

Abstract
Functional exhaustion of antigen-specific T cells is a defining characteristic of many chronic infections, but the underlying mechanisms of T cell dysfunction are not well understood. Epigenetics plays an important role in the control of T cell development, differentiation, and function. To examine if epigenetics also plays a role in T cell exhaustion, we analyzed chromatin remodeling in CD8+ T cells from mice with chronic lymphocytic choriomeningitis virus infection. We observed downregulation of diacetylated histone H3 in both virus-specific and total CD8+ T cells, and functional defects not only in virus-specific CD8+ T cells but also within the total CD8+ T cell population. In vitro treatment of these exhausted CD8+ T cells with histone deacetylase inhibitors restored diacetylated histone H3 levels, and improved their immune functions. Upon adoptive transfer, these treated CD8+ T cells developed into functional memory T cells in vivo that enhanced protective immunity. These results define a role of epigenetics in T cell exhaustion and suggest epigenetic manipulation as a novel molecular therapy to restore immune functions.
Introduction
Following viral infections, naїve antigen-specific CD8+ T cells become activated, proliferate and differentiate into effector CD8+ T cells. These cells are part of an immune response that clears the viral infection. After clearance of the primary infection, most of the effector CD8+ T cells die by apoptosis, but 5–10% survives and differentiates into resting memory CD8+ T cells.1,2,3,4 During many chronic viral infections where the immune responses fail to clear the virus, antigen-specific CD8+ T cells are continuously activated and do not develop into resting memory T cells. Instead, they eventually become exhausted as a result of prolonged antigenic and inflammatory stimulation. The exhausted CD8+ T cells are characterized by poor proliferation, a progressive loss of the ability to produce cytokines (e.g., interleukin 2 (IL-2), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN)-γ), reduced cell-mediated cytotoxicity2 and increased expression of multiple inhibitory receptors.5,6,7,8 CD8+ T cell exhaustion was first characterized in the murine lymphocytic choriomeningitis virus (LCMV) model after infection with LCMV clone 13 (Cl-13). This functional impairment phenotype has now been observed in other murine models, as well as in human chronic viral infections, such as human immunodeficiency virus, hepatitis B virus, and hepatitis C virus.7 The dysfunctional T cell response is a primary reason for the inadequate immunological control of these persisting pathogens, yet the mechanisms underlying these functional deficiencies are not fully understood.
Epigenetics plays a significant role in regulating gene expression in many important biological processes.9 Activation or silencing of gene expression by epigenetic changes, such as DNA methylation and histone acetylation/methylation play a crucial role in T cell development, differentiation, and function—including their ability to produce various cytokines following antigenic stimulation. In CD4+ T cells, chromatin remodeling at the Ifng and Il4 loci is critically involved in the differentiation into Th1 versus Th2 polarized effectors.10,11,12 Naїve CD8+ T cells, which produce little IFN-γ following brief stimulation, have highly methylated DNA and low levels of diacetylated histone H3 (diAcH3) at the Ifng locus relative to levels observed in effector and memory CD8+ T cells, which rapidly produce high levels of IFN-γ.13,14 Moreover, higher levels of histone acetylation are observed at the Ifng locus of functional memory CD8+ T cells, compared to dysfunctional memory CD8+ T cells that are generated without CD4+ T cell help.13 Thus, epigenetic modifications are critical for optimal expression of cytokines and other effector functions by effector and memory CD8+ T cells. In this study, we examined the role of epigenetics, specifically histone acetylation, in CD8+ T cell exhaustion during chronic LCMV infection and tested the potential of epigenetic manipulation as a molecular therapy to restore immune functions.
Results
The diAcH3 level decreases progressively in virus-specific CD8+ T cells, in parallel to their functional exhaustion, during chronic viral Infection
To investigate the role of histone acetylation in regulating CD8+ T cell functions during chronic viral infection, we used the well-established model of mice infected with either LCMV Arm, which is cleared within 8–10 days of infection, or LCMV Cl-13, which results in chronic infection with 2–3 months of viremia and long-term viral persistence in tissues. We assessed diAcH3 expression by fluorescence-activated cell sorting (FACS) in CD8+ T cells specific to the Db/GP33-41 epitope from the LCMV glycoprotein, identified by staining with the Db/GP33 tetramer (Figure 1a). At day 7 after infection, the diAcH3 level was higher in Db/GP33-specific CD8+ T cells from Cl-13-infected mice than in those from Arm-infected mice. At day 40 after infection, however, Db/GP33-specific CD8+ T cells from Cl-13-infected mice had significantly lower levels of diAcH3 compared to those from Arm infection (P < 0.01). Similar results were observed in IFN-γ+CD8+ T cells following ex vivo stimulation with the GP33-41 peptide (Figure 1b). At day 7 postinfection, Db/GP33-specific CD8+ T cells from Cl-13-infected mice had higher levels of diAcH3 and produced more IFN-γ+ on a per cell basis (IFN-γ mean fluorescence intensity (MFI) = 6,533), compared to those (IFN-γ MFI = 2,104) from Arm-infected mice. At day 40, CD8+ T cells from Cl-13-infected mice became functionally defective. Specifically, they were unable to produce IFN-γ, TNF-α, and IL-2 after GP33-41 peptide stimulation, and exhibited hallmark features (PD-1hi/CD62Llo/CD127lo) of exhaustion (Supplementary Figure S1). These defective CD8+ T cells had a significantly lower level of diAcH3 compared to functional memory CD8+ T cells from Arm-infected mice (P < 0.01) (Figure 1b). These results show a direct correlation between the diAcH3 level and the ability of virus-specific CD8+ T cells to produce cytokines in chronically infected mice.
