

Abstract
The development of robust nonviral vectors could facilitate clinical gene therapy applications and may overcome some of the immune complications of viral vectors. Nevertheless, most nonviral gene deliver approaches typically yield only transient and/or low gene expression. To address these caveats, we have explored piggyBac transposons to correct hemophilia B by liver-directed factor IX (FIX) gene therapy in hemophilic mice. To achieve this, we combined the use of: (i) a hyperactive codon-optimized piggyBac transposase, (ii) a computationally enhanced liver-specific promoter, (iii) a hyperfunctional codon-optimized FIX transgene (FIX R338L Padua), and (iv) a modification of the transposon terminal repeats. This combination strategy resulted in a robust 400-fold improvement in vector performance in hepatocytes, yielding stable supraphysiologic human FIX activity (>1 year). Liver-specific expression resulted in the induction of FIX-specific immune tolerance. Remarkably, only very low transposon/transposase doses were required to cure the bleeding diathesis. Similarly, PB transposons could be used to express supraphysiologic factor VIII levels using low transposon/transposase doses. PB transposition did not induce tumors in a sensitive hepatocellular carcinoma-prone mouse model. These results underscore the potency and relative safety of the latest generation PB transposons, which constitutes a versatile platform for stable and robust secretion of therapeutic proteins.
Introduction
Clinical evidence demonstrates that gene therapy is effective to treat patients suffering from a wide range of hereditary diseases. This could be attributed largely to the improvement in viral vector–based gene transfer technologies. Despite these encouraging results, viral vectors can be inactivated by preexisting neutralizing antibodies that preclude gene transfer.1 This is compounded by the induction of cytotoxic T-cell responses directed against the gene-modified cells, consequently preventing long-term gene expression. Moreover, manufacturing of clinical-grade viral vectors is challenging, and their restricted cargo capacity can hamper clinical translation. Nonviral gene transfer may overcome some of these limitations and offer some advantages, especially considering the capacity to accommodate larger inserts, the lack of preexisting host immunity, and reduced costs and time for the production of clinical-grade nonviral DNA plasmid–based vectors. Nevertheless, they typically do not stably integrate into the genome and consequently yield only transient expression of the gene of interest.2 We and others have demonstrated that this could be overcome by using DNA transposons that stably integrate their therapeutic cargo into the target cell genome, enabling stable expression of the gene of interest.3,4 The piggyBac (PB) transposon system, originally derived from the cabbage looper moth Trichoplusia ni,5 is among the most promising transposons for gene therapy applications. To convert the PB transposon into a gene delivery tool, a binary system is required that is composed of an expression plasmid that encodes the PB transposase and a donor plasmid containing the gene of interest, which is flanked in cis by the transposon terminal repeat sequences required for transposition. PB transposons can efficiently transpose in mammalian cells6,7 and can deliver large transgenes,8 which cannot be readily accommodated into most viral vectors due to their intrinsic packaging constraints. PB has been efficiently applied as a tool for functional genomic studies, ex vivo genetic modification of somatic and embryonic cells and generation of induced pluripotent stem cells.9,10 However, there are only few studies that focus on direct in vivo gene transfer with PB and most of these relied on reporter genes.11,12,13,14,15 In this study, we explored the use of PB transposons for liver-directed gene therapy of hemophilia B, which has not yet been investigated. Hemophilia B is a hereditary bleeding disorder caused by a defective factor IX (FIX) gene. It is currently treated by repeated clotting factor infusions but this treatment is not curative.16 Moreover, some patients develop neutralizing antibodies against the administered recombinant FIX protein that renders the therapy ineffective and bleeding episodes difficult to manage. Hence, hemophilia B is an attractive target disease to validate PB transposon-based gene therapy approaches, which has implications for other hereditary disorders,3 including liver-borne diseases. The main objective of this study therefore consisted of establishing proof-of-concept that PB transposons encoding FIX can be used for liver-directed gene delivery to cure hemophilia B and to assess their overall efficacy and safety in appropriate mouse models. Maximizing the therapeutic index of a given gene transfer vector is a crucial step toward implementation of successful clinical trials. Hence, we wanted to augment the efficiency of transposon-mediated gene therapy using a multilayered strategy by optimizing each one of its components including the PB transposase and the transposon, the liver-specific promoter used to drive FIX and the FIX transgene itself. Our results demonstrated that PB transposons in conjunction with a mouse codon-optimized PB transposase (mPB) resulted in prolonged FIX expression and cure hemophilia B in FIX-deficient mice, which had not been shown previously. Moreover, we showed that the efficiency of PB-mediated gene therapy could be enhanced by using the latest generation hyperactive PB transposase (hyPB) and by modifying the transposon terminal repeats.17,18,19 In addition, the use of a liver-specific promoter coupled to in silico designed cis-regulatory modules (CRMs) further increased FIX expression levels.20,21,22 Finally, we demonstrated that the overall efficacy could be further increased by using a codon-optimized FIX containing a hyperfunctional gain-of-function mutation (i.e., FIX Padua R338L).23,24 This combinatorial strategy resulted in robust FIX activity at supraphysiologic levels, substantially reducing the vector dose requirement for reaching therapeutic efficacy. Moreover, it enabled induction of immune tolerance to FIX and prevented the generation of anti-FIX antibodies on immunization with recombinant FIX. We also validated the use of the hyperactive PB platform for delivery of relatively large transgenes, such as FVIII as a gene therapy approach for hemophilia A. Using a highly sensitive tumor-prone hepatocellular carcinoma mouse model, we did not observe any significant increase in oncogenic risk upon PB transposition, suggesting that this improved hyperactive PB platform has a favorable safety profile.
