor model. We next examined the effects of constitutively overexpressed precursor miR-124 (pre-miR-124), which ensure biologically relevant interactions with endogenous processing machinery and regulatory partners, in CT-26 cells (CT-26/pre-miR-124) on tumor growth. We compared tumor progression between the CT-26/pre-miR-124-cell injection groups to that of the CT-26/vector-alone group. In vivo tumor progression was significantly delayed in the CT-26/pre-miR-124 group, compared to that of the control group (Figure 5a,b). Mature miR-124 expression level was significantly increased by twofold (Figure 5c), with a corresponding decrease in the expression level of KITENIN (Figure 5d) in tumor tissues from the CT-26/pre-miR-124 group, compared to those from the control group. Tumor tissues were isolated 17 days after cell injection, and mice in the CT-26/pre-miR-124 group had significantly less tumor burden compared to those of the control group (Figure 5e). Thus, as in the in vivo mature miR-124 induction by tetracycline-inducible system, constitutively overexpression of pre-miR-124 in CT-26 cells suppresses tumor growth in a syngeneic mouse model.
Figure 5.
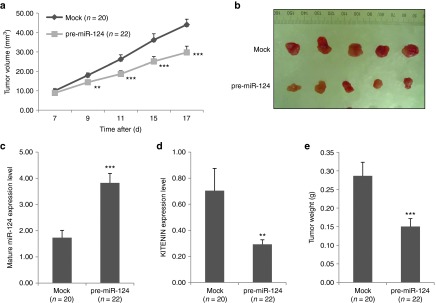
Constitutively overexpressed pre-miR-124 inhibits the in vivo tumorigenicity of CT-26 cells. (a) Effect of constitutively overexpressed pre-miR-124 on tumor formation of CT-26 cells in a syngeneic mouse xenograft model. Significantly slower tumor growth was observed from syngeneic Balb/c mice (n = 22) injected with CT-26/pre-miR-124 cells compared to those of CT-26/vector cells (Mock, n = 20). Tumor volumes are represented as mean ± SEM. An asterisk indicates a significant difference between the Mock and the pre-miR-124 group. (b) Photographs of isolated tumor tissues represent those of five mice from each group. (c,d) Expression of mature miR-124 (c) and its effects on KITENIN expression (d) in resected tumor tissues. Expression levels of mature miR-124 (c) and KITENIN mRNAs (d) were determined (as in Figure 4) in isolated tumor tissues of both groups, obtained at 17 days after CT-26 cell injection. (e) Effects of miR-124 on tumor growth in a syngeneic mouse tumor model. Tumor weight was measured in isolated tumor tissues from both groups obtained at 17 days. KITENIN, KAI1 C-terminal interacting tetraspanin
miR-124 suppresses in vivo colorectal tumorigenesis by targeting MYH9 and SOX9 as well as KITENIN
As a single miRNA can target multiple mRNAs to regulate gene expression,2 it is probable that other targets of miR-124 in addition to KITENIN may contribute to the tumor-suppressive effect of miR-124 on colorectal tumorigenesis. We therefore asked whether ectopically overexpressed pre-miR-124 targets other oncogenes to suppress in vivo colorectal tumorigenesis. We performed mRNA microarray analysis using isolated tumor tissues from mice of both CT-26/pre-miR-124 and CT-26/vector cell injection groups to determine candidate oncogenes as possible targets of miR-124. We found that the expression levels of MYH9 and SOX9, which are predicted targets of miR-124 by bioinformatic analyses, decreased by a greater extent in the tumor tissues from the CT-26/pre-miR-124 group than in those from the control group (Figure 6a). MYH9 has been shown to be responsible for the invasiveness of human MCF-7/6 breast cancer cells22 and SOX9 is known for its crucial role in the progenitors of several adult tissues.23 As SOX9 was reported as a direct target of miR-124,24 we performed experiments of MYH9 3′UTR luciferase reporter assay with miR-124 target site mutation to specify microRNA targeting (Supplementary Figure S3a). Cotransfection of synthetic miR-124 with a reporter construct suppressed luciferase activity in HEK293 cells, however, the repressive effects of miR-124 on luciferase activity was abrogated by mutations in binding sites within the seed region of miR-124 (Supplementary Figure S3b), confirming MYH9 as a direct target of miR-124.
Figure 6.
