

The rapidly expanding field of T-cell immunotherapy has experienced clinical successes along with some serious toxicities. “T Cell Immunotherapy: Optimizing Trial Design,” a workshop sponsored by the National Institutes of Health's (NIH's) Office of Biotechnology Activities (OBA), brought together researchers to discuss the scientific advances and share new data on key trial design issues, including the selection of new targets, optimizing the T-cell population, preconditioning regimens, strategies to promote persistence of cells, and analysis and management of acute reactions to T-cell infusions with the goal of identifying best practices and a research agenda that will facilitate further development and maximize the safety of this promising approach.
Introduction
T-cell immunotherapy for cancer is a rapidly growing field for gene therapy. Broadly, this field can be divided into two approaches—the use of gene-modified T-cell receptors (TCRs) in which recognition of the tumor antigen is in the context of human leukocyte antigens (HLAs) or use of chimeric antigen receptors (CARs) that typically link a single-chain variable region domain of an antibody (scFv) to one or more signaling elements of a TCR complex to allow T-cell activation.1 The decision to use one approach vs. the other may depend on several factors. For example, CARs offer the ability to bind antigens that are not restricted by HLA recognition, and the ability to modify the T-cell signaling moieties may offer “a broader functional effect than transduced” TCRs.2 TCRs, however, have the ability to recognize intracellular proteins, in addition to cell surface antigens, providing a broader array of target tumor-associated targets.
In 2010, the OBA hosted a meeting to examine the state of the science and key trial design questions for this emerging field.3 At the time, some clinical benefit and unexpected toxicities highlighted both the therapeutic potential as well as the need to share data and expertise to optimize the safety of trial design. Since 2010, several promising and clinically successful developments have been reported in leading scientific and medical journals4,5,6,7 as well as national media. Given these developments, the OBA and the NIH Recombinant DNA Advisory Committee concluded that it was an opportune time to reconvene the leading experts in the field from the United States to continue to foster sharing of data across protocols and discuss the key issues in trial design, including optimal management of the cytokine release syndrome (CRS) seen in some research participants in response to the expansion of these active T cells.
The following summary of the OBA workshop represents the views of the individual authors and not the NIH. The full presentations and slides are available at the OBA's website.8
State of the science
The number of CAR and TCR protocols registered with the OBA has continued to increase rapidly (Figure 1); as of the meeting in September 2013 there were 111 protocols, 104 of which targeted cancer, with more than 500 subjects dosed. More than 40 protocols address hematological malignancies, with CD19 being the most common target in these protocols. Among protocols for solid tumors, the melanoma antigens (gp100, MART-1) and cancer-testis antigens predominate for TCRs; for CARs there are multiple targets, with a slight predominance of Her2/neu, GD2, and mesothelin (Figures 2 and 3). Approximately 90% of TCR trials have targeted solid malignancies; approximately 50% of CAR trials have targeted hematological malignancies.
Figure 1.
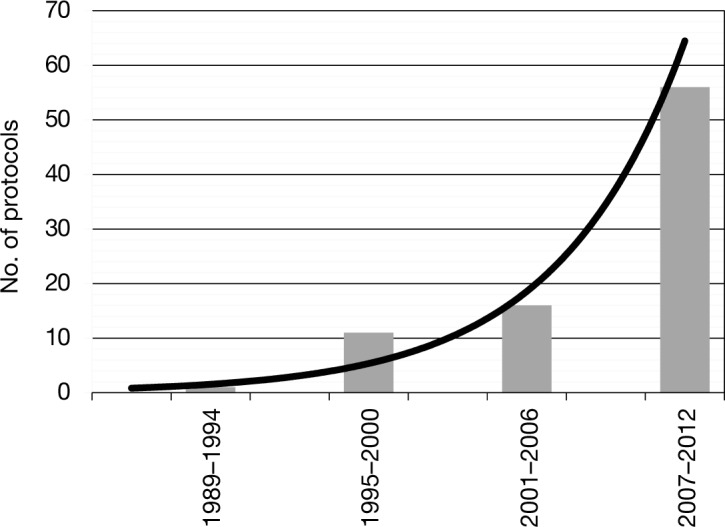
Number of chimeric antigen receptor protocols registered with the National Institutes of Health's Office of Biotechnology Activities by year.
Figure 2.
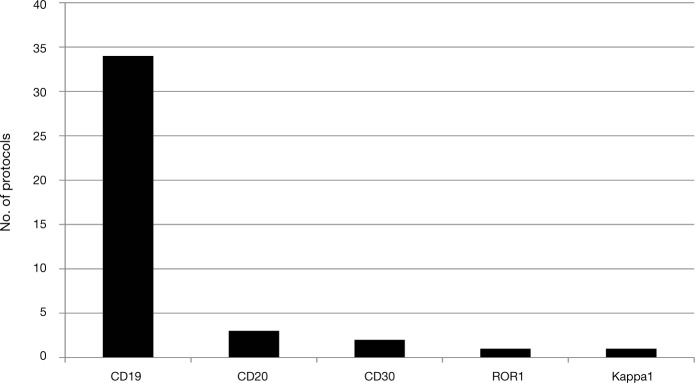
Chimeric antigen receptor targets for hematological-malignancy protocols registered with the National Institutes of Health's Office of Biotechnology Activities.
Figure 3.
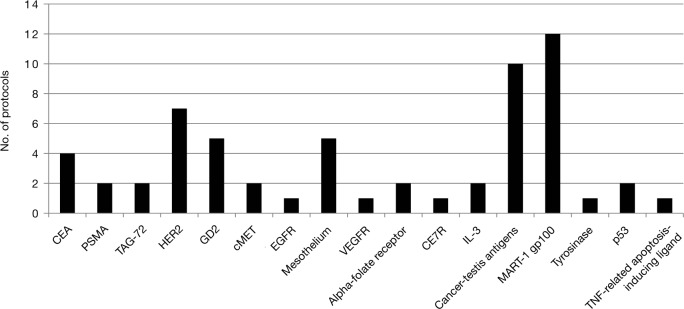
Chimeric antigen receptor targets for solid-malignancy protocols registered with the National Institutes of Health's Office of Biotechnology Activities. CEA, carcinoembryonic antigen; EGFR, epidermal growth factor receptor; IL-3, interleukin-3; PSMA, prostate-specific membrane antigen; TAG-72, tumor-associated antigen 72; TNF, tumor necrosis factor; VEGFR, vascular endothelial growth factor receptor.
Steven Rosenberg reviewed the extensive portfolio of National Cancer Institute (NCI) research in this area, beginning with a summary of his research using unmodified tumor-infiltrating lymphocytes (TILs) against melanoma in 1988. He began using lymphodepletion before administration of TILs in 2002 and demonstrated increased efficacy.9 Dr. Rosenberg has continued to apply this approach to melanoma, including ocular melanoma, as well as metastatic gastrointestinal and human papillomavirus–induced cancers. These studies have demonstrated that in a subset of patients (about 20%), administration of T cells can result in prolonged remissions of five years or longer. The results led to a program of research dedicated to gene-modified T cells that accounts for almost 20% of T-cell immunotherapy protocols registered with the OBA to date. The results of the Rosenberg group's first trials with gene-modified TCRs for melanoma were published in 2006 in Science.10 In a recent TCR study targeting the cancer-testis antigen NY-ESO-1, the overall response rate was 50% in the 19 subjects with melanoma, including 4 with complete remissions, and a 67% overall response for those with synovial sarcoma, including one complete remission, in a population that had multiple prior chemotherapy regimens.11 These results contrasted with the MAGE-A3 trial in which an unexpected off-target neurological toxicity was seen.12 Rosenberg's group has also developed an extensive portfolio of CAR protocols, focusing primarily on solid tumors, with novel targets such as vascular endothelial growth factor receptor 2 (VEGFR2), epidermal growth factor receptor variant III (EGFRvIII), and mesothelin, as well as new targets in development, such as chondroitin sulfate proteoglycan 4 (CSP4).