Results
PB provides long-lasting therapeutic hFIX expression levels and phenotypic correction in hemophilia B mice
The codon usage optimized transposase (mPB)25 was evaluated in combination with PB-hFIXIA (Figure 1d) that carried a wild-type hFIX minigene (hFIXIA)2 and PB-hFIXco (Figure 1e) with a codon-optimized hFIX (hFIXco).24,26,27 In addition, the PB-hFIXco transposon contained a small intron upstream of the hFIXco to boost FIX expression.2,26 Both the hFIXIA and hFIXco were driven from a novel and potent chimeric liver-specific promoter that contained an in silico designed hepatocyte-specific cis-regulatory module, designated as HS-CRM8. This element conferred high liver-specific expression.20,21,22 Liver-directed hydrodynamic cotransfection of the PB-hFIXIA transposon (10 μg) along with 2 μg mPB, resulted in stable therapeutic hFIX antigen and activity levels for >12 months in hemophilic FIX-deficient mice (Figure 2a). Similarly, liver-directed cotransfection of the PB-hFIXco transposon and mPB resulted in a significant ≈12-fold higher (P < 0.001) hFIX protein and activity level that stabilized in the supraphysiologic range (Figure 2b). In contrast, hFIX expression and activity gradually declined to basal levels in control mice that received either PBS or only the transposons in the absence of transposase (Figure 2a,b). Anti-hFIX antibodies could not be detected (data not shown). Transposition is therefore necessary for sustained expression. The initial high peak of FIX expression is likely due to the presence of nonintegrated PB-hFIXIA or PB-hFIXco episomes due to the fact that we used an excess amount of transposon plasmid (10 μg). These episomes are gradually lost and are also epigenetically silenced (Figure 2), consistent with previous observations.2,28,29
Figure 1.

Schematic representation of PB transposon and transposase constructs. (a) Transposase constructs encoding for the native PB transposase mouse codon-optimized (mPB) driven by the CMV promoter cloned upstream of a β-globin intron (βGI). (b) The hyperactive PB transposase mouse codon-optimized (hyPB). The hyperactivating mutations are indicated. (c) The empty control plasmid contains a multiple cloning site (MCS) between the promotor and polyadenylation signal. (d) The PB-hFIXIA transposon is flanked by wild-type inverted repeats (IRwt) and contains the wild-type hFIX cDNA harboring the truncated 1.4 kb hFIX intron A (intron IA) between the first hFIX exon (exon 1) and the following exons (exons 2–8). The hFIXIA minigene is driven by a chimeric promoter composed of the transthyretin minimal promoter (TTRmin) coupled to a hepatocyte-specific cis-regulatory module (designated as HS-CRM8). (e) In the PB-hFIXco transposon, the hFIXIA minigene was replaced by the synthetic codon-optimized hFIX (hFIXco) including a partial 3′ untranslated region. In addition, the minute virus of mice small intron (MVM) was introduced downstream of the chimeric HS-CRM8/TTRmin promoter. The PB transposons were flanked by three different inverted repeats: (e) wild-type minimal inverted repeats (IRwt) (PB-hFIXco/IRwt); (f) truncated “micro” inverted repeats (IRmicro) (PB-hFIXco/IRmicro), and (g) minimal mutant containing the T53C-C136T point mutations (IRmut16) (PB-hFIXco/IR mut16). (h) The PB-hFIXco-R338L/IRmicro transposon is flanked by IRmicro and contains a codon-optimized hFIX sequence with an R338L substitution. (i) The PB-hFVIIIΔBco/IRmicro contains a codon-optimized B-domain deleted FVIII sequence (hFVIIIΔBco) driven by the chimeric HS-CRM8/TTRmin promoter coupled to a minute virus of mice small intron (MVM). (l) PB-c-Myc is flanked by the IRw and contains the c-Myc oncogene under the control of the liver specific chimeric promoter. All the PB transposons were flanked with loxP sites (loxP) and contain the bovine growth hormone polyadenylation signal (bGHpA).
Figure 2.

PB provides long-lasting therapeutic hFIX expression levels and phenotypic correction in hemophilia B mice. Hemophilia B mice were hydrodynamically transfected with equimolar amounts (10 μg) of (a) PB-hFIXA or (b) PB-hFIXco transposon plasmids flanked by IRwt in conjunction with an equimolar amount (2 μg) of mPB-encoding plasmid (+ mPB) (full lines) or an empty control plasmid (+ empty) that does not contain any transposase gene (dashed lines). (a,b) hFIX antigen expression (black squares) and FIX activity (gray squares) were measured on plasma samples collected at the indicated times by enzyme-linked immunosorbent assay and a functional hFIX activity assay, respectively. (c) Transposon copies per diploid genome hFIX mRNA levels relative to (d) FIXIA mRNA levels were determined on liver biopsies 12 months posttransfection. Results were presented as mean ± SEM. n.s. indicates not significant; *P < 0.05; **P < 0.01; ***P < 0.001 (n = 3 mice/group).
Though the transposon copies per genome content were similar in the liver of animals that received equal doses of the PB-hFIXIA or PB-hFIXco transposons (Figure 2c), we observed a significant (P < 0.001) increase in hFIX mRNA level for the hFIXco than its wild-type counterpart (Figure 2d). This demonstrated that the optimized expression cassette, containing a small intron and a codon-usage optimized hFIX transgene (hFIXco), significantly increased mRNA levels concomitant with a robust increase in FIX protein expression level. Though codon optimization may in some cases enhance immunogenicity,30 we found no evidence of an increased antibody response to the codon-optimized FIX protein. A low level of transposon copies (Figure 2c) was apparent in the absence of any transposase-expressing plasmid 12 months posttransfection (designated as “empty” in Figure 1c). Since a relatively high dose of PB transposons was used (10 μg), this likely contributed to the residual presence of episomally silenced unintegrated DNA, as shown previously.2,28,29
Increased therapeutic efficiency using the hyperactive PB platform
To further improve the efficiency of the PB platform, allowing the use of lower transposon/transposase doses, we subsequently explored the use of a hyPB.17 This hyPB contained several mutated residues compared to the original codon usage optimized mPB (Figure 1a,b). Liver-directed hydrodynamic transfection of severe combined immune deficient (SCID) mice with 500 ng of PB-hFIXco transposon along with 1,000 ng hyPB resulted in stable supraphysiologic hFIX levels corresponding to 200% of normal hFIX levels (Figure 3a). These FIX levels were significantly higher (P < 0.001) than what could be achieved with the original mPB transposase. Similarly, liver-directed transfection of SCID mice with 50 ng of PB-hFIXco transposon plasmid along with 100 ng hyPB resulted in a dose-dependent effect, yielding therapeutic hFIX levels corresponding to 20% of normal levels. This represented a significant 20-fold increase (P < 0.001) in FIX levels compared to when the mPB transposase was used (Figure 3a,b).
Figure 3.