Figure 1.

Downregulation of diacetylated histone H3 (diAcH3) in virus-specific CD8+ T cells during chronic Cl-13 infection. B6 mice were infected with LCMV Arm or Cl-13 viruses to establish acute or chronic infection. The diAcH3 level on Db/GP33+ or IFN-γ+ CD8+ T cells was analyzed in peripheral blood by FACS on day 7 (D7) and day 40 (D40) postinfection. (a) Representative data show the frequency of Db/GP33-specific CD8+ T cells (gated on CD8+ T cells). Histograms display diAcH3 staining of Db/GP33-specific CD8+ T cells (Arm: gray filled; Cl-13: black filled), and staining with preimmune control antibodies (Arm: dashed line; Cl-13: black line). Bar graphs display the corresponding MFI values of diAcH3 expression with significant difference (**P < 0.01). (b) The capacity of virus-specific CD8+ T cells to produce IFN-γ was determined by ICS following stimulation with GP33-41 peptide. Flow plots are gated on CD8+ T cells and numbers indicate the frequency of IFN-γ+ cells in CD8+ T cells. Histograms show diAcH3 staining of Db/GP33-specific CD8+ T cells (Arm: gray filled; Cl-13: black filled), and staining with preimmune control antibodies (Arm: dashed line; Cl-13: black line). Bar graphs display the corresponding MFI values of diAcH3 expression (**P < 0.01). Data are representative of more than three independent experiments with three to five mice per group. MFI, mean fluorescence intensity.
To further define the correlation between the diAcH3 level and functionality of virus-specific CD8+ T cells, we performed a kinetic analysis of diAcH3 expression in the virus-specific CD8+ cells on days 7, 20, 30, and 40 post-Cl-13 and Arm infection (Figure 2a). The diAcH3 level of Db/GP33+ cells from Arm-infected mice increased slightly as these cells differentiated into functional memory T cells from days 7 to 40. In Cl-13-infected mice, the diAcH3 level of Db/GP33-specific CD8+ T cells decreased over time. This decrease in diAcH3 was accompanied by a progressive loss of the ability to produce IFN-γ, as shown by decreased percentages of Db/GP33+ cells that made IFN-γ between days 7 and 40 postinfection (Figure 2b). This close correlation in kinetics between the diAcH3 level and T cell exhaustion suggests that reduced histone acetylation may play a role in impaired functionality of virus-specific CD8+ T cells from chronically infected mice.
Figure 2.

Close kinetic correlations between impaired function and reduced diacetylated histone H3 (diAcH3) level in virus-specific CD8+ T during chronic Cl-13 infections. B6 mice were infected with Arm or Cl-13 to establish acute or chronic infection. (a) Expression of diAcH3 in Db/GP33+ or IFN-γ+ CD8+ T cells was determined by FACS at indicated time points after infection. Histograms were gated on Db/GP33+ or IFN-γ+ CD8+ T cells from blood of Arm (unfilled) or Cl-13 (black) infection. (b) Each line represents the percentage of IFN-γ+ CD8+ T cells (circle), or the MFI of diAcH3 of Db/GP33+CD8+ T cells (triangle) relative to day 7, at indicated time points after Cl-13 infection. Data are representative of more than three independent experiments with three to five mice per group.
Most CD8+ T cells in chronically infected mice are functionally impaired and have reduced levels of diAcH3
Previous studies have shown that exhausted CD8+ T cells from chronically infected mice have altered expression of surface receptors and signaling components (such as PD-1) that contribute to their functional defects.6,15 In addition to these known mechanisms, our above results suggest that reduced diAcH3 may also play a role in contributing to the functional defects. To distinguish the contribution of reduced diAcH3 from other mechanisms involving surface receptors and signaling components, we asked whether the functional defects can be rescued by stimulation with phorbol 12-myristate 13-acetate and ionomycin (PMA/I), which bypass surface receptors and proximal signaling components by directly activating downstream protein kinase C and calcium-dependent signaling pathways, respectively. In both Arm- and Cl-13-infected mice at day 7 postinfection, 40–50% of activated CD44hiCD8+ T cells were IFN-γ+ following ex vivo PMA/I stimulation, with no significant difference in MFI between the two groups (Figure 3a). By day 40 post-Cl-13 infection, however, only a very small percentage (<5%) of CD44hiCD8+ T cells were IFN-γ+ with a very low MFI of 547, despite the fact that almost all CD8+ T cells had an activated CD44hi phenotype (~89%). In contrast, >50% of CD44hiCD8+ T cells from Arm-infected mice at day 40 were IFN-γ+, with an MFI of 1,650 (Figure 3b). Thus, brief exposure to PMA/I that bypasses surface-proximal signaling events is able to induce robust cytokine production from T cells responding to an acute infection, but not from T cells responding to a chronic infection, suggesting that the latter have defects far downstream of TCR signaling.