Antoni Ribas, who uses a vector developed by Rosenberg's lab, described his work on melanoma using a TCR-targeting MART-1 given with lymphodepletion. He has observed a high frequency of tumor responses (9 of 14 subjects with tumor-size reductions), but few responses were durable. He has also recently started enrolling research participants into a trial using a TCR-targeting NY-ESO-1. He noted that one of the aspects being tested is whether fresh cells are potentially more active than cryopreserved cells.
Other highlights included clinical results from several investigators targeting CD19 in leukemia and lymphoma. In addition to Dr. Rosenberg's summary of his work in this area,13 Carl June, Renier Brentjens, Laurence Cooper, Stephen Forman, Michael Jensen, Helen Heslop, and Crystal Mackall summarized their results in ongoing trials using CD19-specific CARs in leukemia and lymphoma.4,6,14 Dr. Heslop noted that in a trial comparing first- and second-generation CARs, her group found that the second-generation CAR demonstrated both improved expansion and persistence.15 In addition, several protocols have established that administration of CAR T cells after stem cell transplant does not interfere with engraftment of the transplant. The investigators presented examples of clinical remissions, but, because the goal is often to establish remission so as to proceed with a curative transplant, the durability of remissions from CAR T cells without subsequent transplant has not yet been determined. However, even in the setting of multiple previous therapies, CD19-specific CARs have shown efficacy. Dr. Brentjens reported that in his protocol with relapsed or refractory B-cell acute lymphoblastic leukemia (ALL), 14 of 16 subjects achieved molecular chronic remissions as assessed by deep-sequencing PCR analysis to search for the malignant clone.16 Another emerging theme was the responsiveness of ALL to this approach, which was also highlighted in Dr. June's and Dr. Mackall's presentations. Dr. Cooper presented data from ongoing trials infusing CD19-specific CAR+ T cells after autologous and allogeneic hematopoietic stem cell transplantation. The intent was to augment the graft-vs.-tumor effect, recognizing that the current clinical practice for many patients with B-cell malignancies is to infuse tumor-specific T cells as a bridge to transplantation. These trials have advanced a new approach to human gene therapy based on the electrotransfer of DNA plasmids encoding a second-generation CAR stably expressed following transposition from the Sleeping Beauty (SB) system.
In parallel to work on CD19-specific CARs, Brian Till highlighted the results of his trials targeting CD20, including a trial that used a third-generation CAR with CD28 and 4–1BB costimulatory domains. Unlike the other trials, which use retroviral vectors or SB transposons, he used an electroporated DNA plasmid. In general, the T cells were well tolerated, with some immediate febrile reactions, and two of the three subjects had prolonged remissions with persistence of the T cells for up to a year.17 However, the DNA plasmid vector was not an efficient vector, and the IL-2 used to promote persistence also led to an increase in T regulatory cells (Tregs).
Philip Greenberg highlighted his group's work using a TCR targeting another hematological malignancy antigen, Wilms tumor antigen 1 (WT1), which is highly expressed in leukemia and some solid tumors but is also expressed on some normal tissues. Their trial built on a previous trial using naturally isolated, cloned T cells targeting WT1, which did not show toxicity but had limited efficacy. Using virus-specific T cells, they have recently initiated a trial to test a TCR based on a high-avidity, natural clone.
In the solid-tumor area, Dr. Heslop presented a summary of her group's trials for neuroblastoma, targeting GD2 using both virus-specific and non-virus-specific T cells.18,19 Their data have demonstrated an association between persistence of T cells and reduced tumor progression. In addition, in research participants with prolonged detection of activated T cells, the presence of central memory T cells was important, raising the question of what the optimal T-cell product is.
Other solid-tumor trials discussed included CARs targeting HER2/neu for sarcoma and glioblastoma, including a trial using tri-virus-specific T cells and another trial that combines the CAR with a dominant-negative TGF-β receptor. Data were also presented on first- and second-generation CARs targeting carcinoembryonic antigen (CEA) and prostate-specific membrane antigen. Again, some early indications of clinical efficacy were promising, but an ongoing challenge will be to refine strategies to improve T-cell persistence and efficacy. In some cases, on-target, off-tissue toxicities may ultimately limit the use of certain targets; for example, colitis developed in protocols using CEA-specific TCR and CAR T cells.20
Finally, Dr. Jensen reported his work in glioblastoma using a novel CAR called a zetakine. Instead of an antibody, single-chain target domain, he used a human cytokine, IL-13, with a mutation in the sequence that gave high affinity for IL-13 receptor α2. These cells were infused intracranially, establishing the safety of intracranial administration with some antitumor responses.
These talks provided an overview of a field that continues to expand rapidly, in terms of both targets and diseases. Most protocols involve administration of the cells in the setting of lymphodepletion, and some groups, predominantly in protocols for solid tumors, use IL-2 to promote cell persistence. In addition to identifying effective targets that have minimal off-tumor effects, finding the ideal balance between persistence and expansion of T cells without triggering systemic cytokine reactions is a key issue for the field. This may be achieved by such strategies as including the design of the cells, the type of T cells infused, the dose, the immune status of the recipient, and the use of cytokine support. Finally, as with many cancer therapies, some toxicity is likely. Establishing protocols to limit toxicity so that the risk-to-benefit ratio remains favorable is a high priority.
Promoting T-cell persistence
Persistence of the gene-modified T cells is associated with prolonged remission in subjects,18 and the field has developed strategies to promote persistence. One approach is to create a host environment that is conducive to expansion of the T cells. Expansion should not only promote a rigorous antitumor effect but also lead to the development of a stable population of tumor-specific T cells that can be reactivated in case of recurrence of tumor antigen. Use of selected central memory T cells may be another strategy to promote an enduring T-cell population.
The majority of T-cell protocols registered with the OBA to date involve administration of the cells to subjects when they are lymphopenic. For solid-tumor protocols, this involves administration of the T cells after administration of lymphodepleting chemotherapy, such as cyclophosphamide, whereas the protocols for hematological malignancies have most commonly called for administering cells in the posttransplant setting or the use of disease-specific chemotherapy regimens. However, it is important to note that lymphodepletion has not been universally applied, notable exceptions being studies administering virus-specific T cells, or the successful neuroblastoma protocols targeting GD2, which used both virus-specific and non-virus-specific T cells.18
Dr. Rosenberg reviewed his group's clinical data, as well as the animal data that support lymphodepletion for promoting antitumor efficacy. As stated earlier, in the TIL melanoma studies, despite administration of 109 to 1010 T cells, the cells did not persist and there were minimal objective responses.21 However, when nonmyeloablative (NMA) chemotherapy using cyclophosphamide and fludarabine was added, and the TIL product was generated with a shorter culture time, providing a more diverse TIL population that contained both CD4+ and CD8+ T cells, T-cell persistence was enhanced and 6 of 13 subjects showed objective cancer responses.22 Dr. Rosenberg's group went on to investigate whether the addition of 2 or 12 Gy of total-body irradiation (TBI) to the NMA chemotherapy would further increase efficacy of TIL transfer in melanoma patients. The response rate for those who received chemotherapy alone was about 49%; the addition of 2 Gy resulted in objective response in 52% of subjects, and 12 Gy of TBI resulted in a 72% objective response rate, with a complete response rate of 40%.23 The addition of TBI to NMA chemotherapy was generally well tolerated, with the exception of one death in a subject with an undetected diverticular abscess in the 12-Gy group. A drawback of escalation to 12 Gy of radiation is the need for autologous peripheral blood stem cell support. An ongoing randomized trial is comparing NMA chemotherapy against NMA and TBI, although preliminary results indicate that the challenges of adding TBI may not be balanced by the improved response.