Increased therapeutic efficiency using the hyperactive PB platform. CB17/IcrTac/Prkdcscid mice were hydrodynamically transfected with (a,c,e) 500 ng or (b,d,f) 50 ng of PB-hFIXco (a–f), (c,d; triangle) PB-hFIXco/IRmicro or (e,f, triangle) PB-hFIXco/IR mut16 transposon plasmids along with (a,c) 1,000 ng or (b,d) 100 ng mPB (triangle a,b) or hyPB-expressing plasmid (square a,b) or empty control plasmid (hatched lines). hFIX expression was measured on plasma samples collected at the indicated times by a specific enzyme-linked immunosorbent assay assay. Results were presented as mean ± SEM. n.s. indicates not significant; *P < 0.05; **P < 0.01; ***P < 0.001 (n = 3 mice/group).
We next evaluated whether the terminal repeats IRmicro and IRmut16 in combination with the hyPB could further improve the in vivo potency of the PB transposons. A significant 1.5-fold increase in hFIX expression was apparent when the IRmicro was used compared to IRwt (Figure 3c,d). Liver-directed transfection in SCID mice of the PB-hFIXco/IRmicro (Figure 1g) transposon (500 ng) along with 1,000 ng hyPB transposase-encoding plasmid resulted in stable FIX levels reaching ~300% of normal hFIX levels (Figure 3c). This represents a 100-fold improvement over the 3% FIX activity that could be attained when the hemophilia B was transfected with matched doses of PB-hFIXIA and mPB plasmids (Figure 4c). Similarly, at 10-fold lower PB-hFIXco/IRmicro and hyPB doses, a dose-dependent decrease in hFIX expression was apparent, yielding 30% of normal hFIX levels. In contrast, FIX expression was not or only slightly increased (Figure 3e,f) when the IRmut16 was used (Figure 1g) compared to IRwt (Figure 1e).
Figure 4.

Enhancing PB performance for hemophilia A and B. Hemophilia B mice were hydrodynamically cotransfected with (a) 500 ng or (b) 50 ng of PB-hFIXco-R338L/IRmicro transposon with (a) 1,000 ng or (b) 100 ng of hyPB-expressing plasmid or an equimolar empty control plasmid (hatched lines). hFIX antigen levels (black) and FIX activity (gray) were measured on plasma samples collected at the indicated times by a specific enzyme-linked immunosorbent assay (ELISA) and a functional FIX assay, respectively (a–c). (c) Comparison of the original PB transposon system (PB-hFIXIA + mPB) versus the improved PB platform (PB-hFIXco + hyPB; PB-hFIXco-R338L/IRmicro + hyPB) in hemophilic mice using either 500 or 50 ng PB transposon (PB-hFIXIA, PB-hFIXco, or PB-hFIXco-R338L/IRmicro) and 1,000 ng or 500 ng PB transposase, respectively (mPB or hyPB, as indicated). A ratio of 1:2 transposon/transposase was maintained. (d) Three months after transfection with PB transposons mice were subjected to immunization with recombinant hFIX antigen and adjuvant as indicated (a,b). Anti-hFIX specific antibodies were measured by ELISA at week 2 (black) and week 4 (gray) postimmunization (p.i). PBS-injected hemophilia B mice that were immunized with recombinant hFIX and adjuvant were used as positive control. (e) Tail-clipping assay on hemophilia B mice treated with the indicated transposon constructs and doses (black bars). Blood loss was determined by measuring the absorbance at 575 nm of hemoglobin content in the saline solution in which the tail was placed. Wild-type and untreated hemophilia B (Hemo B) mice were used as controls. (f) Transposon copies per diploid genome were determined on liver biopsies 6 months posttransfection. (g) FVIII antigen level in CB17/IcrTac/Prkdcscid mice hydrodynamically transfected with 500 ng PB-hFVIIIΔBco/IRmicro transposon plasmids along with 1,000 ng hyPB-expressing plasmid or an equimolar empty control plasmid (hatched lines). Physiologic hFVIII concentration (100% = 200 ng/ml) is indicated. hFVIII antigen levels were detected by ELISA. Results were presented as mean ± SEM. n.s. indicates not significant, *P < 0.05; **P < 0.01; ***P < 0.001 (n = 3 mice/group).
Enhancing PB performance using hyperfunctional hFIX-R338L Padua
Subsequently, we tested whether the performance of this optimized hyPB and PB-hFIXco/IRmicro platform could be further enhanced using a synthetic codon-optimized FIX transgene harboring a hyperactivating R338L (Padua) mutation.23 We therefore constructed a PB-hFIXco-R338L/IRmicro transposon (Figure 1h) that was cotransfected along with a hyPB transposase-encoding construct into hemophilia B mice. Sustained FIX antigen levels could be obtained (Figure 4a,b) with similar expression kinetics as in SCID mice (Figure 3). The hFIXco-R338L protein exhibited a >6-fold higher specific FIX activity with respect to protein levels, yielding up to 1,200% FIX activity at the highest hFIXco-R338L/IRmicro transposon dose (500 ng). Most importantly, this represents a robust 400-fold improvement over the 3% FIX activity that could be attained when the hemophilia B were transfected with matched doses of PB-hFIXIA and mPB plasmids (Figure 4c). Moreover, at the lowest dose of only 50 ng of PB-hFIXco-R338L/IRmicro transposon and 100 ng hyPB transposase plasmid, supraphysiologic FIX activity could be attained (≈200%), whereas matched doses of PB-hFIXIA and mPB plasmids yielded no detectable FIX activity (Figure 4c).
Interestingly, none of the hemophilia B mice treated with the PB transposons expressing hFIX or hFIX-R338L (Padua) developed an anti–hFIX-specific antibody response even in the face of immunization with recombinant hFIX protein and adjuvant (Figure 4d). This is consistent with the induction of hFIX-specific immune tolerance. Since FIX antigen levels and expression kinetics were similar in hemophilia B (Figure 4a,b) and SCID mice (Figure 3a–f), this further confirmed the lack of hFIX-specific immune responses.
The superior potency of the hFIXco-R338L/IRmicro transposon and hyPB transposase was confirmed by assessing phenotypic correction 4 months posttransfection in hemophilia B mice (Figure 4e). In particular, hemophilic mice treated with only 50 ng of PB-hFIXco-R338L/IRmicro transposon and 100 ng hyPB transposase plasmid achieved hemostatic correction and consequently survived the tail-clip assay. In contrast, none of the hemophilic mice injected with matched doses of the PB-FIXIA transposon and mPB transposase plasmids survived the injury (Figure 4e).