Figure 3.
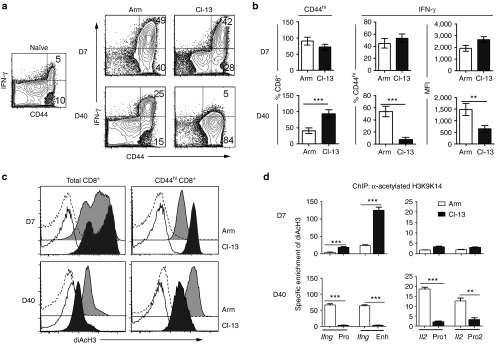
Impaired function and down-regulation of diacetylated histone H3 (diAcH3) in total CD8+ T cells during Cl-13 infection. B6 mice were infected with Arm or Cl-13 viruses and analyzed on day 7 (D7) and day 40 (D40) after infection. (a) IFN-γ production by CD8+ T cells, assessed by ex vivo stimulation with PMA/I. The data from an uninfected naïve mouse is shown as a control. Flow plots are gated on CD8+ T cells. (b) Bar graphs show % of CD8+ T cells that are CD44hi (left), % of CD44hiCD8+ T cells that are IFN-γ+ (middle), and MFI of IFN-γ staining (right). (c) Histograms indicate the diAcH3 expression of total CD8+ T cells and CD44hiCD8+ T cells (Arm: gray filled; Cl-13: black filled), and staining with preimmune control antibodies (Arm: dashed line; Cl-13: black line). (d) Bar graphs display the specific enrichment for diAcH3 at the Ifng promoter, enhancer and Il2 promoters in CD8+ T cells. Arm: unfilled; Cl-13: filled. **P < 0.01, ***P < 0.001. Data are representative of at least three independent experiments with three to five mice per group. PMA/I, phorbol 12-myristate 13-acetate and ionomycin.
While functional exhaustion of virus-specific CD8+ T cells during chronic infection is well established, it is surprising to observe that there was little, if any, IFN-γ production by total CD8+ T cells from chronically infected mice following ex vivo PMA/I stimulation (Figure 3a). On day 7 after infection, ~90% of CD8+ T cells from Arm-infected and ~70% from Cl-13-infected mice had an activated CD44hi phenotype. A similar percentage of CD44hi CD8+ T cells (~55% for Arm and ~60% for Cl-13) produced IFN-γ upon PMA/I stimulation. By day 40 after Arm infection, ~40% of CD8+ T cells were CD44hi and ~62% of CD44hi CD8+ T cells produced IFN-γ. In contrast, few CD44hi CD8+ T cells from Cl-13-infected mice produced IFN-γ despite the fact that ~90% of CD8+ T cells had an activated CD44hi phenotype (Figure 3b). These results indicated that almost all CD8+ T cells became activated and functionally impaired at the later stages of the chronic Cl-13 infection. Consistent with their functional impairment, the diAcH3 levels of CD44hiCD8+ T cells from Cl-13-infected mice were higher on day 7 but much lower on day 40, as compared to that of CD44hiCD8+ T cells from Arm-infected mice (Figure 3c). To further evaluate this finding, we sorted CD44hiCD8+ T cells from Arm or Cl-13-infected mice and performed chromatin immunoprecipitation (ChIP) to measure the diAcH3 level at the Ifng and Il2 loci (Figure 3d). At day 7 after infection, CD8+ T cells from Cl-13-infected mice had a higher (four- to fivefold) level of diAcH3 at the Ifng locus than those from Arm-infected mice, while a similar low level of diAcH3 at theIl2 locus was observed in both Arm- and Cl-13-infected mice. At day 40, however, CD8+ T cells from Cl-13-infected mice had lower levels of AcH3 at both the Ifng locus (10-fold lower) and the Il2 locus (four- to fivefold lower). Thus, chronic Cl-13 infection results in reduced diAcH3 levels and functional exhaustion, not only in virus-specific CD8+ T cells but also within the entire CD8+ T cell population.