A significant amount of animal work has been done to elucidate the mechanisms that underlie the improved antitumor responses observed with lymphodepletion. These data indicate that lymphodepletion augments the antitumor response by eliminating Tregs, cellular “sinks” for cytokines such as IL-7 and IL-15, and by enhancing antigen-presenting cell activation and availability.24,25,26 This activation of the immune system may be due in part to translocation of bacteria from the gut. It was shown in a mouse model that administration of ciprofloxacin, which is effective against Gram-negative bacteria commonly found in the gut, to an irradiated animal reduced the activated dendritic cells in the spleen and reduced the effectiveness of adoptive cell transfer. Of note, it has been demonstrated that the effect of lymphodepletion is on the host rather than on the tumor. Thus, if one shields the host—in this case, the mouse—and treats the tumor, no effect is seen in these melanoma models.
One dilemma is that Tregs are the first T cells to recover after lymphodepletion, and therefore lymphodepletion may foster an environment that works against the antitumor effect. Dr. Rosenberg noted that the NCI group has some data demonstrating an inverse relationship between the recovery of Tregs and objective antitumor response, supporting the importance of eliminating Tregs. However, others questioned whether we clearly understand the role of Tregs, because suppression of a tumor response may depend on whether the Tregs are actually activated and tumor-specific. Therefore, the presence of Tregs may not be absolutely undesirable, as they may also organize the immune response.
Gene delivery and design of T cells
In addition to host preparation, the design of the T-cell vectors is a critical area of research. Dr. Cooper noted that the ability to stably express transgenes, such as CARs, in T cells has revolutionized adoptive immunotherapy for certain malignancies. Recombinant fusion genes constructed to recognize tumor-associated antigens (e.g., TCR and CAR) have been constitutively expressed in T cells using Moloney murine leukemia virus (MMLV)-based retroviruses, HIV-based lentiviruses, and DNA plasmids, including the SB transposon/transposase system.
Until recently, retroviral transduction by recombinant MMLV-derived vectors has been the most common method for delivery of transgenes intended to be integrated into the T-cell genome. Lentiviral vectors have also been successfully used in the clinic. Both approaches are appealing, and at this time there appears to be equipoise regarding the therapeutic potential of these two viral systems for genetic modification of T cells to express CARs. Transduction using retroviral and lentiviral vectors can be highly efficient, and it is possible to integrate multiple copies of a transgene in a given T cell, which provides for a high level of expression of the transduced gene product. The manufacture of clinical-grade retroviral and lentiviral vector virions is quite similar, although retroviral vectors may be produced from stable packaging cell lines, whereas to date most lentiviral vectors have been produced by transient transfection.
Overall, transduction of T cells with recombinant retrovirus and lentivirus involve similar packaging protocols, utilize similar integration mechanisms, and lead to similar transduction efficiencies. Thus, both viral-based approaches to gene transfer are appealing for the human application of CAR+ T cells, although some individual investigators have strong preferences.
DNA transposons now offer an alternative to viral-based gene transfer. Supercoiled plasmids can be directly electroporated into T cells using commercial devices, thus eliminating much of the labor and safety concerns associated with generating recombinant viral particles. DNA transposons, such as those derived from the SB system, insert into the genome via a copy-and-paste mechanism when a transposase is (transiently) available to catalyze the reaction. Dr. Cooper's group has successfully used SB to integrate a CD19-specific CAR into human T cells in four human trials under investigational new drug applications. Unlike retroviral/lentiviral integration into transcriptionally active sites, the SB transposon appears to randomly integrate at TA dinucleotide repeats and is typically present at one or two copies per T-cell genome. As with viral-based gene transfer, there is the possibility that a transposon may cause genotoxicity resulting in oncogenesis. However, because the SB system does not readily target transcriptional or promoter elements, it appears suitable for human application. Furthermore, the relatively low cost of generating DNA plasmids for use in compliance with current good manufacturing practice (GMP), in contrast to the cost and complexity of producing clinical-grade virus, renders the SB system an attractive and nimble approach to generate and modify vectors for delivery of therapeutic genes.
In summary, the investigator has available multiple approaches to genetically modify T cells. The use of a particular approach will depend on resident expertise and the desired T-cell product.
Design of CARs. CARs are recombinant receptors for antigens that retarget and eventually reprogram T-cell function. Unlike the physiological TCR for antigens, which signals T-cell activation through the associated CD3 complex, CARs possess in a single molecule the ability to trigger multiple antigen-specific T-cell functions. The CARs that have recently shown impressive clinical outcomes in research participants with B-cell malignancies are “second-generation CARs,” to distinguish them from earlier forms of activating fusion receptors, which only initiate T-cell activation and are now referred to as “first-generation CARs.”27
Michel Sadelain described how the incorporation of co-stimulatory receptor signaling domains into the cytoplasmic tails of CAR (“embedded costimulation”) greatly increased the potency of CAR-modified T cells in preclinical models.4,28,29 Several costimulatory domains have been incorporated in CARs over the past decade, including CD28 (ref. 28), 4–1BB (ref. 30), OX40 (ref. 31), and others (ref. 2). Different costimulatory molecules play roles in T-cell activation, proliferation, survival, cytokine secretion, antitumor cytolytic activity, and reactivation upon secondary stimulation. The second- and third-generation CARs have varying activities by recruiting multiple T-cell signaling pathways.2 Dr. Sadelain emphasized that small nuances in structural design of different CAR molecules can eventually exert a significant effect on the relative activity of CARs encoding the same signaling domains, depending on epitope position, CAR affinity, physical parameters of the extracellular domains, and transmembrane elements. Levels of CAR expression also affect overall function, making it an important parameter to consider when comparing different CARs. Forced expression of co-stimulatory ligands in the CAR T cells themselves can produce auto- or trans-costimulation and increase T-cell potency.32
Clinical efficacy has been reported in trials from several institutions for B-lineage malignancies using CAR-modified T cells.4,5,6,14,16,33,34 Many features of the trials differ, including CARs (origin of scFv, epitope of CD19 targeted, antigen affinity, signaling domains), enhancer/promoters (varied expression levels, propensity to silencing), T-cell manufacturing techniques (activation of T cells with antibodies to CD3 with or without anti-CD28, different culture media, duration of culture), cell products (cell dose, CD4/CD8 ratio, central memory T cells), lymphodepletion conditioning regimens (cyclophosphamide vs. cyclophosphamide/fludarabine vs. bendamustine), and patient selection (chemosensitive vs. chemoresistant disease). Future trials will need to define the relative importance of these differences to improve response rates. It is noteworthy that the outcomes of CD19 CAR therapy may vary depending on the disorder. Thus, results reported to date show greater efficacy in ALL than in chronic lymphocytic leukemia (CLL), for reasons that remain to be elucidated.
Design of T-cell receptors. TCRs are the physiological recognition system of T cells and react to a major histocompatibility complex–antigen complex. Their two chains, α and β, are necessary and sufficient for T cells to recognize their targets, including cancer cells. Engineering of T cells with genetically modified TCR α- and β-chains redirects their antigen specificity and has been used in the clinic in adoptive cell transfer strategies. Clinical trials expressing TCRs for MART-1, gp100, and NY ESO-1 have demonstrated antitumor activity in subjects with metastatic melanoma and sarcoma. However, these early clinical trials suggest that durable tumor responses seem to occur at lower frequency than with TILs or with CAR-engineered T cells.