Transposon copies were similar in the liver of animals that received equal doses of PB transposons expressing hFIXco or hFIX-R338L (Padua) in the presence of hyPB (Figure 4f) but were significantly higher than in mice injected PB transposons expressing hFIXIA in presence of mPB (P < 0.01) at both tested doses. Remarkably, hemophilic mice treated with only 50 ng of PB-hFIXco-R338L/IRmicro transposon and 100 ng hyPB transposase plasmid achieved supraphysiologic hFIX activity (Figure 4c) and rescue of the bleeding phenotype (Figure 4e). Transposon copies were further reduced (<0.1 copies/diploid genome) decreasing the risk of insertional oncogenesis.
Validating the hyperactive PB platform for FVIII delivery
We then extended these findings using the codon-optimized B-domain deleted FVIII (FVIIIΔBco).31 Liver-directed hydrodynamic transfection of 500 ng of PB-hFVIIIΔBco/IRmicro transposon along with 1,000 ng hyPB in SCID mice resulted in stable supraphysiologic hFVIII levels corresponding to 300% of normal hFVIII levels (Figure 4g). Conversely, in the absence of the hyperactive hyPB transposase, expression declined gradually to basal levels (Figure 4g). This confirms that stable genomic integration by transposition is required for stable hepatic FVIII gene expression. These results are consistent with the results obtained with the PB-FIX transposons.
Safety assessment in a tumor-prone hepatocellular carcinoma model
To assess the risk of insertional oncogenesis, we evaluated the safety profile of the PB platform in the diethylnitrosamine (DEN)-induced hepatocellular carcinoma (HCC) mouse model that resembles human HCC. One month after DEN initiation32 mice were transfected hydrodynamically with either 10 μg PB-hFIXIA transposon or 500 ng of PB-hFIXco in combination with 2 μg mPB plasmid (Figure 5a). These doses were chosen to obtain similar FIX expression levels and compare the intrinsic safety of both PB constructs. The tumor burden was assessed 36 weeks post-DEN initiation. We did not observe any significant differences in the number of HCC nodules between mice transfected with PB-hFIXIA or PB-hFIXco and mPB and PBS-injected mice (Figure 5b). In contrast, tumor incidence and progression was significantly (P < 0.001) increased in control mice that were hydrodynamically transfected with a PB-c-Myc transposon (Figure 5c), (number of nodules (>50), max tumor size (>50 mm) compared to PBS-injected mice (number of nodules (5.83 ± 1.42), max tumor size (5.70 ± 0.29 mm). This ultimately resulted in significant and accelerated mortality (i.e., median survival of 157 days) (Figure 5c), consistent with the well-established role of c-Myc in liver cancer.33 It is noteworthy that a minimal yet significant increase in the number of HCC nodules was apparent in DEN-treated control mice that had undergone hydrodynamic transfection (with PBS) compared to noninjected DEN-treated mice. This suggests that the hydrodynamic procedure per se may provoke tumorigenesis, possibly by triggering a transient liver damage and inflammation associated with hepatocyte proliferation34 (Figure 5b), consistent with previous observations.35 This underscores the high sensitivity of the DEN-induced HCC model.
Figure 5.
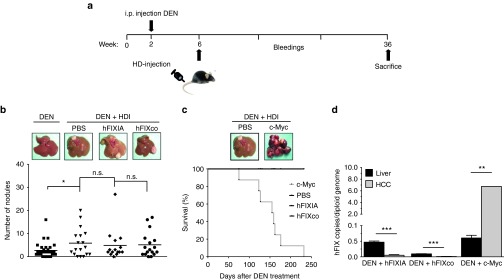
Safety assessment in a tumor-prone hepatocellular carcinoma model. (a) Diagram showing the timeline and experimental procedure. C57BL/6 male mice were subjected to a preweaning protocol of diethylnitrosamine (DEN) initiation. DEN (12.5 mg/kg) was injected intraperitoneally (i.p) in 14-days-old-mice. 4-weeks later, mice were hydrodynamically transfected with 10 μg PB-hFIXIA or 500 ng PB-hFIXco transposons and 2 μg of mPB-encoding plasmid. hFIX expression was measured on plasma samples collected at different time points (bleedings) by enzyme-linked immunosorbent assay. As controls, mice received PBS injection or a PB-c-Myc transposon (10 μg) 2 μg of mPB-encoding plasmid. Tumor burden was assessed 36-weeks post-DEN initiation. (b) Representative images showing the macroscopic liver appearance 36-weeks post-DEN initiation in mice that received DEN only or DEN in combination with hydrodynamic injection (HDI). Liver nodules were counted for each group. (c) Representative picture showing the macroscopic tumor burden associated with PB mediated c-Myc expression and Kaplan–Meier survival curve of PB-c-Myc transposon and PBS-injected mice (c-Myc, n = 9, median survival 157 days). (d) Transposon copy number quantification from genomic DNA isolated from normal liver biopsy (hFIXIA; n = 15; hFIXco, n = 17; c-Myc, n = 9) and HCC nodules (hFIXIA, n = 30; hFIXco, n = 34; and c-Myc, n = 18). Results are presented as mean ± SEM. n.s. indicates not significant; *P < 0.05; **P < 0.01, ***P < 0.001.
To further rule out that PB transposons contributed to HCC in the DEN-induced mouse model, we assessed the PB copy number in normal liver tissue versus HCC nodules. Interestingly, the copy number of the PB-hFIXIA or PB-hFIXco transposons was higher in normal liver biopsies compared to the PB copy number in HCC nodules. Given the significant decrease in PB-hFIXIA or PB-hFIXco transposon copy number in HCC samples, this suggests that HCC initiation and/or progression was not associated with stable genomic transposon integration (Figure 5d). We hereby demonstrated that PB transposition in itself does not significantly increase tumorigenicity in this sensitive HCC tumor-prone mouse model, which underscores the relative safety of the PB-FIX platform. Conversely, the PB-c-Myc copy number was significantly increased in HCC nodules versus normal liver biopsies. This strongly suggests that c-Myc expression contributed to malignant transformation, consistent with natural selection of transformed hepatocyte clones that contained an increased number of PB-c-Myc vector copies.
