

Abstract
Spinocerebellar ataxia type 7 (SCA7) is a late-onset neurodegenerative disease characterized by ataxia and vision loss with no effective treatments in the clinic. The most striking feature is the degeneration of Purkinje neurons of the cerebellum caused by the presence of polyglutamine-expanded ataxin-7. Ataxin-7 is part of a transcriptional complex, and, in the setting of mutant ataxin-7, there is misregulation of target genes. Here, we designed RNAi sequences to reduce the expression of both wildtype and mutant ataxin-7 to test if reducing ataxin-7 in Purkinje cells is both tolerated and beneficial in an animal model of SCA7. We observed sustained reduction of both wildtype and mutant ataxin-7 as well as a significant improvement of ataxia phenotypes. Furthermore, we observed a reduction in cerebellar molecular layer thinning and nuclear inclusions, a hallmark of SCA7. In addition, we observed recovery of cerebellar transcripts whose expression is disrupted in the presence of mutant ataxin-7. These data demonstrate that reduction of both wildtype and mutant ataxin-7 by RNAi is well tolerated, and contrary to what may be expected from reducing a component of the Spt-Taf9-Gcn5 acetyltransferase complex, is efficacious in the SCA7 mouse.
Introduction
Spinocerebellar ataxia type 7 (SCA7) is an autosomal dominant neurodegenerative disease and one of nine known polyglutamine (polyQ) diseases. SCA7 patients suffer from loss of vision and motor coordination including dysarthria, dysphagia, and slower reflexes.1 Anticipation is a feature of this disease, reflected in earlier onset in offspring of affected individuals.2 Currently, there are no disease-modifying treatments for SCA7.
SCA7 is caused by an expansion of ≥37 CAG repeats in exon 3 of the ataxin-7 gene (ATXN7).3 While patients with repeats <58 present with cerebellar and brainstem degeneration prior to exhibiting retinal disease, patients with >59 CAG repeats typically present with retinal degeneration and then develop cerebellar ataxia.4 Ataxin-7 is ubiquitously expressed, yet only specific neuronal populations are affected. The most prominent features are the loss of the Purkinje cells (PCs) and the loss of brainstem neurons in the inferior olive (IO) that project to the molecular layer.5
A hallmark of SCA7 is the presence of nuclear inclusions (NIs), which contain aggregates of mutant polyQ expanded ATXN7 and other ubiquitin-proteasome components.6 ATXN7 is a core component of the Spt-Taf9-Gcn5 acetyltransferase complex,7 a transcriptional coactivator complex that confers histone acetyltransferase activity to its target genes. Mutant polyQ ATXN7 alters Spt-Taf9-Gcn5 acetyltransferase recruitment to target genes, and disrupts its activity.8 Thus, reducing the levels of mutant polyQ ATXN7 could reduce the downstream effects induced by mutant ATXN7.
Previous studies have shown that ~50% reduction of mutant ataxin-7 in a SCA7 mouse model using Cre-medicated excision early after disease onset, alleviates motor symptoms.9 In human patients, targeting a disease associated SNP is one way to specifically reduce mutant ATXN7, leaving the wild-type protein intact. Taking advantage of a SNP on mutant ATXN7 found in 50% of South African SCA7 patients, Scholefield et al. designed allele-specific short hairpin RNAs (shRNAs) to reduce mutant ataxin-7 and accomplished allele-specific decreases in ATXN7 aggregate formation in vitro.10 However, targeting mutant alleles via SNPs is impractical from a drug development perspective. We therefore set out to test if partial reduction of both the mutant and normal alleles could be beneficial in the setting of SCA7.
The BAC Prp SCA7-92Q mouse model expresses human mutant ataxin-7 cDNA containing 92 pathogenic CAG repeats.11 The onset of PC degeneration is ~23 weeks of age, after which motor abnormalities become apparent. Ataxia is progressive and becomes severe as seen by clasping, rotarod, and gait deficits and worsening scores on the ledge test.9,12 Here, we show in proof-of-concept studies that nonallele specific silencing of ataxin-7 by RNAi is safe and well tolerated long-term in the SCA7 mouse. We observed a delayed disease onset, significant improvement of the ataxic and molecular phenotypes, and dramatic reduction in NIs in cerebellar PCs with the recovery of cerebellar transcripts that are known to be dysregulated in this disease.
Results
Silencing ataxin-7 expression
Small interfering RNAs targeting ataxin-7 were generated using a low off-target prediction algorithm13 and were cloned into an artificial miRNA expression vector.14 Plasmids expressing the candidate miRNAs were tested for their ability to reduce ataxin-7 expression in vitro, and one construct, miS4 was chosen for further evaluation in vivo (data not shown). To target the mouse cerebellar PCs, cassettes directing expression of miS4 and a control sequence, miC, were subsequently cloned into adeno-associated viral (AAV) shuttle vectors to generate AAV2/1 viruses also expressing eGFP for visualization of transduced cells (Figure 1a). AAV2/1 has been previously reported to transduce cerebellar PCs.15
Figure 1.