The clinical trials thus far have used TCRs with physiological peptide affinities, and most have used intact TCRs. However, studies with NY ESO-1 and MAGE-A3 as targets used TCRs with altered affinities due to targeted mutations in their complementarity-determining region 2 or 3 (CDR2 or CDR3), the variable regions of the TCR that interact with the major histocompatibility complex–antigen complex. However, care must be taken because a CDR2-modified MAGE-A3 TCR led to cardiac toxicities, due to loss of specificity with cross-reaction to an off-target peptide.35
Other means to increase antitumor activity of TCR-modified T cells are being developed preclinically, such as additional genetic engineering of the T cells to express other immune-activating genes, engineering the signaling pathways downstream of the TCR, or blocking negative regulatory receptors. These approaches would provide simultaneous genetic redirection of T cells with increased T-cell functionality that may no longer be blocked by physiological immune regulatory processes.
A problem with some transgenic TCRs is that, when expressed in T cells that have their own endogenous TCR α- and β-chains, there can be heterologous pairing between the transgenic and endogenous TCR chains. This may decrease the expression of the transgenic TCR and even lead to altered specificities that may potentially result in autoimmune toxicities. Several means to improve self-pairing of the transgenic TCR chains include the use of picornavirus-derived highly efficient self-cleaving 2A-like sequences to allow stoichiometric protein expression, including additional cysteine motifs allowing formation of an increased number of disulfide bonds between the α- and β-chains, partially murinizing the constant region of both TCR chains for preferential pairing, and the use of leucine zippers at the 3′ ends of both α- and β-chains for forced transgenic TCR pairing. As these approaches move into the clinic, it will be important to test them in carefully designed clinical trials to minimize risks but also foster continued improvements in treatment options.
Longer-term antitumor activity may be achievable by targeting hematopoietic stem cells (HSCs), which would continually produce transduced T cells. David Baltimore listed potential advantages of targeting HSCs. Because of the requirement for coexpression of CD3, transgenic TCRs can be expressed only on the surface of T cells derived from the transduced HSCs. The TCRs introduced by the vector should allelically exclude the rearrangement of endogenous TCR genes to yield monoclonal cells. However, one potential limitation of this approach may be that highly active T cells from HSCs that contain highly avid TCRs for self-antigens may be selected out by the thymus. In the trials using a MART-specific TCR, clinical effect was observed when the avidity of the natural TCR was increased several-fold, but such highly active T cells may be negatively selected by the thymus.
HSCs transduced with CAR vectors produce CAR-expressing myeloid and natural killer cells in addition to T cells, and thus may provide more rapid and broader antitumor activity.36 In a mouse model with an EL4 tumor expressing the ovalbumin gene, an antitumor effect was observed using HSCs transduced with lentiviral vectors expressing TCR reactive to ovalbumin. A clinical trial involving autologous CD34+ cells transduced with a lentiviral vector expressing a CD19+ CAR in subjects with non-Hodgkin's lymphoma is being developed at UCLA and the City of Hope Medical Center.
Target selection
Dr. Rosenberg reviewed the status of target selection, which he viewed as the critical challenge confronting immunotherapy. He considered the targets identified thus far to fall into five categories. The category that has been most extensively studied with TCRs is differentiation antigens that are overexpressed on cancers compared with normal tissues (e.g., MART-1, gp100, CEA, HER-2). As with conventional chemotherapy, this approach requires identifying a window of toxicity against the tumor cells without unacceptable damage to normal tissue. In the studies using the melanocyte differentiation antigens, an approximately 25% objective response rate was obtained; however, normal melanocytes were also attacked, causing skin rashes, uveitis, and auditory and vestibular problems, all of which could be reversed by steroid treatment.10,37 Similar problems occurred with targeting of CEA, which is expressed at low levels on colonic epithelium, resulting in temporary but almost complete destruction of that tissue,20 and with HER-2 targeting, which results in severe adverse effects on the pulmonary epithelium and death of the subject.38 T-cell therapy is highly potent but also so sensitive that the T cells can recognize even extremely low target expression in normal tissues. This potential for on-target, off-tumor toxicity has limited the development of certain targets as an effective cancer treatment.
The second class of targets includes antigens expressed on tumor cells and relatively nonessential normal tissues. This includes CD19 as targeted by CARs and thyroglobulin, targeted by TCRs for thyroid cancer. This approach is promising but requires identification of additional tissue-specific proteins on other nonessential organs such as the prostate, ovary, or breast. Another class is shared antigens expressed on multiple tumor types, which includes cancer-testis antigens. An example of this class is NY-ESO-1 that is not expressed on normal tissue but also not on many solid tumors at high concentrations. More than 100 cancer-testis antigens have been identified; however, careful screening studies must be conducted to determine whether normal tissues are also targeted. A modified MAGE-A3 TCR led to cardiac toxicity due to unexpected cross-reaction with a cardiac muscle protein titin.39 A TCR that recognized epitopes from MAGE-A3/A9/A12 led to unexpected neurological toxicity due to cross-reactivity with an epitope on MAGE-A12 expressed in the white matter of the brain.40
The fourth category is antigens resulting from mutations unique to a particular cancer. One such candidate antigen is EGFRvIII, which is expressed in 30–50% of glioblastomas but not normal tissues. Loss variants are unlikely in this class because the mutation is essential for the malignant phenotype. Highly specific antibodies are available for the development of CARs.41 Finally, rather than tumor antigens, the fifth category targets critical components of the tumor stroma such as VEGFR2 (ref. 42). Targeting VEGFR2 may result in the destruction of tumor vasculature. However, because it is also expressed in normal vessels, clinical studies will need to be designed with protracted dose escalations to avoid toxicity.
The future of immunotherapy may be a personalized approached in which tumor samples from each patient are analyzed to identify the specific antigens to target using retroviral vectors introduced into autologous lymphocytes. Exomic sequencing is being applied to identify somatic mutations through the comparison of tumor and matched normal cellular DNA. HLA-binding algorithms are used to identify candidate epitopes.43 This approach can be used to develop either patient-specific TCRs or tumor-infiltrating lymphocytes.
Dr. Sadelain presented a novel method to increase the specificity of T cells for unique combinations of antigens on tumor cells in which two separate chimeric proteins are expressed in T cells, one consisting of a low-affinity first-generation CAR targeting one tumor-associated antigen, the other consisting of a costimulatory domain fused to an antigen-binding domain (termed a chimeric costimulatory receptor, which does not activate T cells and is therefore not a CAR) targeting a second antigen.44 Only when both components are triggered are the T cells sufficiently activated to achieve cytolytic activity. The activity of first-, second-, and third-generation CARs increases via induction of multiple T-cell signaling pathways.2
Dosing considerations
In 2010, when the NIH first hosted a conference on clinical trial design for T-cell immunotherapy, considerable attention was focused on finding the correct dose. Now almost three years later, there is much more experience in dosing. Dr. Brentjens noted that, in published studies using CD19-specific CARs, the doses used were in the range of 3 × 106 to 3 × 107 cells/kg. Of course, with some novel antigen targets, the initial dose may need to be more conservative, starting as low as 106 cells and then escalating to target doses of 109 or 1010 cells. Not surprisingly, the initial dose and escalation protocol must take into account multiple factors, including which disease is being targeted, the CAR design, the conditioning, and the gene transfer technology.