Discussion
In this study, we established proof of concept that the PB platform can be used to obtain sustained expression (>1 year) of a therapeutically relevant transgene (i.e., FIX) leading to a phenotypic correction of the bleeding diathesis in hemophilia B mice. To our knowledge, this is the first report that shows phenotypic correction of a genetic disorder using the PB transposon platform with broad implications for the field at large. Transposition was necessary to ensure sustained FIX expression and stable correction of the bleeding phenotype, whereas FIX expression was transient in the absence of the PB transposase. Furthermore, we demonstrated that a robust 400-fold improvement in vector performance could be achieved using a multilayered approach by using a hyPB, by trimming the transposon terminal repeats (IRmicro) and by optimizing the transgene expression cassette itself. The expression cassette encoded a codon-optimized FIX or its hyperactive FIX R338L (Padua) version that were driven from a robust chimeric liver-specific promoter. Consequently, only very low amounts of hyPB and PB transposons were needed to achieve supraphysiologic FIX activity levels in hemophilic mice paving the way towards preclinical large animal studies.
The robust hepatocyte-specific FIX expression levels in the PB transposons could be attributed, to the use of the chimeric liver-specific TTR promoter coupled to the in silico designed hepatocyte-specific cis-regulatory modules (HS-CRM).20,21,22 These CRMs contained evolutionary conserved clusters of transcription factor binding site motifs that conferred high liver-specific gene expression. Moreover, the modified vector design of the PB-hFIXco and PB-hFIXco-R338L transposons compared to PB-hFIXIA contributed to increased steady-state FIX mRNA levels. The increased activity of hFIXco-R338L (Padua) is consistent with our recent studies showing five- to sevenfold increase FIX activity after liver-directed gene therapy of integration-competent or defective lentiviral vectors,24 which was confirmed independently based on adeno-associated viral vectors.36
The in vivo performance of the PB-FIX transposons and hyPB transposase is consistent with previous reports showing sustained expression of reporter genes after liver-directed hydrodynamic gene delivery11,12,13,14,15 and confirm the superiority of the hyperactive hyPB compared to the conventional mPB transposase. Moreover, in a recent study, PB transposons were shown to stably express α1-antitrypsin (AAT) in transfected hepatocytes in vivo.14 Nevertheless, the PB platform had not been validated in a disease model, and the AAT expression levels attained were relatively modest. In this study, we also demonstrated that the hyPB platform could be used to express supraphysiologic levels of FVIII. The superior activity of the hyPB compared to mPB is likely due to the synergistic effect of the point mutations that enhance the transposition efficiency by increasing the protein levels and improve the mechanism and/or kinetics of transposition.17 The increase in hFIX protein levels associated with the use of IRmicro is likely due to increased PB transposition. However, an effect at the transcriptional level cannot be excluded. Trimming the terminal repeats may indeed prevent the binding of transcriptional repressors that may influence expression of the gene of interest. This hypothesis is consistent with the elevated FIX expression levels in mice transfected with the PB-hFIXco/IRmicro transposon in the absence of the hyPB transposase.
The FIX and FVIII levels that were attained compared favorably to other nonviral gene therapy approaches.37,38,39,40 In particular, hydrodynamic transfection of Sleeping Beauty-based transposons encoding FIX in conjunction with the early SB1041 or latest SB100X transposase4 resulted in lower FIX expression levels and required 50 to100-fold higher doses of SB-FIX transposons (25–50 µg) compared to when the PB-hFIXco-R338L/IRmicro transposon was used in conjunction with hyPB. This implies that the current optimized transposon vectors are least 50 to 100-fold more efficient than the state of the art. Alternative methods for nonviral liver-directed gene therapy relied on hydrodynamic plasmid or minicircle delivery, but these methods yielded lower and/or transient FIX expression.2,42 The FIX levels that were obtained also compare favorably with what can typically be achieved with viral vectors, particularly lentiviral and AAV vectors.24,43,44,45
The sustained hepatocyte-specific FIX expression in hemophilia B mice was consistent with the induction of hFIX-specific immune tolerance, which was maintained even on immunization with hFIX and immune adjuvant. This is consistent with the induction of immune tolerance following liver-directed gene therapy with AAV or lentiviral vectors when hepatocyte-specific promoters were employed, avoiding ectopic expression in antigen-presenting cells.24,43,44,45 This indicates that the ability to induce FIX-specific immune tolerance is irrespective of the vector type and could be attributed to the specific microenvironment in the liver. The exact mechanism that accounted for the induction of immune tolerance in the liver is not fully understood but possibly involves the induction of CD4+CD25+Foxp3+ regulatory T cells.
The PB-hFIXIA (Figure 1d) and PB-hFIXco (Figure 1e) transposons used in this study differ with respect to (i) codon-optimization of the FIX cDNA and (ii) the use of a mini-intron from the MVM virus upstream of the FIX cDNA versus the use of intron A of FIX maintained in the natural position in between exons 1 and exon 2 (Figure 1d). The combined effect of these changes resulted in a 10-fold difference in FIX protein expression levels (Figure 2a,d). We had previously shown that the net effect of FIX codon optimization by itself typically resulted in a two- to threefold increase in FIX protein expression.24 This is likely due to an increase in translation initiation. Consequently, the effect of codon optimization by itself is not sufficient to account for the 10-fold difference in FIX protein expression levels. This could be explained by also taking into account the increase in steady-state FIX mRNA levels (Figure 2d). The increase in FIX mRNA levels could be due to the effect of the MVM intron. Furthermore, some of the nucleotide changes that were introduced in the FIX codons that were intended to boost translation may also have inadvertently increased transcription. The increase in FIX mRNA did not result in a proportionate increase in FIX protein levels. This suggests that posttranscriptional events may be limiting the control of FIX biosynthesis.