Efficient cerebellar transduction and reduction of ataxin-7 expression in vivo. (a) Cartoon of the AAV construct. The U6 promoter drives expression of the artificial miRNA, followed by a cytomegalovirus promoter driving expression of eGFP. (b) Sagittal section of miS4 SCA7 cerebellum showing transduction of the Purkinje cells (PC) and deep cerebellar nuclei. (c) miS4 expression validated by stem-loop PCR after RT reaction with miS4 specific primers in cerebellar extracts at 40 weeks. miC was used as a negative control. (d) Relative human (left panel) and mouse (right panel) ataxin-7 mRNA levels at 40 weeks from cerebellar tissue analyzed by RT-qPCR. Results are represented as mean ± SEM (n = 8 saline; n = 4 miC, n = 8 miS4; n = 7 wildtype), ***P < 0.001. (e) Western blot to assess ataxin-7 protein knockdown in the cerebellum of miS4-injected mice relative to miC. Arrow denotes human ataxin-7. Right panel, quantitation of knockdown by densitometry. (n = 3), **P < 0.01
We injected AAV2/1-miS4, AAV2/1-miC or saline into the deep cerebellar nucleus of the BAC Prp SCA7-92Q mouse to target human mutant ataxin-7 expression in cerebellar PCs. AAV2/1 injections into the deep cerebellar nucleus resulted in trafficking of the viral particles to the PC cell bodies and dendrites by retrograde transport.15,16,17 We observed widespread transduction of the PCs and the deep cerebellar nuclei as observed by eGFP expression (Figure 1b), and expression of the S4 RNAi trigger by stem loop PCR (Figure 1c). To assess the long-term effects of nonallele specific silencing of ataxin-7, we injected presymptomatic BAC Prp SCA7-92Q mice at 7 weeks and followed them until 40 weeks of age (SCA7 mice die at ~50 weeks of age). For this, SCA7 mice were injected bilaterally into the deep cerebellar nucleus with saline, AAV2/1-miC or AAV2/1-miS4. Untreated wild-type littermates were used for assessing relative recovery of disease phenotypes. At 33 weeks postinjection (40 weeks of age), we observed sustained reduction in both human and mouse ataxin-7 transcript levels (~50%, P < 0.001; Figure 1d) and overall protein levels (~35%, P < 0.01; Figure 1e).
miS4 improves motor phenotypes
We evaluated SCA7 mice motor phenotypes at baseline (6 weeks; data not shown) and postinjection at 25 and 40 weeks of age (Figure 2a). The polyQ SCA-7 disease mice clasp their hindlimbs when suspended by the tail, unlike wild-type mice, which splay their hindlimbs.15,18,19 The BAC PrP SCA7-92Q mouse also exhibits hindlimb clasping relative to their wild-type littermates,9 but in miS4-treated animals, there was a significant improvement in this phenotype, while saline or miC injected SCA7 mice continued to exhibit severe clasping (P < 0.001; Figure 2b). miS4-treated mice also improved on the ledge test, while untreated or control treated SCA7 mice showed progressive decline, as reported earlier9,20 (P < 0.001; Figure 2c).
Figure 2.
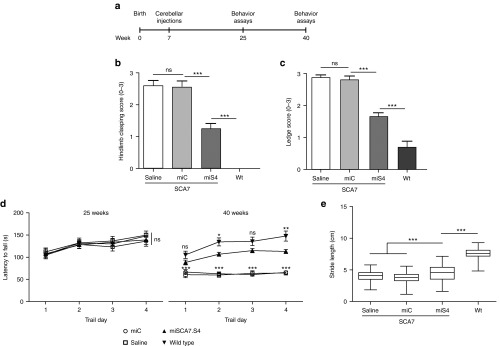
miS4 treatment improves ataxia phenotypes. (a) Experimental scheme for long-term cerebellar studies. (b) Hindlimb clasping score at 40 weeks, where 0 represents no clasping and 3 represents severe clasping. Results are represented as mean ± SEM (n = 10 saline; n = 13 miC, n = 14 miS4; n = 15 wildtype), ***P < 0.001. (c) Ledge score at 40 weeks, where 0 represents good balance and coordination and 3 represents very poor or no balance and coordination. Results are represented as mean ± SEM (n = 10 saline; n = 13 miC, n = 14 miS4; n = 17 wildtype), ***P < 0.001. (d) Rotarod analysis at 25 and 40 weeks with the Y-axis representing the latency to fall. Results are represented as mean ± SEM (n = 10 saline; n = 13 miC, n = 14 miS4; n = 15 wildtype), ***P < 0.001, **P < 0.01; two-way ANOVA with Bonferroni posthoc tests. (e) Box plots representing stride length at 40 weeks measured by footprint analysis. The median of 15–20 steps per mouse were included in the analysis (n = 10–13 mice per group), ***P < 0.001.