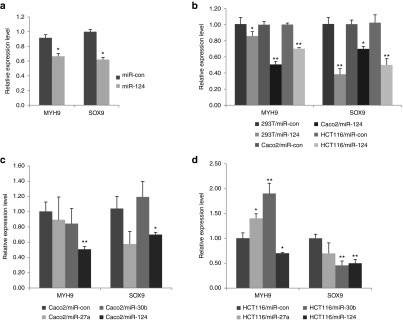
miR-124 suppresses in vivocolorectal tumorigenesis via negatively regulating the expression levels of MYH9 and SOX9, as well as KITENIN. (a) Overexpression of ectopic pre-miR-124 also targets MYH9 and SOX9 to suppress in vivo colorectal tumorigenesis. To identify downregulated oncogenes as possible targets of miR-124, mRNA microarray analysis was performed and compared using isolated tumor tissues from CT-26/pre-miR-124 and CT-26/vector cell injection groups. An asterisk indicates a significant difference between the miR-con and the pre-miR-124 group. (b) MYH9 and SOX9 are real targets of miR-124 in CRC cells. The mRNA expression levels of MYH9 and SOX9 were examined by quantitative RT-PCR in HEK293T, Caco2, and HCT116 cells after transfection of miR-con or mature miR-124. (c,d) MYH9 and SOX9 are not targets of miR-27a and miR-30b in CRC cells. The mRNA expression levels of MYH9 and SOX9 were examined in Caco2 (c) and HCT116 (d) cells after transfection of miR-con, mature miR-27a, miR-30b, or miR-124. CRC, colorectal cancer; KITENIN, KAI1 C-terminal interacting tetraspanin.
We examined again whether MYH9 and SOX9 are real targets of miR-124 in CRC cells using expression analyses. As shown in Figure 6b, expression levels of MYH9 and SOX9 mRNA in Caco2 and HCT116 cells decreased after transfection of mature miR-124. However, miR-27a or miR-30b did not repress MYH9 expression in Caco2 (Figure 6c) and HCT116 cells (Figure 6d). miR-27a showed the tendency to repress SOX9 expression in Caco2 (Figure 6c) and HCT116 (Figure 6d) cells, but not statistically significant, and miR-27a is not predicted to bind on SOX9 3′UTR by bioinformatic analyses. Thus, we ruled out the possibility that miR-27a works as a SOX9-targeting miRNA. In Figure 6c, miR-30b showed the tendency to increase SOX9 expression in Caco2, but significantly represses SOX9 expression in HCT116 cells (Figure 6d). Thus, due to inconsistent results in two CRC cells, we also ruled out the possibility of miR-30b as a SOX9-targeting miRNA in CRC cells. These results indicated that MYH9 and SOX9 are targets of miR-124 in CRC cells. Thus, miR-124 suppresses in vivo colorectal tumorigenesis by negatively regulating the expression of MYH9 and SOX9, in concert with repression of KITENIN.
Discussion
In this study, we developed a strategy to initially screen for miRNAs that effectively target a clinically established oncogene and to subsequently identify therapeutically valuable antioncomirs among them. To identify applicable antioncomirs involved in the suppression of colorectal tumorigenesis, we chose to target the KITENIN oncogene.10,11,12 We identified KITENIN-targeting miRNAs by miRNA library screening and bioinformatic analyses, and selected three miRNAs, namely, miR-27a, miR-30b, and miR-124, for further analysis. These miRNAs suppressed the migration and invasion of several CRC cell lines, as shown by functional studies with synthetic miRNA precursors and inhibitors. We also screened for a final candidate via conditional expression of mature miRNAs in a mouse xenograft tumor model using a tetracycline-inducible system, and selected miR-124 as a potential antioncomir. Using a constitutive pre-miRNA overexpression system, which leads to properly cleaved miRNA and ensures biologically relevant interactions with regulatory partners, we confirmed that miR-124 functions as an effective suppressor of colorectal tumor, and as other targets of miR-124 may be responsible for its suppressive effects on colorectal tumorigenesis, we subsequently performed mRNA microarray analysis on isolated tumor tissues from a xenograft tumor model in which pre-miR-124 was overexpressed to identify other targets of miR-124. As a result, expression levels of MYH9 and SOX9 were found to be significantly lower in the pre-miR-124-delivered tumor tissues compared to control-transfected tissues. Thus, our results demonstrated that miR-124 suppresses in vivo colorectal tumorigenesis by targeting MYH9 and SOX9 as well as KITENIN, and suggest that miR-124 among the KITENIN-targeting microRNAs is a therapeutically valuable antioncomir for CRC.