In vitro treatment with an HDAC inhibitor improves the functionality of exhausted CD8+ T cells
We next tested whether functional impairment of exhausted CD8+ T cells can be reversed by treatment with valproic acid (VPA), an inhibitor of histone deacetylase (HDAC) that increases the histone acetylation level. Splenocytes from mice chronically infected with Cl-13 (>day 40) were treated in vitro with graded doses (0.12–4.8 mmol/l) of VPA in the presence of anti-CD3/CD28 mAbs for 48 hours, and the effect of VPA treatment on diAcH3 level and cytokine production were assessed (Supplementary Figure S2) . The diAcH3 level in CD8+ T cells increased with elevated concentrations of VPA, but the high concentrations of 2.4 and 4.8 mmol/l became toxic as evident by a substantial decrease in cell numbers. Treatment with 0.6, 1.2, and 2.4 mmol/l VPA resulted in marked increases in IFN-γ and TNF-α production. On basis of these data, 1.2 mmol/l VAP was chosen as the optimal dose for subsequent experiments, since this dose resulted in maximal cytokine production with low toxicity.
Treatment of exhausted CD8+ T cells from chronically infected mice with 1.2 mmol/l VPA resulted in increased diAcH3 levels as detected by FACS (Figure 4a) and elevated histone acetylation at the Ifng and Il2 loci as measured by ChIP (Figure 4b). After VPA treatment, there was an average sevenfold increase in the diAcH3 level at both the Ifng promoter and Ifng enhancer relative to the level in untreated CD8+ T cells. Similarly, the Il2 promoter exhibited a twofold increase in its diAcH3 level after VPA treatment. Consistent with an increased diAcH3 level being indicative of open chromatin that facilitates rapid gene expression, Ifng and Tnfa transcripts increased 50-fold and fourfold, respectively, in VPA-treated compared to the untreated cells as measured by real-time PCR (Figure 4c). More CD8+ T cells from the VPA-treated group produced IFN-γ (Figure 4d) and TNF-α (data not shown) after stimulation with GP33-41or PMA/I. Additionally, VPA treatment increased the diACH3 level of exhausted T cells from Cl-13 infected to levels comparable to that in functional memory T cells from Arm-infected mice (Supplementary Figure S3). These results show that in vitro treatment with an HDAC inhibitor increases histone acetylation levels in exhausted CD8+ T cells from chronically infected mice and improves the functionality of these exhausted CD8+ T cells.
Figure 4.

Improved functions of exhausted CD8+ T cells after in vitro VPA treatment. Splenocytes from Cl-13-infected mice (day 40 p.i.) were cultured in vitro with anti-CD3 and anti-CD28 mAbs in the absence or presence of VPA (1.2 mmol/l) for 48 hours, then washed and rested in medium overnight. The diacetylated histone H3 (diAcH3) level and cytokine expression were measured: (a) the diAcH3 level in CD8+ T cells by FACS; (b) specific enrichment of diAcH3 at the Ifng promoter, enhancer and Il2 promoters of CD8+ T cells by ChIP; (c) mRNA level of Ifng and Tnfa by RT-PCR analysis of CD8+ T cells after stimulation with PMA/I; (d) IFN-γ production by FACS (gated on CD8+) after stimulation with GP33-41 peptide or PMA/I. The data shown are representative of more than three separate experiments (*P < 0.05, **P < 0.01, ***P < 0.001). CFU, colony-forming units; PMA/I, phorbol 12-myristate 13-acetate and ionomycin; VPA, valproic acid.
Exhausted CD8+ T cells treated with an HDAC inhibitor develop into functional memory T cells in vivo
Since VPA treatment increased diAcH3 levels and restored the function of exhausted CD8+ T cells in vitro, we asked if the epigenetic changes due to VPA treatment could be maintained over time and allow for development of functional memory T cells in vivo. Splenocytes from mice chronically infected with Cl-13 were treated with or without VPA in vitro in the presence of anti-CD3/CD28 mAbs for 48 hours, and purified CD8+ T cells containing equal numbers of Db/GP33-specific cells were then adoptively transferred into congenic Ly5.2 naїve hosts. At day 4 after cell transfer, similar numbers of donor Db/GP33+ CD8+ T cells (Ly5.1+) were detected in the blood of mice that received VPA-treated or untreated donor cells, and there was no significant difference between the VPA-treated and untreated donor cells in their ability to produce IFN-γ (Figure 5a). In the ensuing days 29 and 37, however, mice that received VPA-treated cells had more Ly5.1+ donor Db/GP33+ CD8+ T cells than the group that received untreated donor cells. Furthermore, more IFN-γ+ T cells were detected in VPA-treated than untreated donor CD8+ T cells in response to PMA/I stimulation (Figure 5b). No obvious signs of autoimmunity were observed in mice receiving VPA-treated splenocytes. Thus, in vitro VPA treatment of exhausted CD8+ T cells had a positive impact on the ability of these cells to survive in the adoptive host and to produce effector cytokines upon restimulation.
Figure 5.