Motor skills and gait assays were also assessed, as BAC Prp SCA7-92Q mice exhibit progressive deficits in these tasks.9 At 25 weeks, there was no difference in rotarod performance between the SCA7 and wild-type mice, but at 40 weeks, miS4-treated mice had a significantly improved rotarod phenotype compared to control-treated mice and performed at levels closer to wildtype on day 4 (Figure 2d; P < 0.05 or P < 0.01). miS4 mice also demonstrated a significantly improved stride length relative to control-treated SCA7 mice (Figure 2e; P < 0.001), however, wild-type mice did significantly better relative to all groups (Figure 2e; P < 0.001).
Reduction of ataxin-7 in the PCs does not induce toxicity
As one measure of assessing the tolerability of reducing both wild-type and mutant ataxin-7 expression, we evaluated injected cerebella for microglial activation at 40 weeks using Iba-1 immunohistochemistry. We found no notable increase in microglia activation in miS4 tissue sections relative to controls (Figure 3a). We also quantified the expression of glial fibrillary acidic protein (GFAP), an astrocyte marker whose expression increases in the presence of injury or inflammation. Untreated and control-treated SCA7 mice exhibit higher mRNA (~25%, P < 0.05;) and protein (~50%, P < 0.01) levels of GFAP relative to wild-type tissues,21 while GFAP levels improved in miS4-treated cerebella and were no longer significantly different to those in wild-type animals(Figure 3b, Supplementary Figure S1a,b). Thus, miS4 treatment did not exacerbate glial activation, but rather improved this progressive SCA7 neuroinflammation phenotype.
Figure 3.
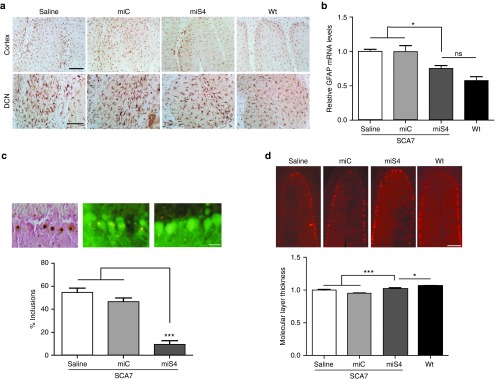
Histological and QPCR analysis of cerebellar tissue at 40 weeks. (a) Representative photomicrograph showing Iba-1 immunoreactivity in the DCN and cortex of the cerebellum (n = 3 mice per group). (b) Relative GFAP mRNA levels in the injected cerebella by QPCR analysis. Results are represented as mean ± SEM (n = 3 per group), *P < 0.05. (c) Representative photomicrographs showing the extent of nuclear inclusions observed by immunohistochemistry for ataxin-7 in saline injected (left panel), or in eGFP+ AAV.miC (middle panel) and AAV.miS4-treated animals (right panel). Quantitation was done as described in the methods (lower panel). Results are represented as mean ± SEM (n = 3 per group), ***P < 0.001. (d) Representative photomicrographs of cerebellar lobules from SCA7 mice treated as indicated, or from untreated Wt mice. Below, quantitation of molecular layer thickness in the various groups. All groups were normalized to saline-treated SCA7 mice. Results are represented as mean ± SEM (n = 3 per group), ***P < 0.001, *P < 0.05.
miS4 has a therapeutic impact on the SCA7 cerebellum
We next assessed the effect of ataxin-7 gene silencing on NIs. NIs are a hallmark of SCA7 and other polyQ diseases and cells laden with NIs increase in number with progressing disease.9 We scored NIs, detected by immunohistochemistry for ataxin-7, in miC- and miS4-transduced PCs at 40 weeks, and compared these to the numbers of inclusions in matched lobules from saline-treated or wild-type mice. miS4-transduced PCs had a dramatic ~80% reduction in NIs relative to miC or saline-treated PCs (P < 0.001) supporting decreased levels of nuclear ataxin-7 protein (Figure 3c).
BAC Prp SCA7-92Q mice show progressive cerebellar lobule thinning.9 To assess potential benefit from ataxin-7 silencing, the molecular layer width was measured in miS4 versus control-treated cerebellar lobules at 40 weeks. miS4-treated mice showed improved cerebellar morphology (increase of ~10%, P < 0.001) than control-treated SCA7 mice (Figure 3d). Wild-type mice retained significantly larger molecular layer widths compared to all groups.
Lastly, we assessed if the reduction of mutant ataxin-7 affected cerebellar transcript levels that have been shown in earlier work to be dysregulated in cell and animal models of SCA7.21,22,23,24 One example is calbindin1, a PC specific marker that is also downregulated in other SCA mice.25,26 Here, we found that calbindin1 mRNA levels were reduced by ~40% in the BAC PrP SCA7-92Q mice relative to wild-type mice (P < 0.001). miS4 treatment partially restored calbindin1 mRNA and protein to wild-type levels (P < 0.05 and P < 0.01, respectively) and were significantly (~20%) higher than those of control-treated SCA7 mice (P < 0.01) (Figure 4a, Supplementary Figure S1a,c). In addition to calbindin1, the insulin-like growth factor pathway was shown to be dysregulated in a knock-in model of SCA7, with Igfbp5 mRNA levels downregulated relative to normal.22 Igfbp5 transcript levels were also lower in BAC PrP SCA7-92Q mice relative to wildtype (Figure 4e; P < 0.05). These transcripts trended upward with miS4 expression, but were not significantly improved relative to control-treated SCA7 mice. Expression of Eaat4, a PC-expressed glutamate transporter, is reduced in SCA7-90Q mice,24 and miS4 treatment restored Eaat4 mRNA and protein levels to normal levels (Figure 4d, Supplementary Figure S1a,d; P < 0.05). Other cerebellar specific transcripts such as Pcp2, Reln, Itpr1, Glast, and Grid2, which are downregulated in other SCA7 models,21,23,24,27 were not reduced in the BAC PrP SCA7-92Q mice, and there were no effects of treatment on their expression levels (Figure 4b,c,f–h).