The regulation of CRC cell motility by miRNAs, and the subsequent modulation of CRC progression, is a novel therapeutic target for overcoming CRC. Both upregulation and downregulation of specific miRNAs have been described in CRC carcinogenesis and progression, such as upregulated miR-135, miR-21, miR-17–92, miR-95, and miR-196a; and downregulated miR-143, miR-145, miR-101, miR-34, miR-200, miR-195, and miR-212.25 Based on expression studies in CRC patients, miR-143 and miR-145 were described to be the first miRNAs possessing tumor-suppressor activity in colon cancer,26 however, the roles and potential mechanisms of miRNAs in the regulation of CRC cell motility are not fully understood.9 Furthermore, oncomirs are implicated in the regulation of CRC cell motility and development of CRC, such as miR-103/107, miR-135a, and miR-21 by targeting the metastasis suppressors DAPK and KLF, metastasis suppressor 1 and PTEN, respectively, whereas anti-oncomirs, such as miR-143, miR-30a, miR-141, and miR-345, suppress cell invasion through downregulation of MACC1, PIK3CD, Smad-interacting protein 1, and BCL2-associated athanogene 3, respectively.8,9,25,27
In this study, we found that miR-27a, miR-30b, and miR-124 suppressed the migration and invasiveness of several CRC cell lines via modulation of KITENIN expression. Specifically, transfection of an antagomir alone to each miRNA in CRC cells that expresses high levels of KITENIN-targeting miRNAs increased cell invasiveness as well as KITENIN expression. Our results indicate that these KITENIN-targeting miRNAs play a biological role in the mechanisms responsible for maintaining the invasiveness of CRC cells, and act as endogenous inhibitors of CRC cell motility. We propose that the modulation of KITENIN expression by KITENIN-targeting miRNAs could play a significant role in the maintenance of an invasive phenotype in CRC cells. Previous reports have shown that each miRNA has positive or negative effects on CRC cell motility by targeting tumor suppressor or oncogene.9 By contrast, our present results demonstrated that several miRNAs, which share the KITENIN oncogene10,11 as a common target, might act as negative regulators of the invasiveness of CRC cells. Previously, we observed that the functional KITENIN complex is also indispensable for the invasion of DLD1 and HCT116 cells harboring KRAS mutation.12 Such findings indicate that the KITENIN complex is also required as a downstream effector for increased invasiveness by KRAS, and suggest that inhibitors of the KITENIN complex may be effective in the treatment of CRC patients with KRAS mutations. Therefore, downregulation of KITENIN expression in CRC tissues through the modulation of KITENIN-targeting miRNAs may improve treatment responses in metastatic CRC patients, regardless of KRAS status.
Deregulated miRNAs are involved in the pathogenesis of cancers mainly by regulating the expression of oncogenes and tumor suppressors.2,28 For example, the expression of miR-124 is very low in many types of cancers, including gastric,29 medulloblastoma,30 cervical,31 breast,32 hepatocellular,33 pancreatic,34 and colorectal35 cancers, which may be caused by DNA methylation-based silencing. These reports indicate that miR-124 plays a significant role in the pathogenesis of various cancers and suggest that lower expression level of miR-124 is an independent bad prognostic factor. Accordingly, direct target genes of miR-124 in cancer cells have been reported, such as cyclin-dependent kinase 6 in medulloblastoma,36 integrin β-1 in oral squamous cancer cells,37 rho-kinase2 and an enhancer of the zeste homologue 2 genes in hepatocellular cancer,33 SPHK1 in gastric cancer,38 androgen receptor in prostate cancer,39 CD151 in breast,40 and Rac1 in pancreatic cancer.34 Thus, these data demonstrate the potential tumor-suppressor role of miR-124. However, it was not answered whether miR-124 also plays a suppressor role in the colorectal tumorigenesis and which target genes are responsible for this action. Moreover, some miRNAs, such as let-7, miR-21, miR-135, miR-143, miR-196a, miR-145, miR-95, miR-137, and miR-212 have been studied for their roles in colorectal carcinogenesis, but it remains unclear whether these miRNAs have therapeutic value for the treatment of CRC patients.8,9,25 In this study, we found that among the KITENIN-targeting miRNAs, miR-124 effectively inhibited colorectal tumor growth in a mouse xenograft model. These results indicate that the miR-124-KITENIN axis contributes to the invasiveness of CRC cells and that the suppressor role on colorectal tumorigenesis is related to targeting SOX9 and MYH9 in addition to KITENIN, which is consistent with reported suppressor roles of miR-124 in various carcinomas.29,30,31,32,33,34 By contrast, the downregulation of miR-124 observed in CRC tissues35 may explain the aberrant expression of KITENIN in advanced CRC.12 Because re-expression of miRNAs silenced during cancer development provides new targets for cancer gene therapy,41,42 restoration of anti-oncomir miR-124 expression may be useful for the clinical treatment of CRC patients. This finding also suggests that the metastasis of KITENIN-overexpressing CRC cells to regional lymph nodes or liver, observed in advanced CRC patients,12 may be inhibited by the upregulation of miR-124.