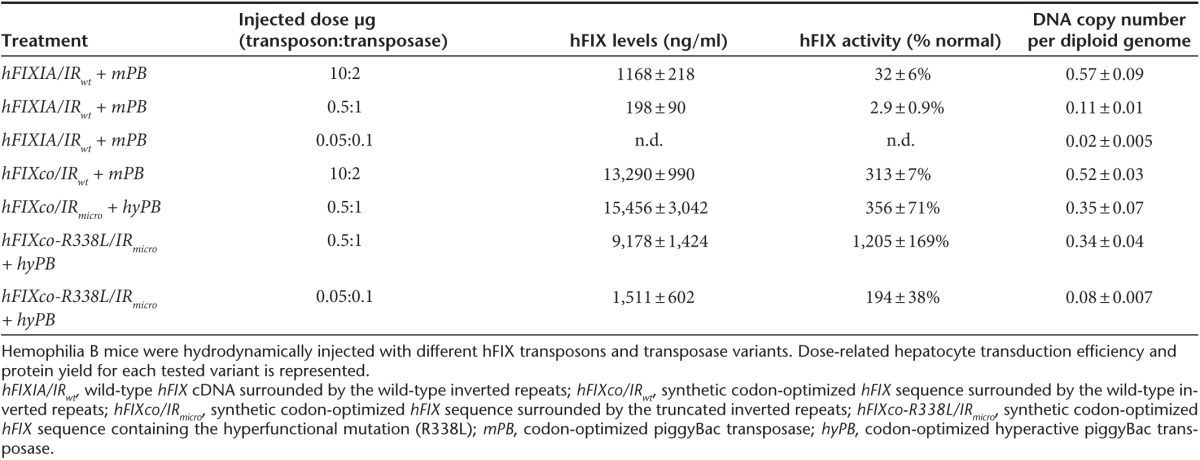
We subsequently assessed the safety of this PB platform in the highly sensitive DEN-treated HCC-prone mice. In this model, DEN induces DNA damage, chronic liver injury and triggers local and systemic inflammatory reactions followed by compensatory hepatocyte proliferation.46 Macroscopic and molecular analysis strongly suggests that PB transposition in itself does not enhance the tumorigenic risk. As a corollary, it would appear that the HS-CRM8 element does not increase the risk of insertional oncogenesis. Moreover, our stepwise improvements of the PB platform required less transposon integrations to obtain sustained therapeutic FIX levels (Table 1), further reducing the risk of insertional oncogenesis. Indeed, we showed that reducing the amount of plasmid used for hydrodynamic gene transfer correlates with a reduction in hepatocyte transfection in terms of transposon copy number (Table 1). Hydrodynamic transfection typically results in up to 10% of transfected hepatocytes in the mouse liver with a preference for the pericentral region of the hepatic acinus.47,48 Consequently, this translates to 10-fold the number of integrants per cell that actually took up the PB transposons. In particular, a transposon copy number of 0.08 ± 0.007 per diploid genome (i.e., following transfection with 50 ng of PB-hFIXco-R338L/IRmicro transposon and 100 ng hyPB transposase plasmid) (Table 1 and Figure 4c) would correspond to less than one copy per transfected hepatocyte. Such a low copy number minimizes the risk of insertional oncogenesis. While the level of expressed FIX is more relevant than the percent of FIX expressing hepatocytes for correcting the bleeding phenotype, it is important to consider that phenotypic correction of other diseases may require a significantly greater fraction of gene-modified hepatocytes.
Table 1. Variants related differences in hepatocyte transduction efficiency, hFIX protein yield and activity per injected dose in piggyBac treated hemophilis B mice.
Exhaustive analysis of genomic integration sites of the hyPB platform in transfected hepatocytes in vivo will be the subject of future studies. Nevertheless, it is reassuring that integration site analyses of transfected human cells revealed previously that PB transposons do not favor integration into genes and showed a reduced frequency of integrations near transcriptional start sites.14 To augment the specificity of PB integration into predefined loci, PB transposases were genetically fused to zinc finger49,50 or transcription activator-like effector51 domains specifically designed to target specific DNA sequences. However, the overall targeting efficiency was still limiting. Alternatively, genome-engineering approaches based on zinc finger nucleases and CRISPR/Cas9 are currently being explored as alternatives to achieve targeted integration in hepatocytes.52,53 To further increase in vivo targeting efficiencies, high vector doses are typically required that are prohibitive for clinical translation.52
The first-in-human transposon-based clinical studies have been initiated which further underscores its therapeutic potential.54 The encouraging efficacy and safety profile shown in this study further supports the use of this PB platform for ultimate clinical applications. Liu and colleagues55 have recently developed and validated an image-guided and computerized hydrodynamic gene delivery technique for hepatic gene delivery using large animal models. This improved method enables relatively efficient gene delivery by localized hydrodynamic liver delivery under controlled conditions, thereby minimizing liver damage compared to conventional hydrodynamic transfection methods. The efficacy and safety of this approach would need to be tested in canine hemophilia models. Ultimately, nanoparticle transfection technology could be used as an alternative to overcome some of the limitations of naked DNA hydrodynamic transfection, including TLR-mediated activation of the innate immune system.37 Future research is required to validate the safety and efficacy of the PB platform in preclinical large animal models.
Materials and Methods
For details on the various materials and methods employed in this study, see Supplementary Data.
Constructs. The mPB and hyPB were custom synthesized and expressed from a cytomegalovirus promoter (Figure 1a,b). An identical expression plasmid devoid of PB (“empty”) was used as control (Figure 1c). The terminal inverted repeats (IRs) of the transposon-containing plasmids (Figure 1) corresponded to truncated wild-type IRs56,57 (IRwt), contained T53C and C136T point mutations (IRmut16),18 or were further truncated (IRmicro).19 The IRmut16 and IRmicro were selected since they increased transposition efficiency based on in vitro studies. Wild-type, codon-optimized or hyperfunctional human factor IX cDNA (Figure 1) was expressed from a chimeric liver-specific promoter that was composed of a liver-specific minimal transthyretin (TTRmin) promoter in conjunction with a potent hepatocyte-specific cis-regulatory module (designated as HS-CRM8).20,21,22 Similarly, the PB-FVIII (Figure 1i) or PB-c-Myc transposon (Figure 1j) used the same promoter to express the B-domain deleted FVIII or c-Myc, respectively.
Animal experiments. C57Bl/6JRj, CB17/IcrTac/Prkdcscid (SCID), or FIX-deficient hemophilia B mice in a C57Bl/6 background were employed. All animal procedures were approved by the institutional animal ethics committees. Mice were transfected by hydrodynamic liver transfection. We collected whole blood (≈200 µl) by phlebotomy of the retro-orbital plexus. The citrated plasma was stored at −80 °C. A tumor-prone HCC mouse model was established by DEN initiation.32 One month later, mice were hydrodynamically injected. Survival was monitored, and HCC nodules were examined at 36 weeks. hFIX antigen and antibody levels were determined by enzyme-linked immunosorbent assay as described.24 FVIII antigen levels were determined by enzyme-linked immunosorbent assay, as described.31 FIX activity was determined using a functional assay, and phenotypic correction was assessed in hemophilic mice using a tail-clip assay. Transposon copy number and transgene mRNA expression was determined by quantitative PCR and reverse transcriptase PCR, respectively.