Figure 4.
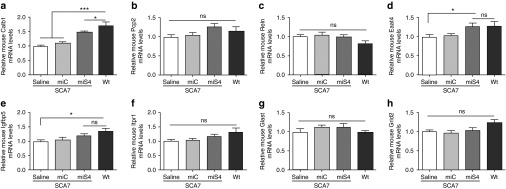
Changes in cerebellar transcripts postinjection at 40 weeks. RT-qPCR experiments representing the relative mRNA expression levels of mouse (a) Calb1, (b) Pcp2, (c) Reln, (d) Eaat4, (e) Igfbp5, (f) Itpr1, (g) Glast, and (h) Grid2. Results are represented as mean ± SEM (n = 6 saline; n = 6 miC, n = 6 miS4; n = 4 wildtype), *P < 0.05; ***P < 0.001.
Discussion
In this study, we provide evidence that nonallele specific silencing of ataxin-7 by RNAi is well tolerated in a SCA7 mouse model. Earlier studies in SCA7 mice demonstrated that excision of ~50% mutant ataxin-7 expression via Cre endonuclease immediate to disease onset partially alleviated ataxic symptoms.9 Here, we observed significant and robust improvements in the ataxic and neuropathological phenotypes as well as a delayed disease onset in SCA7 mice upon sustained reduction of both mutant and wildtype ataxin-7 expression in the cerebellar PCs
SCA7 is a core component of the Spt-Taf9-Gcn5 acetyltransferase co-activator complex, which confers Gcn5-mediated histone acetyltransferase activity when recruited to its target genes.28 Mutant ATXN7 causes a deregulation of histone acetyltransferase activity and decondensation of chromatin, interfering with target gene activation.8 While other alleles causative of SCA result in viable mice upon gene knock-out,29,30,31 it is not known if knock out of ataxin-7 expression is detrimental for survival. Thus it was important to understand the consequences of reducing the levels of ataxin-7 expression in vivo. Here, we found that reducing both mutant and wild-type ataxin-7 expression decreased neuroinflammation, improved ataxin-7-induced neuropathology and had a positive impact on dysregulated transcription. Together these data suggest that partial reduction of ataxin-7 expression by RNAi was tolerated and beneficial in the context of SCA7.
NIs are a hallmark of SCA7 and consist of the truncated mutant ATXN7 protein along with ubiquitin components that increasingly accumulate with age.32,33,34 In primary rat embryonic neuronal cultures, ATXN7-100Q expression caused toxicity and neuronal cell death.35 Although it is debated as to whether NIs per se are protective or toxic in vivo in SCA7, reducing levels of misfolded polyQ-containing proteins should result in decreased NIs and aggregation of mutant polyQ proteins in the nucleus. Vos et al. demonstrated that overexpressing the heat shock protein HSPB7, a potent suppressor of polyQ aggregation, decreased polyQ induced toxicity and rescued retinal degeneration in a Drosophila polyQ disease model.36 Another study in mice showed that reducing nuclear accumulation of mutant ATXN3 using calpain inhibitors reduced neuronal degeneration and dysfunction.37 Similarly, induction of PGC-1α expression in HD mice virtually eliminated huntingtin protein aggregation in the CNS and yielded a marked rescue of motor function.38 Here, we observed a dramatic ~80% reduction in the number of NIs in the miS4-treated SCA7 mice, confirming a decrease in mutant protein aggregation in PC nuclei. Thus, reducing nuclear-resident mutant ATXN7 likely contributes to the beneficial effects that we and others have noted.17,39
RNAi therapy in other polyQ diseases have shown that sustained reduction of mutant polyQ alleles can alleviate disease phenotypes. RNAi-induced knockdown of the mutant SCA1 allele in PCs resulted in a complete rescue of motor and neuropathological phenotypes in a PC specific and a knock-in model of SCA1.15,40 On the other hand, RNAi-induced knockdown of the mutant SCA3 allele in PCs resulted in a rescue of the neuropathological phenotypes but surprisingly, no rescue of motor phenotypes in a YAC model.17,41 The robust rescue of motor phenotypes in SCA1 and lack of rescue in SCA3 could arise from the fact that SCA1, like SCA7, primarily affects the PCs, while in SCA3, significant neuronal loss occurs in the substantia nigra and basal ganglia,42,43 areas that were not targeted in their study. In other work, knockdown of mutant ATXN3 in the PCs in a PC specific SCA3 mouse model rescued motor and neuropathological phenotypes,44 while knockdown in the striatum of a striatal specific rat model improved neuropathology,45 suggesting that RNAi therapy for SCA3 has potential if applied to the appropriate affected areas in combination. In our study, we used a BAC transgenic model with ubiquitous mutant ATXN7 expression. Upon RNAi treatment, there was significant improvement in motor phenotypes and a delay in disease onset. In addition, we observed partial protection from the noted cerebellar molecular layer thinning and normalization of transcripts altered in SCA7. Our main target was cerebellar PCs, but miS4 injected mice also expressed miS4 in the brainstem with modest reduction (~20%, *P < 0.05) of ataxin-7 transcripts in that region (Supplementary Figure S2a), while there was no change in ataxin-7 mRNA levels in the striatum or thalamus (Supplementary Figure S2b,c). Recent work by Furrer et al. showed that the Bergman glia, PCs, and the IO together contribute to the pathology of this mouse model.11 They crossed mice with a floxed mutant ataxin-7 allele to a Cre-Pcp2 line for Cre-mediated excision in PCs and the IO, and showed partial improvement in behavior phenotypes.11 However, when the transgene was excised from the Bergman glia, PCs and IO, a delayed disease onset and more robust improvements in behavior were seen.11 We did not observe complete rescue of motor phenotypes and neuropathology upon presymptomatic RNAi expression, possibly due to the fact that the PCs were the major site of knock-down and that treatment of the Bergman glia and more robust knock down in IO is required.