The SRY-box transcription factor SOX9 is as a target of miR-124 during the transition from the transit-amplifying cell to the neuroblast stage. In addition, SOX9 overexpression has been shown to abolish neuronal differentiation.24 SOX9 is known for its crucial role in the progenitors of several adult tissues and plays a role in embryonic growth, confirmed by the large size of embryos expressing a SOX9 transgene.23 However, its role as an oncogene remains largely unknown. Our results provide the first evidence for the involvement of SOX9 in colorectal tumorigenesis. The miR-124-SOX9 data reported here may help promote further research in this field aimed at elucidating the role of miRNA-mediated modulation of SOX9 on the control of carcinogenesis.
Serum response factor is a global regulator of cytoskeletal gene expression and many of its cytoskeletal targets are controlled through Rho-actin signalling and myocardin-related transcription factors (MRTFs).43 Myocardin-related transcription factors-serum response factor signaling is essential for the effective execution of a number of Rho-dependent cytoskeletal processes, including cell adhesion, motility, and invasion. Nonmuscle myosin heavy chain (MYH9/NMHC-IIA) and its regulatory light chain (MYL9/MLC2), targets of the Rho effector ROCK, are cytoskeletal components involved in actomyosin-based contractility and implicated in the invasive behavior of tumor cells.44,45 MYH9 has been shown to represent myocardin-related transcription factors-dependent expression in breast cancer cells and is required for lung colonization.46 In addition, MYH9 is responsible for the invasiveness of human MCF-7/6 breast cancer cells, which differ from their MCF-7/AZ counterparts.22 It was reported that overexpression of let-7f could inhibit invasion and migration of gastric cancer cells by directly targeting MYH9.47 Our results, which show that the miR-124-MYH9 axis reduces CRC cell invasion and colorectal tumorigenesis, provide the first evidence that the tumor metastasis-associated gene MYH9 is a target of miR-124 and that MYH9 may be a novel therapeutic candidate for CRC.
As the level of repression achieved is dependent on both the amount of mRNA and the amount of available miRNA complexes, the ratio of copy numbers between a specific miRNA and its target may influence its functions.14,19 Even using the same fibronectin transwell invasion assay, we here identified that ectopic expression of miR-30b markedly suppressed cell invasion of Caco2 and HCT116 colorectal cancer cells (Figure 2a), whereas Gaziel-Sovran et al.48 found that ectopic expression of miR-30b and 30d strongly stimulated the invasive capacity of two metastatic melanoma cell lines, 113/6-4L and 131/4-5B1, and a prometastatic role for miR-30d has also been shown in hepatocellular carcinoma.49 However curiously, miR-30e, which shares a seed region with miR-30b/d, has shown an antimetastatic role in breast cancer.50 Although a plausible explanation for this paradox was proposed, these and our observations underscore the context-dependence of miRNA functions in cancer.20 Thus, despite their biological importance, the current understanding the cell-type specific functions of miRNAs is not fully understood.
In conclusion, we have demonstrated a crucial role for the miRNA-mediated modulation of KITENIN expression, such as miR-27a, miR-30b, and miR-124, in maintaining the invasiveness of CRC cells. The identification of miR-124 as a suppressor of colorectal tumorigenesis by targeting KITENIN, SOX9, and MYH9 represents a leap forward in our understanding of controlling CRC progression by miRNAs and offers new opportunities for therapeutic intervention in CRC.
Materials and Methods
Cells and oligonucleotides. Human CRC cell lines (Caco2, DLD1, HCT116, SW480, and SW620), and 293T cells were obtained from the American Type Culture Collection (Rockville, MD) and were grown as described.12 For the generation of stable cells, electroporation was performed according to the manufacturer's instructions. Selected clones expressing each construct were obtained in the presence of antibiotics and were assayed. For most assays using stable cell lines, we used mixed polyclonal cells to exclude out clonal variation. The sequences of all oligonucleotide primers and miRNA-mimic are given in Supplementary Table S1. All miRNA mimics and miRNA inhibitor (anti-miR) were synthesized by Bioneer (Daejeon, Korea) and transfected by using RNAiMAX (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The final concentration of miRNA mimics and inhibitors in the transfection system was 30 nmol/l. Fluorescein-labeled miRNA mimic negative control (miR-con), which is not targeting KITENIN mRNA and contains fluorescein-isithiocyanate, was used for microRNA transfection control and transfection efficiency for the single and cotransfected studies was determined by fluorescence microscope.