Exhausted CD8+ T cells treated with VPA develop into functional memory T cells in vivo. Exhausted CD8+ T cells (Ly5.1), after in vitro treatment with or without VPA as in Figure 4, were adoptively transferred into congenic Ly5.2 mice. At days 4, 29 and 37 after cell transfer, (a) Db/GP33-specific CD8+ T cells were examined, and (b) IFN-γ production was measured after stimulation with PMA/I. (c) At day 40 after cell transfer, mice were infected with rLmGP33. Donor Ly5.1+ CD8+ T cell response were examined on day 10 after rLmGP33 infection: percentage and absolute number (mean ± SD) of Db/GP33+ CD8+ T cells (top), of IFN-γ+ CD8+ T cells after stimulation with the GP33-41 peptide (middle) or PMA/I (bottom). Bacterial numbers in spleen and liver were enumerated at day 3 after rLmGP33 infection. Data are representative of three independent experiments with five mice per group. *P < 0.05, ** P < 0.01, ***P < 0.001. PMA/I, phorbol 12-myristate 13-acetate and ionomycin; VPA, valproic acid.
The above results suggest that in vitro VPA treatment of exhausted CD8+ T cells may have allowed them to differentiate into long-term, functional memory T cells in adoptive hosts. To further test this possibility, we examined the in vivo recall response and the ability of these cells to provide protective immunity. Exhausted CD8+ T cells were treated with or without VPA in vitro and transferred into adoptive hosts as above. More than 40 days after adoptive transfer, we challenged these mice with recombinant L. monocytogenes expressing the GP33-41 epitope (rLmGP33), and assessed their recall responses and protective immunity (Figure 5c). On day 10 after rLmGP33 challenge, mice receiving either VPA-treated cells or untreated cells had a comparable response from host (Ly5.1−) CD8+ T cells. However, the VPA-treated donor (Ly5.1+) cells had a significantly greater number of Db/GP33+ CD8+ T cells producing more cytokines compared with the untreated donor cells. The stronger recall response from VPA-treated donor cells provided better protection against rLmGP33. On day 3 after rLmGP33 challenge, there were ~100-fold fewer bacteria in the spleen and liver of mice that received VPA-treated donor cells than in mice that received untreated donor cells. These results indicate that exhausted CD8+ T cells treated with VPA can survive and differentiate into long-term, functional memory T cells that are capable of mounting an enhanced recall response and providing better protective immunity.
In summary, our results have shown that (i) in vitro VPA treatment rescued the functionality of exhausted CD8+ T cells, (ii) these rescued cells are capable of long-term survival in adoptive hosts, and (iii) maintain their improved functionality for an extended period of time, and (iv) differentiate into functional memory T cells that are capable of mounting an enhanced recall response and providing better protective immunity.
Discussion
CD8+ T cell exhaustion is a common feature of many chronic viral infections in both animal models and human diseases.2,7,16 Considerable effort has been focused on elucidating the mechanisms of T cell exhaustion during chronic viral infections. Multiple regulatory factors and pathways have been implicated, including inhibitory receptors (PD-1, LAG-3, Tim-3, CTLA-4, 2B4, and CD160), transcription factors (T-bet and Blimp-1), cytokines (IL-10, IL-21, TGF-β, RANTES, and IFN), Treg and CD4+ helper T cells.6,17,18,19,20,21,22,23,24,25 The function of exhausted CD8+ T cells can be partially restored by blocking many of the inhibitory pathways.21,26,27,28,29,30,31,32,33 However, exhausted CD8+ T cells are unable to revert and differentiate into functional memory T cells even when removed from the chronic infection environment,34 indicating imprints of functional defects within exhausted CD8+ T cells. In this study, we found that progressive loss of CD8+ T cell functions during chronic viral infection was associated with a decreased diAcH3 level, and in vitro treatment of exhausted CD8+ T cells with an HDAC inhibitor restored the diAcH3 level and improved their functionality. Furthermore, treatment of exhausted CD8+ T cells with an HDAC inhibitor allowed them to persist long term and develop into functional memory cells in adoptive hosts. Together, these results suggest that exhaustion is regulated by various signals and controlled by multiple pathways. It remains to be determined what role each signal plays at different stages of infection, and how these pathways interact with each other to cause functional exhaustion. Understanding these interactions may allow us to develop optimal therapies that combine epigenetic manipulations with blockage of external inhibitory signals. To this end, we are currently testing the possibility of treating chronic virus infection by immunotherapy using VPA-treated exhausted T cells, alone or in combination with blockage of inhibitory signals such as PD-1 and/or IL-10.
Our results showed that function of exhausted CD8+ T cells could not be rescued by stimulation with PMA/I, which bypasses surface receptors and proximal signaling components by directly activating downstream protein kinase C and calcium-dependent signaling pathways, respectively. Thus, functions of exhausted CD8+ T cells are not only regulated by external signals but also controlled by internal mechanisms. Youngblood et al have shown that the Pdcd1 regulatory regions remain demethylated, allowing rapid expression of the PD-1 inhibitory receptor in exhausted CD8+ T cells.35 In this study, our results show that the inability to produce IFNγ and IL-2 by exhausted CD8+ T cells is correlated with a low level of diAcH3 in the regulatory regions of the Ifng and Il2 loci. Treatment with an HDAC inhibitors elevated diAcH3 levels and restored the ability of exhausted T cells to produce cytokines such as IFNγ and IL-2 cytokines. Thus, different epigenetic modifications likely play an important role in controlling functions of exhausted CD8+ T cells, with DNA demethylation and histone acetylation involved in regulating expression of inhibitory receptors (such as PD-1) and effector cytokines (such as IFN-γ and IL-2), respectively.