SCA7 is unique among the polyQ diseases as mutant ataxin-7 causes vision loss and ataxia. While other studies have demonstrated partial improvement of some SCA7 motor phenotypes by overexpression of HGF46 and IFN-β,39 here, we chose to target the causative gene directly and assess therapeutic efficacy. It will be interesting in future work to assess the effects of RNAi therapy for SCA7 retinal disease.
Materials and Methods
Animals. The University of Iowa Animal Care and Use Committee (IACUC) approved all animal protocols. PrP-floxed-SCA7-92Q BAC transgenic mice were generated in the La Spada lab and were maintained on the C57BL/6J background. Mice were genotyped using primers specific for the mutant human ataxin-7 transgene.9 Hemizygous and age-matched wild type littermates were used for the experiments. In the therapeutic trials, the treatment groups comprised approximately equal numbers of male and female mice. Mice were housed in a controlled temperature environment on a 12-hour light/dark cycle. Food and water were provided ad libitum.
Viral vectors. The plasmids expressing mouse U6-driven artificial miRNAs (S1-S4, HS5, mm, and C) were cloned as previously described using DNA oligonucleotides.14 U6 is an empty vector control, C (miC), and mm are scrambled RNAi control sequences, S1-S5 were sequences designed to target both mutant and wild-type ataxin-7, while HS5 was designed to target human ataxin-7.10 The following primers were used to construct miS4—S4 forward primer: AAAACTCGAGTGAGCGGGGCTCAGGAAAGAAACGCAAACTGTAAAGCCACAGATGGG, S4 Reverse Primer: AAAAACTAGTAGGCGCGGCTCAGGAAAGAAACGCAAACCCATCTGTGGCTTTACAG). Artificial miRNA expression cassettes were cloned into pAAVmcsCMVeGFP plasmids which co-expressed CMV-driven eGFP.14 Recombinant AAV serotype 2/1 vectors (AAV.miC.eGFP and AAV.miS4.eGFP) were generated by the University of Iowa Vector Core facility, as previously described.47 AAV vectors were dialyzed and resuspended in Formulation Buffer 18 (University of Iowa Gene Transfer Vector Core, Iowa City, IA) and titers (viral genomes/ml) were determined by RT-qPCR.
AAV injections and tissue harvesting. SCA7 transgenic mice were injected with AAV vectors as previously reported.48 For all cerebellar studies, transgenic mice were injected bilaterally into the deep cerebellar nuclei (coordinates −6.0 mm caudal to bregma, ± 2.0 mm from midline, and −2.2 mm deep from cerebellar surface) with 4 μl of AAV1 virus (at 4.26 × 1012 viral genomes/ml) or saline. Following the injections, a topical antibiotic ointment was applied and the mice were allowed to recover according to the University of Iowa Animal Care and Use Committee's (IACUC) guidelines for Postanesthesia monitoring, including monitoring of breathing and muscle tone. To harvest the cerebella, mice were killed with a ketamine/xylazine mix and transcardially perfused with 20 ml of cold saline. Mice were decapitated, and for histological analyses, brains were removed and postfixed overnight in 4% paraformaldehyde. Brains were stored in a 30% sucrose/0.05% azide solution at 4 °C and cut on a sliding knife microtome at 45 μm thickness and stored at −20 °C in a cryoprotectant solution. For RT-qPCR analyses, brains were removed, sectioned into 1-mm thick coronal slices using a brain matrix (Roboz, Gaithersburg, MD) and eGFP expression was verified. Total RNA was isolated from whole cerebellum using the TRIzol (Life Technologies, Grand Island, NY) extraction. RNA quantity and quality were measured using a NanoDrop ND-1000 (NanoDrop, Wilmington, DE).