In the published literature on CD19 CAR protocols, there was not a clear correlation between dose and efficacy. This may reflect in part that the actual dose for an individual patient depends on the degree of expansion of the cells. On the other hand, one must be cautious in extending this observation to solid tumors. In hematological malignancies that express CD19, a considerable number of cells express the target antigen, which may facilitate expansion of the T cells even at lower doses. For solid tumors, there may be a threshold dose that must be reached to obtain antitumor response. Indeed, Dr. Rosenberg noted that in his protocol for glioblastoma targeting EGFRvIII they have not seen antitumor responses in their initial dose cohorts, which may indicate a therapeutic threshold.
Another question is whether split dosing adds to safety. In response to an early toxicity on a CD19 CAR protocol,45 which is now thought not to be directly related to the T cells, the Memorial Sloan-Kettering group instituted split dosing; a fraction of the T cells were given the first day and the remainder on subsequent days, provided there was no initial toxicity. Other groups also instituted split dosing, but it was not uniformly adopted. To date, there is no evidence that this approach improves safety, but the experience is still limited. In part, it may reflect that dose-related toxicities often occur days, not hours, after T-cell administration. On the other hand, some have postulated that split dosing might enhance efficacy by “priming” the immune system, upregulating certain molecules, and altering trafficking of cells.
Finally, one investigator advocated forgoing lymphodepletion with new CARs and testing for toxicity of second-generation CARS without promoting engraftment.46 This approach has not been favored by most investigators.
Managing the unexpected: CRS and other adverse reactions
The OBA received reports for about 40 severe adverse events (SAEs) that were possibly related to the infusion of genetically modified T cells over the period from July 2010 to August 2013. Key issues pertaining to the adverse events associated with CAR and TCR therapy were discussed at the meeting, including: B-cell aplasia; systemic inflammatory release syndrome, also known as CRS; tumor lysis syndrome, central nervous system (CNS) toxicity; macrophage activation syndrome; and off-target toxicities.
B-cell aplasia is an expected on-target result of CD19- or CD20-directed therapies, and has served as a useful surrogate to determine persistence and effectiveness of CAR T cells. B cells recover when engraftment of the CAR T cells is lost. The duration of B-cell aplasia can be at least three years, based on the updated results from the University of Pennsylvania.34 Fortunately, B-cell aplasia is a manageable disorder; patients may be infused with gamma globulin as replacement therapy, although this could become an expensive and difficult treatment to implement across all B-lineage malignancies that may eventually be treated with CAR T cells. Persistent B-cell aplasia may also result in an increased risk of infection, even with prophylactic replacement therapy. In an ideal setting, the CAR T cells would persist long enough to eradicate disease but then allow for recovery of normal B cells and plasma cells such that patients could be revaccinated. However, the long-term persistence of dormant tumor cells in humans creates a conundrum: the desire to minimize B-cell aplasia while retaining the potential to cure patients.47
Severe infusional toxicity, occurring within an hour of T-cell infusion, has rarely been reported following the infusion of genetically modified T cells. Anaphylaxis has occurred following the repeated infusion of CAR T cells expressing a mouse scFv.48 Fatal infusional toxicity occurred when a high dose of CAR T cells specific for Her2/neu was infused.38 However, at the OBA meeting Stephen Gottschalk reported encouraging results with low doses of CAR T cells specific for HER2/neu. His preliminary data suggest that it may be possible to use a “sneak-through” strategy to deliver CAR T cells to a tumor bed distal to the cardiopulmonary system, thereby increasing the therapeutic index through dose reduction.
The most commonly reported adverse event is CRS,49 with about three-quarters of the patients with CRS requiring admission to an intensive care unit. In the case of CAR therapy, the onset of CRS is related to the particular signaling domain in the CAR, with early-onset CRS in the first several days after infusion related to CARs that encode a CD28 signaling domain.4,16 By contrast, CARs encoding a 4-1BB signaling domain tend to have delayed-onset CRS (range, 7 to 50 days) after CAR T-cell infusion.6 CRS has also been reported after the infusion of TCR-modified T cells, with onset typically five to seven days after infusion. The development of CRS is often, but not invariably, associated with clinically beneficial tumor regression. Several cytokines have been reported to be elevated in the serum—most commonly, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and interleukin (IL)-6. Management of CRS has included supportive care, corticosteroids, etanercept, tocilizumab, and alemtuzumab. The role of suicide genes in the management of CRS remains unknown.50
Given the finding that delayed-onset CRS seems to correlate with antitumor activity and proliferation of TCR- or CAR-modified T cells, one question that has emerged is the degree to which the innate immune system contributes to the antitumor efficacy. It is possible that the IL-6 is produced by the dying B cells, dying tumor cells, or activated macrophages that are recruited to digest lysed tumor cells. Although it is straightforward to hypothesize that CAR T cells directly kill tumor cells, it is not entirely clear which cell type produces the vast majority of the cytokines, and whether blockade of cytokines with anti-cytokine therapy such as tocilizumab and etanercept or general immune suppression with corticosteroids affects the antitumor response. It should be noted that steroid-refractory graft-vs.-host disease (GvHD) occurs51 and has been managed by T cell–directed therapies such as infusion of anti-CD3 or antithymocyte globulin, offering another strategy for CRS if anticytokine or general immunosuppression fails.
Does interruption of the cytokine cascade lead to interruption of the antitumor effect? This remains an unanswered question that has direct clinical impact for patients and physicians deciding on when to abort the CRS. Furthermore, there was consensus at the workshop that, although all responding patients have some degree of CRS, it is not yet clear whether its severity is related to antitumor efficacy, although it does appear to be related to the tumor burden.4 If engagement of the innate immune system contributes to the mechanism of action, this could bode well for the use of CAR T cells in solid tumors, where T cells may not preferentially home to and persist at the sites of tumors as efficiently as they do in hematological malignancies.
Several patients in CD19 CAR trials across institutions have experienced obtundation, seizures, aphasia, and mental status changes, all of which have been reversible. Some of these may be related to CRS, but it is not clear whether they result from systemic cytokines crossing the blood–brain barrier and engaging cytokine receptors in the brain or from direct cytokine production in the CNS. IL-6 is known to alter astrocyte function,52 and it is possible that this enhances cytokines directly in the CNS. Many of these patients also develop macrophage activation syndrome (MAS) with striking elevations of serum ferritin levels, and MAS itself is often associated with neurological toxicity.53,54,55 In addition, CAR T cells have been unexpectedly found in the cerebral spinal fluid (CSF) of asymptomatic patients, even when there is no evidence of CD19+ disease in the CNS.5 It is possible that the hyperthermia and IL-6 released during CRS enhances trafficking of CAR T cells to the CSF via an antigen-independent mechanism.56 It is also possible that there is some cross-reactivity or as-yet-undetected expression of CD19 in the brain. Blinatumomab, a type of bispecific T cell–engaging antibody that is a fusion protein between an anti-CD19 scFv and an anti-CD3 scFv, has neurological toxicity and seizures as its dose-limiting toxicity, even though it does not appear to control CNS disease. It is interesting that blinatumomab has also been shown to cause MAS.57 Optimistically, engineered T cells may provide a way of controlling occult or frank CNS malignancy without chemotherapy or radiotherapy, given the trafficking to CSF that has been observed in leukemia and melanoma patients.5,12
On-target, off-tumor toxicity has been reported with CAR and TCR trials. The first incident was hepatotoxicity following infusion of carboxyanhydrase IX–specific CAR T cells, due, in retrospect, to previously unknown expression of the target in the biliary tract.58 Toxicity has also occurred in four cases with TCR-modified T cells, when MAGE-A3-specific T cells reacted with the same epitope expressed on MAGE-A12 in the CNS.12 Off-target reactivity following infusion of T cells engineered to express a MAGE-A3 TCR has also occurred, resulting in severe cardiac toxicity in two cases.35,39 As gene-modified T cells are emerging as powerful therapies capable of effecting dramatic antitumor responses as well as significant toxicities, strategies to incorporate suicide genes or abortive mechanisms may become necessary to manage on-target, off-tumor toxicities.51,59
Suicide-gene strategies
Because T cells that have been genetically modified with artificial receptors may persist and expand in number, any adverse effect such as CRS or GvHD may be prolonged and even worsen as the cells expand. Therefore, there has been considerable interest in including a suicide gene in constructs so that genetically modified cells can be destroyed as necessary by exposure to a specific signal. Although ablation of infused cells may not abrogate all adverse reactions if other immune effector mechanisms have been activated, suicide genes are being included in several constructs as a safety switch. These suicide genes may need to be activated at different stages of an adverse reaction, depending on the properties of each construct, such as speed and potency.