Our results showed that the diACH3 level of effector CD8 T cells on day 7 is higher in Cl-13 than Arm-infected mice, consistent with more IFN-γ produced per cell by effector CD8 T cells from Cl-13 than Arm-infected mice on day 7 (Figure 1b). This initial higher level of diACH3 may reflect a heightened activation status due to more stimulation from higher viral load in Cl-13 than Arm infection. While over-stimulation is known to contribute to functional exhaustion, it is not known if the initial higher level of diACH3 may play a role in the eventual decline of diACH3 level and exhaustion. Levels of histone acetylation are regulated by the combined effects of multiple histone acetylases and HDAC enzymes. Histone acetylation at specific gene loci is controlled by not only the level of histone acetylase/HDAC expression and their enzymatic activity, but also recruitment and formation of active complexes at specific loci. Our data show that exhausted CD8+ T cells have decreased levels of histone acetylation globally and at specific loci encoding effector cytokines, which can be reversed by blocking HDAC enzymatic activity. These results suggest that elevated HDAC expression and/or enzymatic activity might be a consequence of the chronic infection environment. Additionally, recruitment of HDAC to specific loci may be regulated during chronic infection. We are currently studying expression and regulation of different HDAC enzymes in T cells and determining if dysregulation of their expression and recruitment plays a role in T cell exhaustion.
Functional exhaustion of virus-specific CD8+ T cells during chronic infection is well documented. Exhausted CD8 T cells present a paradox of having an activated CD44hi phenotype, yet being defective in cytokine production. They are also defective in proliferative responses following antigenic stimulation and in homeostatic proliferation.36,37 In addition to activated effector T cells, resting memory T cells are CD44hi and they are more capable of rapid cytokine production in response to stimulation. We were surprised to find that ~90% of CD8+ T cells were CD44hi in chronically infected mice yet they all failed to produce cytokines in response to PMA/I stimulation. These results suggest that chronic viral infections cause functional exhaustion of not only virus-specific CD8+ T cells but also bystander memory T cells with specificity to other pathogens. Indeed, ovalbumin-specific memory CD8+ T cells became functionally defective in mice chronically infected with LCMV Cl-13 . These results are consistent with findings that not only persistent antigenic stimulation, but also chronic inflammation, contributes to functional exhaustion.16,38,39 Furthermore, our results show that the CD8+ T cell population as a whole had reduced levels of diAcH3 in chronically infected mice, and that their functions can be restored by in vitro treatment with an HDAC inhibitor. These results suggest a potential mechanism for why chronically infected hosts become susceptible not only to opportunistic infections but also to pathogens that are otherwise protected against by immunological memory induced by vaccines or prior infections. Further studies are needed to test if epigenetic manipulations can repair not only virus-specific T cell function in chronic infection but also the host's general immunity and immunological memory to other pathogens. An analogous approach could be used to rescue exhausted tumor-reactive T cells that could be then reintroduced into cancer patients. Furthermore, systemic delivery of HDAC inhibitors in vivo may improve immune control of chronic viral infection and tumors, while having the added benefit of boosting overall immune function by repairing the host's immunological memory to other pathogens.
Materials and Methods
Mice and infections. Female (8–10 weeks) C57BL/6 mice were purchased from National Cancer Institute (Bethesda, MD) and maintained under specific pathogen-free conditions at the animal facilities of the University of Pennsylvania. All animal experiments were performed in accordance with Institutional Animal Care and Use Committee-approved protocols at the University Of Pennsylvania School Of Medicine Animal Facility (Philadelphia, PA). LCMV strains were propagated, titered, and used as previously described.37 For virus infection, mice were infected with 2 × 105 plaque-forming units of LCMV Arm intraperitoneally or 2 × 105 plaque-forming units of LCMV Cl-13 intravenously.
In vitro activation and VPA treatment. A total of 2 × 106 cells per well were incubated in media containing RPMI 1640 + 15% fetal bovine serum + 100 U/ml Penicillin + 100 ug/ml Streptomycin + 2 mmol/l l-glutamine + 50 μmol/l β-mercaptoethanol. Splenocytes harvested from B6 mice at day ~40 after chronic LCMV Cl-13 infection were activated with precoated anti-CD3 (clone 145-2C11; 1.0 µg/ml) and anti-CD28 (clone 37.51; 0.5 µg/ml) antibodies in the presence of varied dose of VPA (0, 0.12, 0.6, 1.2, 2.4, and 4.8 mmol/l, Cayman Chemical Company, Ann Arbor, MI) for 48 hours. The cells were then washed and incubated in medium without VPA overnight.