Luciferase assay. To construct reporter plasmids, a 2.3-kb DNA fragment of the KITENIN 3′ UTR containing the putative miR-27a, miR-30b, and miR-124 binding site, or the mutated binding sites within the seed region of miR-124, miR-30b, or miR-27a, was prepared by PCR. A 1.3-kb DNA fragment of the MYH9 3′ UTR containing the putative miR-124 binding site or mutated 3′-UTR of MYH9 was also prepared by PCR. DNA fragments were cloned into the pMIR-REPORT luciferase vector (Ambion, Austin, TX) downstream of the reporter gene. The sequences and cloning direction of these PCR products were validated by sequencing. For luciferase assay, cells (4 × 104 per well) were seeded into 24-well plates and cultured for 24 hours. The cells were then cotransfected with reporter plasmids and 100 nmol/l chemically-modified miR-27a, miR-30b, and miR-124 mimics or miRNA-negative control (miR-con). The pRL-SV40 Renilla luciferase plasmid (Promega, Madison, WI) was used as an internal control. Two days later, cells were harvested and lysed with passive lysis buffer (Promega). Luciferase activity was measured using a dual luciferase reporter assay (Promega) and normalized by Renilla luciferase activity.
Western blot analysis. The cells were lysed in a buffer (50 mmol/l Tris–HCl, pH 7.4, containing 150 mmol/l NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, 0.1% deoxycholic acid, 10 mmol/l NaF, 10 mmol/l Na4P2O7, 0.4 mmol/l Na3VO4, and protease inhibitors) for 30 minutes on ice. The total protein content of the cells was determined by the bicinchoninic acid method (Pierce Biotechnology, Rockford, IL). Loading amounts were standardized to the middle protein concentration for all samples. Then the conditioned media was concentrated with 10% trichloroacetic acid precipitation. The precipitate was dissolved in a 1× sample buffer (62.5 mmol/l Tris-HCl, pH 6.8, containing 10% glycerol, 2% sodium dodecyl sulfate, and 0.005% bromophenol blue). Proteins (30 μg) were separated by 10% polyacrylamide gel electrophoresis containing 0.1% sodium dodecyl sulfate and transferred to nitrocellulose membranes (Schleicher and Schuell, Maidstone, UK). The membranes were incubated for 1 hour at room temperature in a blocking buffer (20 mmol/l Tris-HCl, 137 mmol/l NaCl, pH 8.0, containing 0.1% Tween and 3% nonfat dry milk), and probed with antibodies against Vangl1 (KITENIN, Atlas, St Louis, MO) and actin (Sigma, St Louis, MO). The blots were developed with peroxidase-conjugated secondary antibodies and reacted proteins were visualized using an electrochemiluminescence (ECL) system (Pierce Biotechnology).
Cell invasion assay. Cell invasion was measured by using the Transwell migration apparatus as described.12 Briefly, cultured cells pretreated with miR-con, miR-mimic, or miR-inhibitor (for 48 hours) were loaded into the top of a 24-well invasion chamber assay plate (Costar, Cambridge, UK). Conditioned Dulbecco's modified Eagle's medium (DMEM) containing 20 µg/ml fibronectin (Calbiochem, La Jolla, CA) was added to the bottom chamber as a chemoattractant. After 20 hours of incubation, the cells were stained and analysed. The cells on the top surface of the filters were wiped off with a cotton ball, and the migrated cells on the bottom surface were counted in six random squares of 0.5 mm × 0.5 mm for each filter. The results are represented as the mean ± SEM of the number of cells per field for at least three independent experiments.
Cell migration assay. Cells were plated at 80% confluence in DMEM supplemented with 10% FBS. At 24 hours after seeding, the monolayers were wounded by scoring with a sterile plastic 200 µl micropipette tip, washed, and then incubated in DMEM in the absence of serum. At vary hours, cells were photographed using a low-magnification fluorescence microscope.
Cell viability assay. The method for growth and survival of CRC cells is based on the colorimetric quantification of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Briefly, Caco2 cells were plated and cultured in 24-well plates (1 × 105 cells/well), and treated with miR-mimic or miR-con for 48 hours. The assay was performed as described.10
In vivo tumor growth. The experimental protocol was approved by the Chonnam National University Medical School Research Institutional Animal Care & Use Committee. Maintenance of animals and all in vivo experiments were performed according to the Guiding Principles in the Care and Use of Animals (DHEW publication, NIH 80-23).
A Tet-On vector (H1-miRNA plasmid) that expresses a ~100-base murine miR-27a, miR-30b, and miR-124 and the control empty vector were purchased from Genolution (Seoul, Korea). A pEGP-mmu-mir-miR-124 expression vector that expresses a ~100-base murine pre-miR-124 and the pEGP-mmu-mir-null vector were purchased from Cell Biolaps (San Diego, CA). Pseudovirus production and cell transduction were performed following the manufacturer's protocol. The resulting miRNA expressing-cells were selected through antibiotics and expression was confirmed by qPCR. Animal studies were performed in male Balb/c mice (5-week-old). Genetically engineered CT-26 cells (1 × 106 cells/mouse), which express Tet-On vector (H1-miR-27a, -30b, -124 plasmid) or pEGP-mmu-pre-miR-124 constitutively expression vector were subcutaneously injected into BALB/c syngeneic mice, and tumor size was measured as described.11 Two groups of syngeneic mice were prepared and were subcutaneously injected with CT-26/vector, or CT-26/miR cells. At the second week after injection, expressions of mature miRNA and KITENIN were measured by qPCR.