Three suicide genes have been included in constructs infused in the clinic: herpes simplex viral thymidine kinase (Tk) gene, an inducible caspase 9 (iC9), and a truncated epithelial growth factor receptor (EGFR) gene. The latter could be characterized as a targeted way of removing the T cells.
The most widely used suicide gene has been Tk, the product of which phosphorylates ganciclovir or acyclovir to the active moiety that interferes with DNA synthesis.60 In studies where this construct has been transferred into donor T lymphocytes infused following stem cell transplantation, administration of the ganciclovir prodrug has controlled GvHD, and the approach has now progressed to a phase III clinical trial. One limitation of Tk is that the product is itself immunogenic, leading to undesired elimination of the transduced T-cell population.61,62 Moreover, the mechanism of action predominantly targets DNA synthesis in dividing cells and may therefore take days, or even weeks, to produce maximum effects. Even then, killing of the transduced T cells may be incomplete, a problem for treating acute adverse events or when the targeted cells are postmitotic. An advantage of this system is that the activating drug (typically ganciclovir) is commercially available. However, at least in the transplant setting, ganciclovir is often needed to treat cytomegalovirus reactivation, which can't be done without ablating the T cells.
The truncated EGFR gene is included in some constructs but has not yet been used in the clinic to ablate the T cells. Like Tk, it can be used to eliminate transduced T cells with a commercially available drug—in this case, cetuximab, which induces antibody-dependent cell-mediated cytotoxicity over 24–48 hours.
More rapid cell destruction can be obtained using iC9, in which a modification to a component of the caspase pathway that is nonimmunogenic rapidly—in less than an hour—produces apoptosis even in nondividing cells.63 The molecule can be activated by administration of a small-molecule dimerizer (AP1903) that links two nonfunctional iC9 molecules to form the active enzyme. In a clinical trial, iC9-transduced donor cells administered after allogeneic transplant had good engraftment and functionality, but in research participants who developed GvHD these cells were destroyed within minutes of administering the dimerizer drug, with sustained clinical resolution of GvHD.50 Although this rapid action would be beneficial in treating any adverse effects of TCR or CAR gene-modified T cells, AP1903 is an experimental drug and is available only through a collaboration with the AP1903 supplier, Bellicum Pharmaceuticals. At present, because the drug can be accessed only from an investigational pharmacy at a study site, research participants must stay close to the study site until the time of greatest risk of adverse events has passed.
The next step: commercialization
The field is approaching a point of maturation where it is appropriate to consider what steps will be necessary to translate the technology into US Food and Drug Administration (FDA)-approved products. Currently, research has been led for the most part by academic laboratories; however, attaining the ultimate goal of licensed products will, in most cases, require industry partnerships. Several such partnerships already exist, including collaborations between (i) the University of Pennsylvania and Novartis to develop CAR immunotherapies and to establish a joint Center for Advanced Cellular Therapies; (ii) Baylor College of Medicine, Celgene, and bluebird bio; (iii) the cooperative research and development agreement between the Surgery Branch of the NCI, and Kite Pharma; and (iv) Juno Therapeutics, recently formed by scientific founders at the Fred Hutchinson Cancer Research Center, Seattle Children's Hospital, and Memorial Sloan-Kettering Cancer Center. A challenge for the field will be to foster growth while balancing the maintenance of academic independence against the needs of industry partners supporting the later steps to commercialization.
In designing later-stage trials, a key issue may be establishing an acceptable level of toxicity. For example, any life-threatening toxicities are often transient or reversible (e.g., confusion, hypotension). If acceptable to the FDA, the field may need to establish the level of expected toxicities that will not be dose-limiting toxicities, much in the same way chemotherapy is expected to produce myelosuppression that can lead to certain predicted morbidities and even mortality that is balanced against the prognosis and potential benefit.
Another issue for the field is whether a localized or central model for cell production is more suitable. Each institution could establish a GMP facility, presumably in a blood bank; however, this would require each institution to conduct manufacturing studies to obtain FDA approval. Alternatively, a central facility could produce cells for multiple sites. The NIH is developing a cooperative research and development agreement to work with a commercial company. Other institutions, such as Memorial Sloan-Kettering Cancer Center and the University of Pennsylvania, are collaborating to share investigational new drug applications, exchange standard operating procedures, and cross-train investigators. The central model would require cryopreserved products for shipping. The field must conduct further studies to compare the efficacy of fresh vs. frozen cell products. Successful cryopreservation would be one criterion for the development of off-the-shelf (OTS) products.
To date, most research has involved autologous or donor-specific products prepared for a single subject. The development of allogeneic products capable of being stored would allow for production in advance of patient need and avoid real-time manufacturing concerns. One possible approach would be to introduce into T cells both the CAR genetic modification and zinc-finger nucleases to eliminate expression of endogenous TCRs.64 The clinical translation of such OTS T cells is appealing because a third-party donor can be used to generate a biological product ahead of need, enabling T cells to be delivered on demand rather than when available. This approach would lend itself to centralized manufacturing and multicenter trials to establish the maximally tolerated dose and thus a pathway to combining OTS T cells with other therapeutic investigations. Clinical studies using allogeneic cells in multiple recipients include a glioblastoma trial at City of Hope, using cord blood–derived CD8+ cytotoxic T cells expressing IL-13-zetakine in which rejection was expected to be slowed by the intratumoral method of administration and the use of dexamethasone, and the virus-specific cytotoxic T-cell trials conducted at Baylor.
From a regulatory perspective, key issues included collecting information regarding product characterization to provide comparability data to facilitate the transition of manufacturing to commercial facilities. Potency assays will need to be developed.65 The experience of the three autologous cell products that have been licensed to date should be informative for the field. Efficacy data will need to be collected by a registration trial, which may not necessarily have to be a phase III trial. TCR and CAR approaches, particularly for ALL, may be candidates for breakthrough-therapy designation, which would convey fast-track program features and intensive FDA guidance.