Immunohistochemistry and western blotting. Free-floating sagittal cerebellar sections (45 μm thick) were washed in 1× phosphate-buffered saline at room temperature and blocked for 1 hour in 5% serum, 0.03% TritonX100 in 1× PBS. Sections were incubated with primary antibody in 3% serum and 0.03% TritonX in 1× PBS overnight at 4 °C. Primary antibodies used for IHC and western blotting were polyclonal rabbit anti-Iba1 (1:1000; WAKO, Richmond, VA), polyclonal rabbit anti-Calbindin (1:2000; Cell Signaling Technology, Danvers, MA), polyclonal rabbit anti-ATXN7 (1:1000; #PA1-749, Thermo Fisher Scientific, Rockford, IL), anti-EAAT4 (1:2000, Ab41650; Abcam, Cambridge, MA) GFAP (1:1000, zo334, Dako, Glostrup, Denmark), anti-myc antibody (1:1000, Cell Signaling Technology, Danvers, MA) and β actin (1:10000, A5441, Sigma-Aldrich, St Louis, MO), and quantification was performed using Image J software (Rasband, W.S., Image J, U.S. National Institutes of Health, Bethesda, MD). For fluorescent IHC, sections were incubated with goat antirabbit Alexa Fluor 568 (1:1000; Life Technologies) in 3% serum and 0.03% Triton-100 in 1× PBS for 1 hour at room temperature. For 3,3′ diaminobenzidine immunohistochemical staining, sections were incubated in goat antirabbit biotin-labeled secondary antibody (1:200; Jackson Immunoresearch, West Grove, PA) in 3% serum and 0.03% Triton-X at room temperature for 1 hour. Tissues were developed with VECTASTAIN ABC Elite Kit (Vector Laboratories, Burlingame, CA), according to the manufacturer's instructions. All sections were mounted onto Superfrost Plus slides (Fischer Scientific, Pittsburgh, PA) and cover slipped with Fluoro-Gel (Electron Microscopy Sciences, Hatfield, PA). Images were captured on Leica Leitz DMR fluorescence microscope connected to an Olympus DP72 camera using the Olympus DP2-BSW software (Olympus, Melville, NY).
RT-qPCR and stem loop PCR. First-strand cDNA synthesis was performed using 1 μg total RNA (High Capacity cDNA Reverse Transcription Kit; Life Technologies) as per manufacturer's instructions. RT-qPCR assays were performed on a sequence detection system using primers/probe sets specific for human or mouse ataxin-7, mouse GFAP, mouse Calbindin, Pcp2, Reln, Itpr1, Glast, Grid2, Eaat4, Igfbp5, or mouse β-actin (ABI Prism 7900 HT, TaqMan 2Xuniversal master mix and power SYBR green PCR master mix;, Life Technologies). RT-qPCR values were normalized to mouse β-actin. Stem loop PCR was performed as describer earlier.49 Briefly, PCR primers were designed to identify miS4. Reverse transcription was performed with RT-specific primers (S4:GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGGCTCA) using the High Capacity cDNA Reverse Transcription Kit; Life Technologies and cDNA obtained was subject to PCR using a specific forward primer (S4 Fwd: GCCCTTTGCGTTTCTTTCC) and a reverse primer (5′ GTGCAGGGTCCGAGGT).
Behavior analysis. All behavior assays were performed at 6, 25, and 40 weeks of age and 25- and 40-week data are presented here as means ± SEM unless otherwise specified. For all studies, P values were obtained by using one-way analysis of variance followed by Bonferroni posthoc analysis to assess for significant differences between individual groups, unless indicated otherwise. In all statistical analyses, P < 0.05 was considered significant.
Ledge test and hindlimb clasping assays and their scoring parameters are detailed previously.12 To take stride length measurements, mice were allowed to walk across a paper-lined chamber (100 cm × 10 cm with 10 cm walls) and into an enclosed recess. Nontoxic red and blue paint was applied to their fore- and hind paws, respectively. Mice were then tested three times to produce three separate footprint tracings. Stride lengths were measured from the middle of each paw print between the same paws for steps taken during their gait. Steps were discarded in instances where a mouse stopped walking or turned around. Measurements were averaged for all four paws.
Rotarod mice were tested on an accelerated rotarod apparatus (model 47600; Ugo Basile, Comerio, Italy). Baseline testing was conducted at 6 weeks of age to separate SCA7 mice equally into treatment groups. No difference between SCA7 and wild-type mice was seen at 4 weeks of age (data not shown). Mice were first habituated on the rotarod for 4 minutes. Mice were then tested three trials per day (with at least 30 minutes of rest between trials) for four consecutive days. For each trial, acceleration was from 4 to 40 rpm over 5 minutes, and then speed maintained at 40 rpm. Latency to fall (or if mice hung on for two consecutive rotations without running) was recorded for each mouse per trial. The trials were stopped at 400 seconds, and mice remaining on the rotarod at that time were scored as 400 seconds. Two-way analysis of variance followed by Bonferroni posthoc analysis was used to assess for significant differences. Variables were time and treatment.