Quantitative RT-PCR and microarray. For quantitative expression analysis of miRNA, total RNA was isolated from fresh-frozen cultured cells or xenograft tumors using TRIzol reagent (Life Technologies, Carlsbad, CA). The mature miRNA level was measured using a TaqMan miRNA assay kit (Applied Biosystems, Foster City, CA) following the manufacturer's instruction and PCR amplification was carried out in a 7900HT Fast Real-time PCR system (Applied Biosystems). Real-time PCR was performed in triplicate for each sample. The abundance of miRNA was measured using Ct (threshold cycle) following the approach as described previously.11 ΔCt was calculated by subtracting the Ct of U6 RNA from the Ct of the miRNA tested. ΔΔCt was calculated by subtracting the ΔCt of a reference sample from the ΔCt of each sample. For clinical tissues, the reference sample is one CaP specimen tested that expresses the lowest level of miRNA. Fold change was generated using the equation 2−ΔΔCt.
Microarray analyses were done by using WG-6 expression v.2 bead array (Illumina, San Diego, CA) and the manufacturer's protocols. Total RNA was extracted from the isolated tumor tissues from both CT-26/miR-124 and CT-26/vector cell injection groups by using TRIzol (Invitrogen) followed by RNeasy (Qiagen, Santa Clarita, CA) to remove residual DNA.
Statistical analysis. Experimental differences were tested for statistical significance using analysis of variance followed by Tukey HSD post hoc test or Student's t-test. All statistical tests were two-sided, and P values of less than 0.05 were considered statistically significant. Statistics was performed with PASW Statistics 20 (SPSS) software.
SUPPLEMENTARY MATERIAL Figure S1. Identification of several microRNAs that decrease the expression of KITENIN. Figure S2. miR-27a, miR-30b, and miR-124 function as KITENIN-targeting microRNAs in CRC cells, which express these endogenous KITENIN-targeting microRNAs in measurable degrees. Figure S3. Identification of miR-124 as a MYH9-targeting microRNA. Table S1. Primer (Forward/Reverse) and microRNA specific sequences.
Acknowledgments
This work was supported by the National Research Foundation of Korea grant (MRC, 2011-0030132) funded by the Korea government (MSIP). The authors declared no conflict of interest.
Supplementary Material
Identification of several microRNAs that decrease the expression of KITENIN.
miR-27a, miR-30b, and miR-124 function as KITENINtargeting microRNAs in CRC cells, which express these endogenous KITENIN-targeting microRNAs in measurable degrees.
Identification of miR-124 as a MYH9-targeting microRNA.
Primer (Forward/Reverse) and microRNA specific sequences.
References
- Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- Fiorucci G, Chiantore MV, Mangino G, Percario ZA, Affabris E, Romeo G. Cancer regulator microRNA: potential relevance in diagnosis, prognosis and treatment of cancer. Curr Med Chem. 2012;19:461–474. doi: 10.2174/092986712798918798. [DOI] [PubMed] [Google Scholar]
- Al-Sohaily S, Biankin A, Leong R, Kohonen-Corish M, Warusavitarne J. Molecular pathways in colorectal cancer. J Gastroenterol Hepatol. 2012;27:1423–1431. doi: 10.1111/j.1440-1746.2012.07200.x. [DOI] [PubMed] [Google Scholar]
- Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer. 2009;8:102. doi: 10.1186/1476-4598-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetter AJ, Harris CC. Alterations of microRNAs contribute to colon carcinogenesis. Semin Oncol. 2011;38:734–742. doi: 10.1053/j.seminoncol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WK, Law PT, Lee CW, Cho CH, Fan D, Wu K, et al. MicroRNA in colorectal cancer: from benchtop to bedside. Carcinogenesis. 2011;32:247–253. doi: 10.1093/carcin/bgq243. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park SR, Chay KO, Seo YW, Kook H, Ahn KY, et al. KAI1 COOH-terminal interacting tetraspanin (KITENIN), a member of the tetraspanin family, interacts with KAI1, a tumor metastasis suppressor, and enhances metastasis of cancer. Cancer Res. 2004;64:4235–4243. doi: 10.1158/0008-5472.CAN-04-0275. [DOI] [PubMed] [Google Scholar]
