

Abstract
Substituted piperidines are emerging as important medicinally-active structural motifs. Here, we report highly stereoselective carbolithiation reactions of α-aryl piperidine enecarbamates that offer direct access to vicinally-substituted piperidine compounds. We have also demonstrated that the carbanion intermediates can be trapped with a carbon electrophile
The lithiation chemistry of piperidines has been studied for over two decades following the seminal discovery by Beak that N-Boc piperidine can be deprotonated at the α position (i.e., C2 or C6, see 1 for numbering) by the action of sec-BuLi in the presence of a diamine.1 This early discovery has led to a wealth of knowledge on α-lithiated piperidines in terms of both fundamental reactivity and synthetic applications.2 The piperidine structural motif is found in a wide variety of biologically active natural products and pharmaceuticals (for selected examples see Figure 1).3,4 As such, there continues to be sustained effort from the synthesis community to develop new methods to access substituted piperidines. Even though one of the most established methods for the functionalization of piperidines is using lithiation chemistry,1 to date, all the reported lithiation methods for the functionalization of piperidines (which are significantly more challenging to deprotonate compared to pyrrolidines)1b have only achieved the introduction of a single substituent at the activated α-positions (i.e., C2 or C6)5,6,7 or at the unactivated C3 position (through a metal-mediated 1,2-migration from an α-metalated precursor).8 Given the well-established reactivity of benzylic lithiated piperidines,6,7 we sought to develop methods involving lithiated intermediates that could achieve multiple functionalizations. We have been especially drawn to direct methods that would enable vicinal substitution on the piperidine structural motif, specifically at C2 and C3, given the prevalence of biologically active natural products (e.g., 4 and 5) which possess this substitution pattern. Importantly, because of the emergence of C2-arylated piperidines with medicinal potential (e.g., piperidine 1 was identified as a lead compound in an NK1 receptor antagonist program at Merck laboratories)9 we reasoned that the synthesis of C2/C3 vicinally substituted piperidines would expand the structural space for the discovery of novel compounds of medicinal value.
Figure 1.

α-Aryl and vicinally-substituted piperidines.
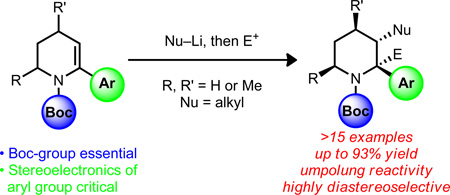
In our view, α-arylated dehydro-piperidines (e.g., 7, Scheme 1) could serve as versatile substrates for the purpose of building highly functionalized C3 substituted, α-arylated, piperidine derivatives. Owing to the ready availability of δ-lactams (e.g., 6), we anticipated that they would be good starting points for the synthesis of dehydro-piperidines such as 7 (see Figure 2) using the method of Occhiato et al.10 Given the inherent nucleophilicity of the C3 position of enamine derivatives related to 7, numerous methods have been developed for the introduction of electrophiles at C3. However, only strong electrophiles (e.g., formyl, or acylium ions using Vilsmeier-Haack formylations and Friedel-Crafts-type acylations) have been successful reacting partners.11 To the best of our knowledge, the direct electrophilic installation of alkyl groups under transition metal-free conditions, which would be beneficial from a late-stage diversification standpoint, have not been reported nor have they been successful in our hands.12 We envisioned an alternative approach to the C3-alkylation of enecarbamates, which would utilize alkyl nucleophiles in what is formally an ‘umpolung’ process.13 The success of this type of addition would exploit the recognized stabilization of lithium carbanions at C2 of N-Boc piperidines by chelation of the lithium ion to the Boc group.14 Several challenges had to be overcome in order for the addition of relatively basic alkyl nucleophiles to dehydropiperidines to be successful. These include: (1) the potential for allylic deprotonation at C4, (2) deprotonation at C6 and at C3 (i.e., directed ortho-lithiation of the enecarbamate15), (3) ortho-lithiation of the α-aryl substituent, or (4) the addition of the nucleophile into the Boc group. Finally, it was our goal to achieve highly diastereoselective vicinal difunctionalizations of piperidines. This latter goal is especially important given that only poor diastereoselectivity is observed in analogous processes attempted with the azepene homologue.16
Scheme 1.

Diastereoselective vicinal functionalization of piperidine enecarbamates.
Figure 2.