Figure preparation. All photographs were formatted with Adobe Photoshop software (Adobe Systems, San Jose, CA), all graphs were made with Prism GraphPad software (GraphPad Software, La Jolla, CA), and all figures were constructed with Adobe Illustrator software (Adobe Systems).
SUPPLEMENTARY MATERIAL Figure S1. Recovery of cerebellar proteins at 40 weeks of age. Figure S2. Mutant ataxin-7 mRNA levels in the SCA7 brain at 40 weeks of age.
Acknowledgments
This work was funded by the National Ataxia Foundation (B.L.D. and P.S.R.), the Roy J Carver Trust (B.L.D.) and the National Institutes of Health (R01 HD44093 and P01 NS50210 to BLD; R21 NS082112 and R01 EY014061 to ARL). B.L.D. is a co-founder of Spark Therapeutics, a gene therapy company.
Supplementary Material
Recovery of cerebellar proteins at 40 weeks of age.
Mutant ataxin-7 mRNA levels in the SCA7 brain at 40 weeks of age.
References
- Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12:2–15. doi: 10.1038/sj.ejhg.5201108. [DOI] [PubMed] [Google Scholar]
- Whitney A, Lim M, Kanabar D, Lin JP. Massive SCA7 expansion detected in a 7-month-old male with hypotonia, cardiomegaly, and renal compromise. Dev Med Child Neurol. 2007;49:140–143. doi: 10.1111/j.1469-8749.2007.00140.x. [DOI] [PubMed] [Google Scholar]
- Michalík A, Del-Favero J, Mauger C, Löfgren A, Van Broeckhoven C. Genomic organisation of the spinocerebellar ataxia type 7 (SCA7) gene responsible for autosomal dominant cerebellar ataxia with retinal degeneration. Hum Genet. 1999;105:410–417. doi: 10.1007/s004390051123. [DOI] [PubMed] [Google Scholar]
- Johansson J, Forsgren L, Sandgren O, Brice A, Holmgren G, Holmberg M. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet. 1998;7:171–176. doi: 10.1093/hmg/7.2.171. [DOI] [PubMed] [Google Scholar]
- Garden GA, La Spada AR. Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum. 2008;7:138–149. doi: 10.1007/s12311-008-0027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg M, Duyckaerts C, Dürr A, Cancel G, Gourfinkel-An I, Damier P, et al. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet. 1998;7:913–918. doi: 10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci USA. 2005;102:8472–8477. doi: 10.1073/pnas.0503505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmüller A, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer SA, Waldherr SM, Mohanachandran MS, Baughn TD, Nguyen KT, Sopher BL, et al. Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet. 2013;22:890–903. doi: 10.1093/hmg/dds495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholefield J, Greenberg LJ, Weinberg MS, Arbuthnot PB, Abdelgany A, Wood MJ. Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype. PLoS One. 2009;4:e7232. doi: 10.1371/journal.pone.0007232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer SA, Mohanachandran MS, Waldherr SM, Chang C, Damian VA, Sopher BL, et al. Spinocerebellar ataxia type 7 cerebellar disease requires the coordinated action of mutant ataxin-7 in neurons and glia, and displays non-cell-autonomous bergmann glia degeneration. J Neurosci. 2011;31:16269–16278. doi: 10.1523/JNEUROSCI.4000-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden and GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010;39:e1787. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Spengler RM, Hylock RH, Kusenda BJ, Davis HA, Eichmann DA, et al. siSPOTR: a tool for designing highly specific and potent siRNAs for human and mouse. Nucleic Acids Res. 2013;41:e9. doi: 10.1093/nar/gks797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Davidson BL. Generation of hairpin-based RNAi vectors for biological and therapeutic application. Methods Enzymol. 2012;507:275–296. doi: 10.1016/B978-0-12-386509-0.00014-4. [DOI] [PubMed] [Google Scholar]
- Keiser MS, Geoghegan JC, Boudreau RL, Lennox KA, Davidson BL. RNAi or overexpression: alternative therapies for Spinocerebellar Ataxia Type 1. Neurobiol Dis. 2013;56:6–13. doi: 10.1016/j.nbd.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med. 2004;10:816–820. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Lebrón E, Costa Md, Luna-Cancalon K, Peron TM, Fischer S, Boudreau RL, et al. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol Ther. 2013;21:1909–1918. doi: 10.1038/mt.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou AH, Yeh TH, Ouyang P, Chen YL, Chen SY, Wang HL. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol Dis. 2008;31:89–101. doi: 10.1016/j.nbd.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Auerbach W, Hurlbert MS, Hilditch-Maguire P, Wadghiri YZ, Wheeler VC, Cohen SI, et al. The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum Mol Genet. 2001;10:2515–2523. doi: 10.1093/hmg/10.22.2515. [DOI] [PubMed] [Google Scholar]
- Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, et al. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–4905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, Watase K, Thaller C, Carson JP, Jafar-Nejad P, Shaw C, et al. The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc Natl Acad Sci U S A. 2008;105:1291–1296. doi: 10.1073/pnas.0711257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough SD, Xu X, Dent SY, Bekiranov S, Roeder RG, Grant PA. Reelin is a target of polyglutamine expanded ataxin-7 in human spinocerebellar ataxia type 7 (SCA7) astrocytes. Proc Natl Acad Sci USA. 2012;109:21319–21324. doi: 10.1073/pnas.1218331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich B, Euler P, Ziegler R, Kuhn A, Landwehrmeyer BG, Luthi-Carter R, et al. Comparative analyses of Purkinje cell gene expression profiles reveal shared molecular abnormalities in models of different polyglutamine diseases. Brain Res. 2012;1481:37–48. doi: 10.1016/j.brainres.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–283. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ström AL, Forsgren L, Holmberg M. A role for both wild-type and expanded ataxin-7 in transcriptional regulation. Neurobiol Dis. 2005;20:646–655. doi: 10.1016/j.nbd.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet. 2004;13:1257–1265. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- Kiehl TR, Nechiporuk A, Figueroa KP, Keating MT, Huynh DP, Pulst SM. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem Biophys Res Commun. 2006;339:17–24. doi: 10.1016/j.bbrc.2005.10.186. [DOI] [PubMed] [Google Scholar]
- Matilla A, Roberson ED, Banfi S, Morales J, Armstrong DL, Burright EN, et al. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci. 1998;18:5508–5516. doi: 10.1523/JNEUROSCI.18-14-05508.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switonski PM, Fiszer A, Kazmierska K, Kurpisz M, Krzyzosiak WJ, Figiel M. Mouse ataxin-3 functional knock-out model. Neuromolecular Med. 2011;13:54–65. doi: 10.1007/s12017-010-8137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Fu YH, Sopher BL, Libby RT, Wang X, Li LY, et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. 2001;31:913–927. doi: 10.1016/s0896-6273(01)00422-6. [DOI] [PubMed] [Google Scholar]
- Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, et al. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37:383–401. doi: 10.1016/s0896-6273(02)01190-x. [DOI] [PubMed] [Google Scholar]
- Helmlinger D, Abou-Sleymane G, Yvert G, Rousseau S, Weber C, Trottier Y, et al. Disease progression despite early loss of polyglutamine protein expression in SCA7 mouse model. J Neurosci. 2004;24:1881–1887. doi: 10.1523/JNEUROSCI.4407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latouche M, Fragner P, Martin E, El Hachimi KH, Zander C, Sittler A, et al. Polyglutamine and polyalanine expansions in ataxin7 result in different types of aggregation and levels of toxicity. Mol Cell Neurosci. 2006;31:438–445. doi: 10.1016/j.mcn.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Vos MJ, Zijlstra MP, Kanon B, van Waarde-Verhagen MA, Brunt ER, Oosterveld-Hut HM, et al. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum Mol Genet. 2010;19:4677–4693. doi: 10.1093/hmg/ddq398. [DOI] [PubMed] [Google Scholar]
- Simões AT, Gonçalves N, Koeppen A, Déglon N, Kügler S, Duarte CB, et al. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain. 2012;135 Pt 8:2428–2439. doi: 10.1093/brain/aws177. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, et al. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chort A, Alves S, Marinello M, Dufresnois B, Dornbierer JG, Tesson C, et al. Interferon β induces clearance of mutant ataxin 7 and improves locomotion in SCA7 knock-in mice. Brain. 2013;136:1732–1745. doi: 10.1093/brain/awt061. [DOI] [PubMed] [Google Scholar]
- Keiser MS, Boudreau RL, Davidson BL. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: implications for spinocerebellar ataxia type 1 therapy. Mol Ther. 2014;22:588–595. doi: 10.1038/mt.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Carmo Costa M, Luna-Cancalon K, Fischer S, Ashraf NS, Ouyang M, Dharia RM, et al. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol Ther. 2013;21:1898–1908. doi: 10.1038/mt.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüb U, Brunt ER, Deller T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado-Joseph disease) Curr Opin Neurol. 2008;21:111–116. doi: 10.1097/WCO.0b013e3282f7673d. [DOI] [PubMed] [Google Scholar]
- Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- Nóbrega C, Nascimento-Ferreira I, Onofre I, Albuquerque D, Hirai H, Déglon N, et al. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS One. 2013;8:e52396. doi: 10.1371/journal.pone.0052396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves S, Nascimento-Ferreira I, Dufour N, Hassig R, Auregan G, Nóbrega C, et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3. Hum Mol Genet. 2010;19:2380–2394. doi: 10.1093/hmg/ddq111. [DOI] [PubMed] [Google Scholar]
- Noma S, Ohya-Shimada W, Kanai M, Ueda K, Nakamura T, Funakoshi H. Overexpression of HGF attenuates the degeneration of Purkinje cells and Bergmann glia in a knockin mouse model of spinocerebellar ataxia type 7. Neurosci Res. 2012;73:115–121. doi: 10.1016/j.neures.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Recovery of cerebellar proteins at 40 weeks of age.
Mutant ataxin-7 mRNA levels in the SCA7 brain at 40 weeks of age.