Appropriate larger-animal models may also need to be developed. Nonhuman primate (NHP) models have proven useful for the study of the engraftment, persistence, and safety of genetically modified cells; however, there are no appropriate NHP tumor models. One useful tumor model is lymphoma in dogs. Adoptive T-cell studies in veterinary trials in companion dogs provide both a preclinical model for human studies and the potential for a therapeutic outcome for the dogs.66
During discussion of resources needed to move the field forward, one resource that was considered critical by the workshop participants was access to GMP materials required for product development. Unlike most other pharmaceutical products, TCRs and CARs, because of their biological nature, require the use of multiple reagents and materials (e.g., medium, RetroNectin for vector transduction, CD3/CD28 beads for the activation and expansion of T cells), and some of these reagents are controlled by single sources. Concern was expressed about exclusive licensing agreements between the manufacturers of required reagents and industry. For example, Life Technologies and Novartis have entered into an exclusive license and supply agreement for Dynabeads CD3/CD28 CTS for use in CAR applications.67 Depending on the terms of these agreements, such arrangements could potentially limit access by academic investigators to reagents beyond initial research trials. Although for certain reagents it might be possible to develop alternative technologies (such as artificial antigen-presenting cells to propagate T cells), the time spent developing alternatives slows the field's progress. For some reagents, such as RetroNectin, developing an alternative may be particularly difficult. The participants noted the need for a means of continued access to reagents that are critical to the manufacture of the T cells.
The participants also expressed interest in the development of additional means to share data. Dr. Rosenberg suggested that it would be useful for the investigators to share information in some type of compendium of results, including the number of partial and complete responses, with durability of responses broken down by adult vs. pediatric populations and whether transplants were subsequently performed. Developing a way to allow for online sharing of information about SAEs was also considered useful. Most of the clinical trials conducted so far have been small, single-site trials, which may have limited the opportunities for sharing of SAE experiences. In particular, it would be beneficial for research participants if information about successful management of similar SAE experiences could be shared among investigators at other sites. One option is for the OBA to use its access to safety and annual reports to set up a mechanism for sharing of data across investigators.
Conclusion
Significant progress has been made in the adoptive T-cell therapy field, with improvements in efficacy, especially in the use of CD19+ CARs for hematological malignancies. However, there are still critical questions, including the optimal design of the receptor, CARs vs. TCRs, the subset of T cells to use, and the effect of freezing T cells on efficacy. Although most protocols administer cells to research participants who are lymphopenic, the optimal host preparation that allows for engraftment and persistence of cells but limits toxicity may be different for solid malignancies than for hematological malignancies. To date, this therapy has been tested in research participants for whom other standard therapies for their advanced malignancies have failed; therefore, there has been a greater tolerance for risk. A common toxicity is CRS, and, although a variety of management strategies are emerging, there is still a limited understanding of whether certain subjects are more susceptible and whether there is an ideal time to intervene to prevent morbidity without limiting the antitumor response. In addition, the role of suicide genes to ameliorate immediate as well as long-term toxicity remains to be established. Although some investigators expressed concerns that regulators may be overly focused on toxicity, which may be comparable to other oncology treatments, determining an acceptable level of toxicity is difficult in the absence of a larger body of data on efficacy. Similarly, although much has been learned about target selection, unexpected off-target toxicity not identified by extensive preclinical testing is a reminder that there are certainly unknown risks, and conservative design of new trials with novel targets is therefore warranted.
A challenge for the field is to identify tumor-associated antigens that can be safely targeted without clinically severe toxicities arising from on-target, off-tissue recognition by genetically modified T cells. The field has already witnessed several major adverse events, and programs that are generating CAR+ and TCR+ T cells with new specificities will need to address the potential for deleterious targeting of normal structures.
As research on these important questions continues, certain cell products show the potential to provide real clinical benefit for patients. In the past few years, the private sector has recognized this potential and is partnering with investigators to bring these therapies into standard medical care. Decisions on how best to commercialize these products, which to date have largely been personalized, as opposed to third-party OTS products, will need to be made, including decisions on centralization of manufacturing. Involvement of private interests may also bring new challenges to a field that emerged in academia, where information and reagent sharing is the norm. Nonetheless, the potential of this field to provide new options for patients appears closer to reality, both in the United States and overseas, and the recent emergence of biotechnology partnerships is anticipated to increase funding and accelerate progress. It has rightly earned its place in Science's top scientific breakthrough for 2013.68
Acknowledgments
The authors thank all the participants for their contributions. The views expressed in this paper are those of the individual authors and do not represent the views of the NIH or the FDA.
C.J. has sponsored research support from Novartis through a grant that supports research in the field of CARs for oncology. He also has intellectual property in the area of adoptive cell therapy that is owned by the University of Pennsylvania and the US government. This has been licensed to Novartis. Both the university and C.J. may receive royalties; this arrangement is managed in accordance with the University of Pennsylvania policies and oversight.
L.C. founded and owns InCellerate, Inc. He has patents with Sangamo BioSciences with artificial nucleases. He consults with Targazyme, Inc. (formerly American Stem cells, Inc.), GE Healthcare, Ferring Pharmaceuticals, Inc., and Bristol-Myers Squibb. He receives honoraria from Miltenyi Biotec. D.P. holds patents for CARs that are licensed to companies or institutions that may gain or lose value from publication of the article. R.J.B., M.S., and P.G. are scientific founders and stockholders in Juno Therapeutics, Inc., as well as consultants for the company.
H.H. has a licensing agreement with Cell Medica, and her institution has a collaborative research agreement with Celgene Corporation. C.M. has a patent pending on CD22-CAR and has received research funding from Neomune. M.J. is a consultant, inventor of licensed patents, recipient of SRA funding, and equity holder in Juno Therapeutics, Inc.
S.G.'s institution has research collaborations with Celgene Corporation and bluebird bio. He has been funded by the DANA Foundation, the James S. McDonnell Foundation, and Cancer Prevention Research Institute Texas grant RP101335. H.H. and S.F. are supported by grants P01CA094237, P50CA126752, and U54HL08100 from the NIH and a SCOR award from the Leukemia and Lymphoma Society.