Statistics. Data were analyzed using GraphPad Software (GraphPad, La Jolla, CA). Statistical analyses were performed using a two-tailed unpaired Student's t-test. Results were presented as mean ± SEM.
Acknowledgments
We thank I. Verma and L. Wang (Salk Institute, La Jolla, CA, USA) and M. Kay (Salk Institute Stanford University, Stanford, CA, USA) for the FIX-deficient mice. This study was supported by grants from FWO (Fonds Wetenschappelijk Onderzoek); EU FP7 (222878, PERSIST), Geconcerteerde Onderzoeksacties (GOA)—EPIGEN (VUB); Strategic Research Program “Grower”—GENEFIX (VUB); Industrieel Onderzoeksfonds (IOF)—Groups of Applied Research (GEAR)—GENECURE (VUB); Industrieel Onderzoeksfonds (IOF)—Proof of Concept grant (PoC) (VUB); Association Française contre les Myopathies, Stichting tegen Kanker to T.V. and M.C. M.D.M. was supported through an FWO fellowship.
The authors declare no competing financial interests.
M.D.M. designed and performed experiments, analyzed data, and wrote the paper. M.C. and T.V.D. designed experiments, analyzed data, coordinated the work, and wrote the paper. E.S.M. designed and performed experiments. N.W., S.W., and J.M. contributed to the factor VIII work.
References
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Miao CH, Ohashi K, Patijn GA, Meuse L, Ye X, Thompson AR, et al. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol Ther. 2000;1:522–532. doi: 10.1006/mthe.2000.0075. [DOI] [PubMed] [Google Scholar]
- Di Matteo M, Belay E, Chuah MK, Vandendriessche T. Recent developments in transposon-mediated gene therapy. Expert Opin Biol Ther. 2012;12:841–858. doi: 10.1517/14712598.2012.684875. [DOI] [PubMed] [Google Scholar]
- Mátés L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet. 2009;41:753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- Cary LC, Goebel M, Corsaro BG, Wang HG, Rosen E, Fraser MJ. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology. 1989;172:156–169. doi: 10.1016/0042-6822(89)90117-7. [DOI] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Wilson MH, Coates CJ, George AL., Jr PiggyBac transposon-mediated gene transfer in human cells. Mol Ther. 2007;15:139–145. doi: 10.1038/sj.mt.6300028. [DOI] [PubMed] [Google Scholar]
- Li MA, Turner DJ, Ning Z, Yusa K, Liang Q, Eckert S, et al. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res. 2011;39:e148. doi: 10.1093/nar/gkr764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandenDriessche T, Ivics Z, Izsvák Z, Chuah MK. Emerging potential of transposons for gene therapy and generation of induced pluripotent stem cells. Blood. 2009;114:1461–1468. doi: 10.1182/blood-2009-04-210427. [DOI] [PubMed] [Google Scholar]
- Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saridey SK, Liu L, Doherty JE, Kaja A, Galvan DL, Fletcher BS, et al. PiggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol Ther. 2009;17:2115–2120. doi: 10.1038/mt.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, Higuchi Y, Kawakami S, Yamashita F, Hashida M. piggyBac transposon-mediated long-term gene expression in mice. Mol Ther. 2010;18:707–714. doi: 10.1038/mt.2009.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JE, Huye LE, Yusa K, Zhou L, Craig NL, Wilson MH. Hyperactive piggyBac gene transfer in human cells and in vivo. Hum Gene Ther. 2012;23:311–320. doi: 10.1089/hum.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnight ER, Staber JM, Korsakov P, Li X, Brett BT, Scheetz TE, et al. A hyperactive transposase promotes persistent gene transfer of a piggyBac DNA transposon. Mol Ther Nucleic Acids. 2012;1:e50. doi: 10.1038/mtna.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JE, Woodard LE, Bear AS, Foster AE, Wilson MH. An adaptable system for improving transposon-based gene expression in vivo via transient transgene repression. FASEB J. 2013;27:3753–3762. doi: 10.1096/fj.13-232090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah MK, Evens H, VandenDriessche T. Gene therapy for hemophilia. J Thromb Haemost. 2013;11 suppl. 1:99–110. doi: 10.1111/jth.12215. [DOI] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste A, Berenshteyn F, Brivanlou AH. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell. 2009;5:332–342. doi: 10.1016/j.stem.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Meir YJ, Weirauch MT, Yang HS, Chung PC, Yu RK, Wu SC. Genome-wide target profiling of piggyBac and Tol2 in HEK 293: pros and cons for gene discovery and gene therapy. BMC Biotechnol. 2011;11:28. doi: 10.1186/1472-6750-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah MK, Petrus I, De Bleser P, Le Guiner C, Gernoux G, Adjali O, et al. 2014Liver-specific transcriptional modules identified by genome-wide in silico analysis enable efficient gene therapy in mice and non-human primates Mol Therepub ahead of print). [DOI] [PMC free article] [PubMed]
- Nair N, Rincon MY, Evens H, Sarcar S, Dastidar S, Samara-Kuko E, et al. Computationally designed liver-specific transcriptional modules and hyperactive factor IX improve hepatic gene therapy. Blood. 2014;123:3195–3199. doi: 10.1182/blood-2013-10-534032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viecelli HM, Harbottle RP, Wong SP, Schlegel A, Chuah MK, Vandendriessche T, et al. 2014Treatment of phenylketonuria using minicircle-based naked-DNA gene transfer to murine liver Hepatologyepub ahead of print). [DOI] [PMC free article] [PubMed]
- Simioni P, Tormene D, Tognin G, Gavasso S, Bulato C, Iacobelli NP, et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua) N Engl J Med. 2009;361:1671–1675. doi: 10.1056/NEJMoa0904377. [DOI] [PubMed] [Google Scholar]
- Cantore A, Nair N, Della Valle P, Di Matteo M, Màtrai J, Sanvito F, et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood. 2012;120:4517–4520. doi: 10.1182/blood-2012-05-432591. [DOI] [PubMed] [Google Scholar]
- Cadiñanos J, Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007;35:e87. doi: 10.1093/nar/gkm446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Sun J, Zhang T, Yin C, Yin F, Van Dyke T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther. 2008;16:280–289. doi: 10.1038/sj.mt.6300355. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, He CY, Meuse L, Kay MA. Silencing of episomal transgene expression by plasmid bacterial DNA elements in vivo. Gene Ther. 2004;11:856–864. doi: 10.1038/sj.gt.3302231. [DOI] [PubMed] [Google Scholar]