α-Aryl dehydro-piperidines utilized in this study.
We initiated our studies by investigating the addition of alkyl lithium nucleophiles to arylated enecarbamate 7a (Figure 3A). n-BuLi was selected as the nucleophile because of its ready availability and the recognized superiority of lithium-based intermediates in carbolithiation chemistry.14,17 Following a survey of several conditions, we were delighted to find that the reaction of 7a with n-BuLi gives the product of carbolithiation (i.e., 9aA, following a methanol quench) along with 10 (resulting from addition of n-BuLi into the Boc group). The carbolithiation process is highly diastereoselective, with the C2 aryl and the C3 alkyl groups anti (see Figure 3B). The nature of the aryl substituent has a marked effect on the extent of competing nucleophile addition into the Boc group. As illustrated in Figure 3B, phenyl, 2-naphthyl, m-methoxyphenyl and m-chloro phenyl enecarbamate substrates readily participate in the carbolithiation without competing ortho-lithiation18 of the aryl group (e.g., in the case of 9cB) or halogen-metal exchange (in the case of 9dC). The use of sec-BuLi as a nucleophile resulted in excellent stereoselectivity with respect to the C2 and C3 positions (see 9aB and 9cB). However, no stereoselectivity was observed with respect to the stereocenter bearing the ethyl and methyl groups in these products. Unambiguous support for the nature of the alkylated products that are formed was obtained by X-ray crystallographic analysis of the p-nitro-benzoyl derivative of 9bC (see CYLview19 of 11; most H’s removed for clarity).
Figure 3.

Scope of the carbolithiation/proton quench. (In the numbering, lowercase letter reflects enecarbamate substrate and upper case letter reflects alkyl nucleophile).
Not all piperidine enecarbamate substrates that we have examined undergo efficient carbolithiation. For example, enecarbamates 7e and 7f (see Figure 2), bearing electron-rich arenes, as well as sterically encumbered 1-naphthyl bearing 7g mainly undergo addition of the alkyllithium into the Boc group, whereas 7h, which possesses a fluorine group on the aryl substituent, yields a complex mixture (presumably arising from competing aryl ortho-lithiation/benzyne formation or dehalogenation).20 In addition, contrary to the observations made in the carbolithiation of a single azepene enecarbamate example,16 our studies on piperidine enecarbamates have revealed that in the latter substrates, aryl lithium nucleophiles do not add easily to C3. This observation highlights the significant reactivity differences in the lithiation chemistry of azacycles of different ring sizes.21,1b
We have also examined the diastereoselectivity of the piperidine enecarbamate carbolithiation processes in the presence of other substituents on the ring. For example, a C4 methyl group (see 7i, Figure 4A) does not adversely impact the efficiency of the carbolithiation and instead leads (following a MeOD quench and Boc cleavage) to a 78% yield of 9iC as a single diastereomer. Similarly, 2-naphthyl substituted enecarbamate 7j provides carbolithiation product 9jC in 79% yield over two steps. The stereochemical outcome of these carbolithiations was confirmed by single crystal X-ray crystallographic analysis of the p-nitrobenzoyl derivative of 9jC (see CYLview of 12). C6-methyl substituted enecarbamates such as 7k and 7l also undergo highly diastereoselective carbolithiations to give alkylated adducts (e.g., 9kA, obtained from 7k and n-BuLi in 70% yield and 9lA, which was obtained from 7l in 86% yield over 2 steps). Furthermore, carbolithiation of enecarbamate 7m bearing two methyl substituents at C4 and C6 yields 9mC as a single diastereomer22 when treated with tert-butyllithium.
Figure 4.

Substrate-controlled diastereoselective carbo-lithiation/proton quench substrate scope.
A DFT computational analysis performed at the B3LYP/6-31G(d) level of theory on the structures of the lithiated intermediates that arise upon carbolithiation (e.g., 14, see Scheme 2) revealed that in all cases, the syn isomer (i.e., bearing the alkyl nucleophile, Nu, and the Li group on the same face) is ~8 kcal/mol more stable than the corresponding anti isomer.23 Additionally, our computations support the pseudo-axial orientation of substituents at the C2 and C6 positions of the N-Boc piperidine intermediates (i.e., carbolithiations proceed via enecarbamate conformers similar to 13 and 16, respectively). The surprising case is 7i/j, where the stereochemical outcome appears to be governed by a pseudo-axial placement of the C4 methyl group (see R in 17). In this case, a lower-lying transition state is presumably accessed by virtue of a stabilizing donor-acceptor interaction between the axially disposed C–C bond and the developing C–C σ* in accord with a Cieplak-type effect24 and an unusual manifestation of the Fürst-Plattner rule.25 This stabilizing effect likely overrides any developing syn-pentane interaction between the C4 and C6 substituents. Attempts to extend the carbolithiations from C2-arylated enecarbamate substrates to the C2-alkyl variants have been unsuccessful. We believe that the success of the carbolithiation in the α-arylated enecarbamate variants may be attributed to the added carbanion stabilization afforded by the aryl group. This assertion is supported by the correlation of the efficiency of carbolithiation with the electronics of the aryl moiety (e.g., electronic-rich aryl substrates 7e and 7f do not undergo efficient carbolithiation).
Scheme 2.