References
- Eshhar Z, Waks T, Gross G., and, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R., and, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl HC, Zaia J, Rosenberg S, June C, Dotti G, Kahn J.et al. (2011Considerations for the clinical application of chimeric antigen receptor T cells: observations from a Recombinant DNA Advisory Committee Symposium held June 15, 2010 Cancer Res 713175–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens R, Davila M, Riviere I, Park J, Wang X, Cowell L.et al. (2013CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia Sci Transl Med 5177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter D, Rheingold R.et al. (2013Chimeric antigen receptor–modified T cells for acute lymphoid leukemia N Engl J Med 3681509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D, Levine B, Kalos M, Bagg A., and, June C. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz S. Engineered T-cell therapy shows efficacy in blood cancer. Nat Biotechnol. 2011;29:853–855. doi: 10.1038/nbt1011-853. [DOI] [PubMed] [Google Scholar]
- National Institutes of Health Office of Biotechnology Activities. Scientific symposium—T cell immunotherapy—optimizing trial design < http://osp.od.nih.gov/office-biotechnology-activities/event/2013-09-10-123000-2013-09-11-161500/scientific-symposium-t-cell-immunotherapy-optimizing-trial-design > ( September 2013
- Rosenberg S., and, Dudley M. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R, Dudley M, Wunderlich J, Hughes M, Yang J, Sherry R.et al. (2006Cancer regression in patients after transfer of genetically engineered lymphocytes Science 314126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins P, Morgan R, Feldman S, Yang J, Sherry R, Dudley M.et al. (2011Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive in NY-ESO-1 J Clin Oncol 29917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R, Chinnasamy N, Abate-Daga D, Gros A, Robbins P, Zheng Z.et al. (2013Cancer regression and neurological toxicity following anti-MAGE-A3 gene therapy J Immunother 26133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer J, Wilson W, Janik J, Dudley M, Stetler-Stevenson M, Feldman S.et al. (2010Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19 Blood 1164099–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens R, Riviere I, Park J, Davila M, Wang X, Stefanski J.et al. (2011Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias Blood 1184817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo B, Ramos C, Liu E, Mims MP, Keating MJ, Carrum G.et al. (2011CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients J Clin Invest 1211822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila M, Riviere I, Wang X, Bartido S, Park J, Curran K.et al. (2014Efficacy and toxicity management of 19–28z CAR T cell therapy in a B cell acute lymphoblastic leukemia Sci Transl Med 6224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till B, Jensen M, Wang J, Qian X, Gopal A, Maloney D.et al. (2012CD20-specific adoptive immunotherapy for lymphomas using a chimeric antigen receptor with both CD28 and 4–1BB domains: pilot clinical trial results Blood 1193940–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis C, Savoldo B, Dotti G, Pule M, Yvon E, Meyers G.et al. (2011Antitumor activity and long-term fate of chimeric antigen receptor–positive T cells in patients with neuroblastoma Blood 1186050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G.et al. (2008Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma Nat Med 141264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst M, Yang J, Langan R, Dudley M, Nathan D, Feldman S.et al. (2011T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis Mol Ther 19620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley M, Wunderlich J, Nishimura M, Yu D, Yang J, Topalian S.et al. (2001Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma J Immunother 24363–373. [DOI] [PubMed] [Google Scholar]
- Rosenberg S., and, Dudley M. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA. 2004;101:14639–14645. doi: 10.1073/pnas.0405730101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley M, Yang J, Sherry R, Hughes M, Royal R, Kammula U.et al. (2008Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens J Clin Oncol 265233–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony P, Piccirillo C, Akpinarli A, Finkelstein S, Speiss P, Surman D.et al. (2005CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells J Immunol 1742591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Finkelstein S, Klebanoff C, Antony P, Palmer D, Spiess P.et al. (2005Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells J Exp Med 202907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulos CM, Wrzesinski C, Kaiser A, Hinrichs C, Chieppa M, Cassard L.et al. (2007Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling J Clin Invest 117492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R., and, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher J, Brentjens R, Gunset G, Riviere I., and, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- Brentjens R, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K.et al. (2007Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts Clin Cancer Res 135426–5435. [DOI] [PubMed] [Google Scholar]
- Imai C, Mihara K, Andreansky M, Nicholson I, Pui C, Geiger T.et al. (2004Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia Leukemia 18676–684. [DOI] [PubMed] [Google Scholar]
- Pule MA, Straathof K, Dotti G, Heslop H, Rooney C, Brenner M.et al. (2005A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells Mol Ther 12933–941. [DOI] [PubMed] [Google Scholar]
- Stephan M, Ponomarev V, Brentjens R, Chang A, Dobrenkov K, Heller G.et al. (2007T cell-encoded CD80 and 4–1BBL induce auto- and transcostimulation, resulting in potent tumor rejection Nat Med 131440–1449. [DOI] [PubMed] [Google Scholar]
- Korchenderfer J, Dudley M, Feldman S, Wilson W, Spaner D, Maric I.et al. (2012B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells Blood 1192709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A.et al. (2011T cells expressing chimeric receptors establish memory and potent antitumor effects in patients with advanced leukemia Sci Transl Med 395ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette G, Stadtmauer E, Maus M, Rapoport A, Levine B, Emery L.et al. (2013Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma Blood 122863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschweng R, De Oliveira S., and, Kohn D. Hematopoietic stem cells for cancer immunotherapy. Immunol Rev. 2014;257:237–249. doi: 10.1111/imr.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, Morgan R, Dudley M, Cassard L, Yang J, Hughes M.et al. (2009Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen Blood 114535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R, Yang J, Kitano M, Dudley M, Laurencot C., and, Rosenberg S. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC.et al. (2013Identification of a titin-derived HLA-A1–presented peptide as a cross-reactive target for engineered MAGE A3–directed T cells Sci Transl Med 5197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R, Chinnasamy N, Abate-Daga D, Gros A, Robbins P, Zheng Z.et al. (2013Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy J Immunother 36133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R, Johnson L, Davis J, Zheng Z, Woolard K, Reap E.et al. (2012Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma Hum Gene Ther 101043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnasamy D, Tran E, Yu Z, Morgan R, Restifo N., and, Rosenberg S. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013;73:3371–3380. doi: 10.1158/0008-5472.CAN-12-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins P, Lu Y, El-Gamil M, Li Y, Gross C, Gartner J.et al. (2013Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells Nat Med 19747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss C, Condomines M, Cartellieri M, Bachmann M., and, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens R, Yeh R, Bernal Y, Riviere I., and, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junghans RP. Strategy escalation: an emerging paradigm for safe clinical development of T cell gene therapies. J Transl Med. 2010;8:55. doi: 10.1186/1479-5876-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKie RM, Reid R., and, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med. 2003;348:567–568. doi: 10.1056/NEJM200302063480620. [DOI] [PubMed] [Google Scholar]
- Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X.et al. (2013T cells expressing chimeric antigen receptors can cause anaphylaxis in humans Cancer Immunol Res 126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ., and, Tang YM. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014;343:172–178. doi: 10.1016/j.canlet.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C.et al. (2011Inducible apoptosis as a safety switch for adoptive cell therapy N Engl J Med 3651673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeg HJ. How I treat refractory acute GVHD. Blood. 2007;109:4119–4126. doi: 10.1182/blood-2006-12-041889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunello AG, Weissenberger J, Kappeler A, Vallan C, Peters M, Rose-John S.et al. (2000Astrocytic alterations in interleukin-6/soluble interleukin-6 receptor alpha double-transgenic mice Am J Pathol 1571485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P., and, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-γ-producing lymphocytes and IL-6-and TNF-α-producing macrophages. Blood. 2005;105:1648–1651. doi: 10.1182/blood-2004-08-2997. [DOI] [PubMed] [Google Scholar]
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S.et al. (2007HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis Pediatric Blood Cancer 48124–131. [DOI] [PubMed] [Google Scholar]
- Allen CE, Yu X, Kozinetz CA., and, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:1227–1235. doi: 10.1002/pbc.21423. [DOI] [PubMed] [Google Scholar]
- Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM.et al. (2011IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells J Clin Invest 1213846–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE.et al. (2013Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine directed therapy Blood 1215154–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C.et al. (2013Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity Mol Ther 14904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR.et al. (2011 A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells Blood 1181255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M.et al. (2009Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I–II study Lancet Oncol 10489–500. [DOI] [PubMed] [Google Scholar]
- Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F.et al. (2007The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies Blood 1094708–4715. [DOI] [PubMed] [Google Scholar]
- Jensen MC, Popplewell L, Cooper LJ, Digiusto D, Kalos M, Ostberg JR.et al. (2010Anti-transgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans Biol Blood Marrow Transplant 161245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tey SK, Dotti G, Rooney CM, Heslop HE., and, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2007;13:913–924. doi: 10.1016/j.bbmt.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torikai H, Reik A, Liu P, Zhou Y, Maiti S, Huls H.et al. (2012A foundation for universal T-cell base immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR Blood 1195697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatsan R, Bross PF, Liu K, Theoret M, De Claro AR, Lu J.et al. (2013Regulation of immunotherapeutic products for cancer and FDA's role in product development and clinical evaluation J Immunother Cancer 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor CM, Sheppard S, Hartline CA, Huls H, Johnson M, Palla SL.et al. (2012Adoptive T-cell therapy improves treatment of canine non-Hodgkin lymphoma post chemotherapy Sci Rep 2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy S.2013Life Technologies enters into an exclusive license and supply agreement for DynabeadsPress release < < http://www.bioportfolio.com/news/article/1584397/Life-Technologies-Enters-into-an-Exclusive-License-and-Supply-Agreement-for-Dynabeads.html > ( 31 July 2013
- Couzin-Frankel J. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]