Flow cytometry and tetramer staining. MHC class I tetramer of H-2Db complexed with LCMV GP33-41 (Db/GP33) was produced as described.40 Biotinylated complexes were tetramerized using phycoerythrin or allophycocyanin-conjugated streptavidin (Invitrogen, Carlsbad, CA). Lymphocytes from spleen or blood of LCMV-infected mice were stained as follows. For surface staining, single-cell suspensions (1–3 × 106 cells) were stained with CD antigen-specific antibodies in RPMI 1640 containing 1% fetal bovine serum (FACS buffer) for 30 minutes at 4 °C, followed with two washes in FACS buffer. Antibodies specific for CD antigens were purchased from eBioscience (San Diego, CA). Cells were acquired using a FACS Canto flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed with FlowJo software (FlowJo, Philadelphia, PA).
Intracellular cytokine staining. Single-cell suspensions were stimulated ex vivo either with the GP33-41 (0.2 µg/ml) peptide or PMA (50 ng/ml; Sigma-Aldrich, St Louis, MO) and ionomycin (1 μg/ml; Sigma-Aldrich) for 5 hours at 37 °C in the presence of BD GolgiStop (BD Biosciences). LCMV GP33-41 encodes an immunodominant epitope that is presented by H-2Db and H-2Kb. Both H-2Db and H-2Kb restricted T cells are stimulated by GP33-41 peptide in this assay.41 Following the staining of the surface antigens as described above, cells were stained for intracellular cytokines using the BD cytofix/cytoperm kit (BD Pharmingen, San Jose, CA) according to the manufacturer's instructions. Antibodies used for intracellular cytokine detection were anti-IFN-γ (clone XMG1.2), anti-IL-2 (clone JES6-5H4), and anti-TNF-α (clone MP6-XT22) were purchased from eBioscience.
Flow cytometric analysis for histone modification. Flow cytometric analysis for histone modification was performed using intracellular staining kit (Cytofix/Cytoperm, BD Biosciences) as previously described.42 In brief, cells were incubated with a rabbit polyclonal antibody specific for diacetylated histone H3 (K9K14; #06-599; Millipore, Billerica, MA) at 10 µg/ml, or pre-immune rabbit serum as control. The cells were then washed and incubated with goat antirabbit-antibody (BD Biosciences). FACS Canto flow cytometer (BD Biosciences) was used to acquire the cells. Data were analyzed with FlowJo software.
Real-time PCR. Total RNA was isolated from magnetic-activated cell sorting (Miltenyi Biotech, San Diego, CA) purified CD8+ T cells using Trizol (Invitrogen), reverse transcription of cDNA was prepared with the SuperScript First Strand kit (Invitrogen) using random hexamer priming. Real-time PCR was performed with primers as described in Supplementary Table S1. SYBR green detection of amplification was performed using the StepOne Plus cycler (Applied Biosystems, Grand Island, NY). Transcript expression values were generated with the comparative threshold cycle (Delta CT) method by normalizing to the expression of the β-actin gene.
Chromatin immunoprecipitation. ChIP with an antiacetyl histone-H3 antibody was performed using an AcH3 ChIP assay kit (Upstate Biotechnology, Lake Placid, NY), as described previously, and the quantity of DNA was purified and assayed by real-time PCR13 (primers shown in Supplementary Table S1). Specific enrichment is calculated using the cycle threshold (Ct): 2(Ct of control ChIP − Ct of control Input)/2(Ct of AcH3 ChIP − Ct of AcH3 Input). Each ChIP analysis was performed three times with chromatin from independently purified CD8+ T cells.
Adoptive transfer and infection. In vitro VPA-treated exhausted CD8+ T cells were harvested and transferred i.v. into congenic wild-type B6 (Ly5.2) recipient mice (2 × 106 cells per mouse) followed infection i.v. with 5 × 104 colony-forming units of rLmGP33.
Statistical analyses. Statistical analysis was assessed by the unpaired, two-tailed Student's t-test (GraphPad, San Diego, CA), and statistical significance was defined as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
SUPPLEMENTARY MATERIAL Figure S1. Phenotypes of exhausted CD8+ T cells during chronic LCMV infection. Figure S2. Dose-dependent effect of in vitro VPA treatment on exhausted CD8+ T cells. Figure S3. VPA treatment increased the diACH3 level of exhausted T cells to levels comparable to that in functional memory T cells. Table S1. PCR primers used for experiments.
Acknowledgments
We thank members of Shen lab for technical support and critical discussion. This work was funded by the National Institute of Health Grant R21 AI079724, and by a grant (2011CB512104) from the National Basic Research Program of China. The authors declared no conflict of interest.
Supplementary Material
Phenotypes of exhausted CD8+ T cells during chronic LCMV infection.
Dose-dependent effect of in vitro VPA treatment on exhausted CD8+ T cells.
VPA treatment increased the diACH3 level of exhausted T cells to levels comparable to that in functional memory T cells.