- Lee JH, Cho ES, Kim MY, Seo YW, Kho DH, Chung IJ, et al. Suppression of progression and metastasis of established colon tumors in mice by intravenous delivery of short interfering RNA targeting KITENIN, a metastasis-enhancing protein. Cancer Res. 2005;65:8993–9003. doi: 10.1158/0008-5472.CAN-05-0590. [DOI] [PubMed] [Google Scholar]
- Kho DH, Bae JA, Lee JH, Cho HJ, Cho SH, Lee JH, et al. KITENIN recruits Dishevelled/PKC delta to form a functional complex and controls the migration and invasiveness of colorectal cancer cells. Gut. 2009;58:509–519. doi: 10.1136/gut.2008.150938. [DOI] [PubMed] [Google Scholar]
- Lee JK, Lim SC, Kim HD, Yoon TM, Kim K, Nam JH, et al. KITENIN represents a more aggressive phenotype in a murine model of oral cavity squamous carcinoma. Otolaryngol Head Neck Surg. 2010;142:747–52.e1. doi: 10.1016/j.otohns.2009.12.032. [DOI] [PubMed] [Google Scholar]
- Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagopoulos I, Isaksson M, Billström R, Strömbeck B, Mitelman F, Johansson B. Fusion of the NUP98 gene and the homeobox gene HOXC13 in acute myeloid leukemia with t(11;12)(p15;q13) Genes Chromosomes Cancer. 2003;36:107–112. doi: 10.1002/gcc.10139. [DOI] [PubMed] [Google Scholar]
- Quagliata L, Matter MS, Piscuoglio S, Arabi L, Ruiz C, Procino A, et al. lncRNA HOTTIP / HOXA13 expression is associated with disease progression and predicts outcome in hepatocellular carcinoma patients. Hepatology. 2014;59: 911–923. doi: 10.1002/hep.26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu ZD, Shen LY, Wang H, Chen XM, Li Y, Ning T, et al. HOXA13 promotes cancer cell growth and predicts poor survival of patients with esophageal squamous cell carcinoma. Cancer Res. 2009;69:4969–4973. doi: 10.1158/0008-5472.CAN-08-4546. [DOI] [PubMed] [Google Scholar]
- Han Y, Tu WW, Wen YG, Li DP, Qiu GQ, Tang HM, et al. Identification and validation that up-expression of HOXA13 is a novel independent prognostic marker of a worse outcome in gastric cancer based on immunohistochemistry. Med Oncol. 2013;30:564. doi: 10.1007/s12032-013-0564-1. [DOI] [PubMed] [Google Scholar]
- Gao FB. Context-dependent functions of specific microRNAs in neuronal development. Neural Dev. 2010;5:25. doi: 10.1186/1749-8104-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet. 2011;43:854–859. doi: 10.1038/ng.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derycke L, Stove C, Vercoutter-Edouart AS, De Wever O, Dollé L, Colpaert N, et al. The role of non-muscle myosin IIA in aggregation and invasion of human MCF-7 breast cancer cells. Int J Dev Biol. 2011;55:835–840. doi: 10.1387/ijdb.113336ld. [DOI] [PubMed] [Google Scholar]
- Grandjean V, Gounon P, Wagner N, Martin L, Wagner KD, Bernex F, et al. The miR-124-Sox9 paramutation: RNA-mediated epigenetic control of embryonic and adult growth. Development. 2009;136:3647–3655. doi: 10.1242/dev.041061. [DOI] [PubMed] [Google Scholar]
- Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. 2009;12:399–408. doi: 10.1038/nn.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetter AJ, Okayama H, Harris CC. The role of microRNAs in colorectal cancer. Cancer J. 2012;18:244–252. doi: 10.1097/PPO.0b013e318258b78f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael MZ, O' Connor SM, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- Ye JJ, Cao J. MicroRNAs in colorectal cancer as markers and targets: Recent advances. World J Gastroenterol. 2014;20:4288–4299. doi: 10.3748/wjg.v20.i15.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Ando T, Yoshida T, Enomoto S, Asada K, Tatematsu M, Ichinose M, et al. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: its possible involvement in the formation of epigenetic field defect. Int J Cancer. 2009;124:2367–2374. doi: 10.1002/ijc.24219. [DOI] [PubMed] [Google Scholar]
- Li KK, Pang JC, Ching AK, Wong CK, Kong X, Wang Y, et al. miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum Pathol. 2009;40:1234–1243. doi: 10.1016/j.humpath.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Wilting SM, van Boerdonk RA, Henken FE, Meijer CJ, Diosdado B, Meijer GA, et al. Methylation-mediated silencing and tumour suppressive function of hsa-miR-124 in cervical cancer. Mol Cancer. 2010;9:167. doi: 10.1186/1476-4598-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv XB, Jiao Y, Qing Y, Hu H, Cui X, Lin T, et al. miR-124 suppresses multiple steps of breast cancer metastasis by targeting a cohort of pro-metastatic genes in vitro. Chin J Cancer. 2011;30:821–830. doi: 10.5732/cjc.011.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Liao YJ, Cai MY, Liu YH, Liu TH, Chen SP, et al. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut. 2012;61:278–289. doi: 10.1136/gut.2011.239145. [DOI] [PubMed] [Google Scholar]