- Schüttrumpf J, Milanov P, Abriss D, Roth S, Tonn T, Seifried E. Transgene loss and changes in the promoter methylation status as determinants for expression duration in nonviral gene transfer for factor IX. Hum Gene Ther. 2011;22:101–106. doi: 10.1089/hum.2009.212. [DOI] [PubMed] [Google Scholar]
- Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12:683–691. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- Ward NJ, Buckley SM, Waddington SN, Vandendriessche T, Chuah MK, Nathwani AC, et al. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood. 2011;117:798–807. doi: 10.1182/blood-2010-05-282707. [DOI] [PubMed] [Google Scholar]
- Vesselinovitch SD, Mihailovich N. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1983;43:4253–4259. [PubMed] [Google Scholar]
- Lin CP, Liu CR, Lee CN, Chan TS, Liu HE. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J Hepatol. 2010;2:16–20. doi: 10.4254/wjh.v2.i1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rácz Z, Godó M, Révész C, Hamar P. Immune activation and target organ damage are consequences of hydrodynamic treatment but not delivery of naked siRNAs in mice. Nucleic Acid Ther. 2011;21:215–224. doi: 10.1089/nat.2010.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard LE, Keravala A, Jung WE, Wapinski OL, Yang Q, Felsher DW, et al. Impact of hydrodynamic injection and phiC31 integrase on tumor latency in a mouse model of MYC-induced hepatocellular carcinoma. PLoS One. 2010;5:e11367. doi: 10.1371/journal.pone.0011367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn JD, Nichols TC, Svoronos N, Merricks EP, Bellenger DA, Zhou S, et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood. 2012;120:4521–4523. doi: 10.1182/blood-2012-06-440123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kren BT, Unger GM, Sjeklocha L, Trossen AA, Korman V, Diethelm-Okita BM, et al. Nanocapsule-delivered Sleeping Beauty mediates therapeutic Factor VIII expression in liver sinusoidal endothelial cells of hemophilia A mice. J Clin Invest. 2009;119:2086–2099. doi: 10.1172/JCI34332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Liu H, Mah C, Fletcher BS. Indoleamine 2,3-dioxygenase attenuates inhibitor development in gene-therapy-treated hemophilia A mice. Gene Ther. 2009;16:724–733. doi: 10.1038/gt.2009.13. [DOI] [PubMed] [Google Scholar]
- Liu L, Mah C, Fletcher BS. Sustained FVIII expression and phenotypic correction of hemophilia A in neonatal mice using an endothelial-targeted sleeping beauty transposon. Mol Ther. 2006;13:1006–1015. doi: 10.1016/j.ymthe.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Ohlfest JR, Frandsen JL, Fritz S, Lobitz PD, Perkinson SG, Clark KJ, et al. Phenotypic correction and long-term expression of factor VIII in hemophilic mice by immunotolerization and nonviral gene transfer using the Sleeping Beauty transposon system. Blood. 2005;105:2691–2698. doi: 10.1182/blood-2004-09-3496. [DOI] [PubMed] [Google Scholar]
- Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet. 2000;25:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- Riu E, Grimm D, Huang Z, Kay MA. Increased maintenance and persistence of transgenes by excision of expression cassettes from plasmid sequences in vivo. Hum Gene Ther. 2005;16:558–570. doi: 10.1089/hum.2005.16.558. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátrai J, Cantore A, Bartholomae CC, Annoni A, Wang W, Acosta-Sanchez A, et al. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology. 2011;53:1696–1707. doi: 10.1002/hep.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annoni A, Brown BD, Cantore A, Sergi LS, Naldini L, Roncarolo MG. In vivo delivery of a microRNA-regulated transgene induces antigen-specific regulatory T cells and promotes immunologic tolerance. Blood. 2009;114:5152–5161. doi: 10.1182/blood-2009-04-214569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61:1733–1743. doi: 10.1136/gutjnl-2011-301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Liu D. Hydrodynamic gene delivery: its principles and applications. Mol Ther. 2007;15:2063–2069. doi: 10.1038/sj.mt.6300314. [DOI] [PubMed] [Google Scholar]
- Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- Kettlun C, Galvan DL, George AL, Jr, Kaja A, Wilson MH. Manipulating piggyBac transposon chromosomal integration site selection in human cells. Mol Ther. 2011;19:1636–1644. doi: 10.1038/mt.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt K, Gogol-Döring A, Miskey C, Chen W, Cathomen T, Izsvák Z, et al. Retargeting sleeping beauty transposon insertions by engineered zinc finger DNA-binding domains. Mol Ther. 2012;20:1852–1862. doi: 10.1038/mt.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens JB, Mauro D, Stoytchev I, Bhakta MS, Kim MS, Segal DJ, et al. Transcription activator like effector (TALE)-directed piggyBac transposition in human cells. Nucleic Acids Res. 2013;41:9197–9207. doi: 10.1093/nar/gkt677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti SN, Huls H, Singh H, Dawson M, Figliola M, Olivares S, et al. Sleeping beauty system to redirect T-cell specificity for human applications. J Immunother. 2013;36:112–123. doi: 10.1097/CJI.0b013e3182811ce9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura K, Suda T, Zhang G, Aoyagi Y, Liu D. Parameters affecting image-guided, hydrodynamic gene delivery to swine liver. Mol Ther Nucleic Acids. 2013;2:e128. doi: 10.1038/mtna.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lobo N, Bauser CA, Fraser MJ., Jr The minimum internal and external sequence requirements for transposition of the eukaryotic transformation vector piggyBac. Mol Genet Genomics. 2001;266:190–198. doi: 10.1007/s004380100525. [DOI] [PubMed] [Google Scholar]
- Li X, Harrell RA, Handler AM, Beam T, Hennessy K, Fraser MJ., Jr piggyBac internal sequences are necessary for efficient transformation of target genomes. Insect Mol Biol. 2005;14:17–30. doi: 10.1111/j.1365-2583.2004.00525.x. [DOI] [PubMed] [Google Scholar]