PCR primers used for experiments.
References
- Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH.et al. (2006Restoring function in exhausted CD8 T cells during chronic viral infection Nature 439682–687. [DOI] [PubMed] [Google Scholar]
- Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL.et al. (2009A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection Immunity 31309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007;19:408–415. doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010;22:223–230. doi: 10.1016/j.coi.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanczyk M, Roszczenko K, Marcinkiewicz K, Bojarczuk K, Kowara M, Winiarska M. HDACi–going through the mechanisms. Front Biosci (Landmark Ed) 2011;16:340–359. doi: 10.2741/3691. [DOI] [PubMed] [Google Scholar]
- Sanders VM. Epigenetic regulation of Th1 and Th2 cell development. Brain Behav Immun. 2006;20:317–324. doi: 10.1016/j.bbi.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Ansel KM, Lee DU, Rao A. An epigenetic view of helper T cell differentiation. Nat Immunol. 2003;4:616–623. doi: 10.1038/ni0703-616. [DOI] [PubMed] [Google Scholar]
- Fields PE, Kim ST, Flavell RA. Cutting edge: changes in histone acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2 differentiation. J Immunol. 2002;169:647–650. doi: 10.4049/jimmunol.169.2.647. [DOI] [PubMed] [Google Scholar]
- Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. 2006;177:1062–1069. doi: 10.4049/jimmunol.177.2.1062. [DOI] [PubMed] [Google Scholar]
- Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM.et al. (2006Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells J Immunol 1764083–4093. [DOI] [PubMed] [Google Scholar]
- Blackburn SD, Crawford A, Shin H, Polley A, Freeman GJ, Wherry EJ. Tissue-specific differences in PD-1 and PD-L1 expression during chronic viral infection: implications for CD8 T-cell exhaustion. J Virol. 2010;84:2078–2089. doi: 10.1128/JVI.01579-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- Koike T. Interferon-γ-independent suppression of Th17 cell differentiation by T-bet expression in mice with autoimmune arthritis. Arthritis Rheum. 2012;64:40–41. doi: 10.1002/art.33331. [DOI] [PubMed] [Google Scholar]
- Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q.et al. (2010Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF Nat Med 161147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, Wherry EJ. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog. 2011;7:e1002098. doi: 10.1371/journal.ppat.1002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, Brooks DG. Decoding the complexity of type I interferon to treat persistent viral infections. Trends Microbiol. 2013;21:634–640. doi: 10.1016/j.tim.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G.et al. (2013Blockade of chronic type I interferon signaling to control persistent LCMV infection Science 340202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, Brooks DG. Interfering with type I interferon: a novel approach to purge persistent viral infection. Cell Cycle. 2013;12:2919–2920. doi: 10.4161/cc.26175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. 2010;129:474–481. doi: 10.1111/j.1365-2567.2010.03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MA, Harrington LE, Zajac AJ. Cytokines and the inception of CD8 T cell responses. Trends Immunol. 2011;32:180–186. doi: 10.1016/j.it.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood B, Wherry EJ, Ahmed R. Acquired transcriptional programming in functional and exhausted virus-specific CD8 T cells. Curr Opin HIV AIDS. 2012;7:50–57. doi: 10.1097/COH.0b013e32834ddcf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E. J. T cell exhaustion. Nat Immunol. 2010;131:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT.et al. (2012Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection Nat Immunol 13188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. 2009;324:1569–1572. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci USA. 2008;105:20428–20433. doi: 10.1073/pnas.0811139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Lee AM, Elsaesser H, McGavern DB, Oldstone MB. IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. J Exp Med. 2008;205:533–541. doi: 10.1084/jem.20071948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG.et al. (2013PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells J Clin Invest 1232604–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. 2013;191:3419–3429. doi: 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. 2012;86:8161–8170. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE.et al. (2011Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells Immunity 35400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Ahmed R. Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination. J Virol. 2005;79:8960–8968. doi: 10.1128/JVI.79.14.8960-8968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci USA. 2004;101:16004–16009. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD.et al. (1998Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection Immunity 8177–187. [DOI] [PubMed] [Google Scholar]
- Hudrisier D, Oldstone MB, Gairin JE. The signal sequence of lymphocytic choriomeningitis virus contains an immunodominant cytotoxic T cell epitope that is restricted by both H-2D(b) and H-2K(b) molecules. Virology. 1997;234:62–73. doi: 10.1006/viro.1997.8627. [DOI] [PubMed] [Google Scholar]
- Dispirito JR, Shen H. Histone acetylation at the single-cell level: a marker of memory CD8+ T cell differentiation and functionality. J Immunol. 2010;184:4631–4636. doi: 10.4049/jimmunol.0903830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotypes of exhausted CD8+ T cells during chronic LCMV infection.
Dose-dependent effect of in vitro VPA treatment on exhausted CD8+ T cells.
VPA treatment increased the diACH3 level of exhausted T cells to levels comparable to that in functional memory T cells.
PCR primers used for experiments.