- Wang P, Chen L, Zhang J, Chen H, Fan J, Wang K, et al. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene. 2014;33:514–524. doi: 10.1038/onc.2012.598. [DOI] [PubMed] [Google Scholar]
- Wang MJ, Li Y, Wang R, Wang C, Yu YY, Yang L, et al. Downregulation of microRNA-124 is an independent prognostic factor in patients with colorectal cancer. Int J Colorectal Dis. 2013;28:183–189. doi: 10.1007/s00384-012-1550-3. [DOI] [PubMed] [Google Scholar]
- Pierson J, Hostager B, Fan R, Vibhakar R. Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J Neurooncol. 2008;90:1–7. doi: 10.1007/s11060-008-9624-3. [DOI] [PubMed] [Google Scholar]
- Hunt S, Jones AV, Hinsley EE, Whawell SA, Lambert DW. MicroRNA-124 suppresses oral squamous cell carcinoma motility by targeting ITGB1. FEBS Lett. 2011;585:187–192. doi: 10.1016/j.febslet.2010.11.038. [DOI] [PubMed] [Google Scholar]
- Xia J, Wu Z, Yu C, He W, Zheng H, He Y, et al. miR-124 inhibits cell proliferation in gastric cancer through down-regulation of SPHK1. J Pathol. 2012;227:470–480. doi: 10.1002/path.4030. [DOI] [PubMed] [Google Scholar]
- Shi XB, Xue L, Ma AH, Tepper CG, Gandour-Edwards R, Kung HJ, et al. Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene. 2013;32:4130–4138. doi: 10.1038/onc.2012.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ZB, Yang Z, Chi Y, Zhang L, Wang Y, Ji Y, et al. MicroRNA-124 suppresses breast cancer cell growth and motility by targeting CD151. Cell Physiol Biochem. 2013;31:823–832. doi: 10.1159/000350100. [DOI] [PubMed] [Google Scholar]
- Schoof CR, Botelho EL, Izzotti A, Vasques Ldos R. MicroRNAs in cancer treatment and prognosis. Am J Cancer Res. 2012;2:414–433. [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Maruyama R, Yamamoto E, Kai M. DNA methylation and microRNA dysregulation in cancer. Mol Oncol. 2012;6:567–578. doi: 10.1016/j.molonc.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T, Mayanagi T, Sobue K. Reorganization of the actin cytoskeleton via transcriptional regulation of cytoskeletal/focal adhesion genes by myocardin-related transcription factors (MRTFs/MAL/MKLs) Exp Cell Res. 2007;313:3432–3445. doi: 10.1016/j.yexcr.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Betapudi V, Licate LS, Egelhoff TT. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 2006;66:4725–4733. doi: 10.1158/0008-5472.CAN-05-4236. [DOI] [PubMed] [Google Scholar]
- Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol. 2006;16:1515–1523. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11:257–268. doi: 10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, He L, Zhao X, Miao Y, Gu Y, Guo C, et al. MicroRNA let-7f inhibits tumor invasion and metastasis by targeting MYH9 in human gastric cancer. PLoS One. 2011;6:e18409. doi: 10.1371/journal.pone.0018409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaziel-Sovran A, Segura MF, Di Micco R, Collins MK, Hanniford D, Vega-Saenz de Miera E, et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer Cell. 2011;20:104–118. doi: 10.1016/j.ccr.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Liang L, Huang S, Ding J, Tan N, Zhao Y, et al. MicroRNA-30d promotes tumor invasion and metastasis by targeting Galphai2 in hepatocellular carcinoma. Hepatology. 2010;51:846–856. doi: 10.1002/hep.23443. [DOI] [PubMed] [Google Scholar]
- Yu F, Deng H, Yao H, Liu Q, Su F, Song E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene. 2010;29:4194–4204. doi: 10.1038/onc.2010.167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identification of several microRNAs that decrease the expression of KITENIN.
miR-27a, miR-30b, and miR-124 function as KITENINtargeting microRNAs in CRC cells, which express these endogenous KITENIN-targeting microRNAs in measurable degrees.
Identification of miR-124 as a MYH9-targeting microRNA.
Primer (Forward/Reverse) and microRNA specific sequences.