Rationalization of the stereochemical outcome.
In the successful carbolithiation cases, the lithiated intermediate likely exists as an ion pair with stabilization of the carbanion by the electron deficient aryl substituents.5b,26,27 This is supported by NBO analysis of 18a/b (Figure 5) where C–Li bonding is not pronounced (see the Supporting Information for a full reaction coordinate/transition state analysis). In contrast, for the C2-methyl intermediate 19, a highly covalent C2–Li bond was computed. For carbanion intermediates 18a/b and 19, computations indicate that the O–Li bond distance (1.843 Å, Ar = Ph) is shortened whereas the C–Li bond distance is lengthened (2.133 Å), consistent with significant interaction between the lithium and Boc carbonyl group.5d,5e,28 During the course of the carbolithiation, reorganization of the coordination sphere is required as reflected in the Li–C2–C7 bond angle, which is compressed when electron-deficient aryl substituents are employed. The lower degree of distortion (as compared to the starting enecarbamate),29,30, leads to a lower-lying transition state and to a lower energy barrier.31
Figure 5.

Computed carbanion intermediates (compounds 18a, b and 19). Color code: gray (C), blue (N), red (O) and purple (Li). H’s removed for clarity.
Finally, in a preliminary study, we have demonstrated that the lithiated carbanion intermediate (e.g., 20, Scheme 3) can be stereospecifically intercepted by other electrophiles such as dimethyl sulfate leading to a product (21) that possesses three contiguous stereocenters of which one (at C2) is tetra-substituted. In this single-step transformation, two C–C bonds are forged on vicinal carbons, thus, highlighting the power of the carbolithiation/trapping protocol described herein. The reactivity of benzylic lithio-carbanion intermediates such as 20 with other electrophiles is the focus of future studies.
Scheme 3.

Diastereoselective carbolithiation/methyl electrophile trap.
Conclusions
In conclusion, we report the first examples of highly diastereoselective carbolithiations of α-arylated dehydro-piperidine enecarbamates with alkyllithium nucleophiles.32 This novel reactivity side-steps the challenge of the direct deprotonation of N-Boc piperidines and has led to the diastereoselective synthesis of piperidine derivatives bearing vicinal (C2, C3) substituents. This short, umpolung-type synthetic sequence provides a straightforward method to access alkylated piperidines that may be applied in the synthesis of compounds of medicinal and biological importance. Efforts to render this unusual umpolung-type coupling enantioselective as well as to extend the scope to include other α-substituted enecarbamates are underway.33
Supplementary Material
Acknowledgments
This research was supported by the U.S. National Institutes of Health (NIGMS RO1 086374). T.K. B. is grateful to the NIGMS for a postdoctoral fellowship (F32GM-103210-02). H. T. thanks The Kyoto University Foundation for a visiting scholar fellowship. M. W. gratefully acknowledges the German Academic Exchange Service (DAAD) for a postdoctoral fellowship. We thank A. DiPasquale for solving the crystal structures of 11 and 12 (displayed with CYLview), supported by NIH Shared Instrumentation Grant No. S10-RR027172. The NSF (CHE-0840505) is acknowledged for funding computational resources at UCB.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/c000000x/
Notes and references
- 1.(a) Beak P, Lee WK. Tetrahedron Lett. 1989;30:1197. [Google Scholar]; (b) Bailey WF, Beak P, Kerrick ST, Ma S, Wiberg KB. J. Am. Chem. Soc. 2002;124:1889. doi: 10.1021/ja012169y. [DOI] [PubMed] [Google Scholar]
- 2.(a) Mitchell EA, Peschiulli A, Lefevre N, Meerpoel L, Maes BUW. Chem.-Eur. J. 2012;18:10092. doi: 10.1002/chem.201201539. [DOI] [PubMed] [Google Scholar]; (b) Vo C-VT, Bode JW. J. Org. Chem. 2014;79:2809. doi: 10.1021/jo5001252. [DOI] [PubMed] [Google Scholar]
- 3.For a recent discussion, see: Duttwyler S, Chen S, Takase MK, Wiberg KB, Bergman RG, Ellman JA. Science. 2013;339:678. doi: 10.1126/science.1230704.
- 4.(a) Watson PS, Jiang B, Scott B. Org. Lett. 2000;2:3679. doi: 10.1021/ol006589o. [DOI] [PubMed] [Google Scholar]; (b) Bailey PD, Millwood PA, Smith PD. Chem. Commun. 1998:633. [Google Scholar]
- 5.(a) Beak P, Lee WK. J. Org. Chem. 1993;58:1109. [Google Scholar]; (b) Coldham I, Raimbault S, Whittaker DTE, Chovatia PT, Leonori D, Patel JJ, Sheikh NS. Chem. -Eur. J. 2010;16:4082. doi: 10.1002/chem.200903059. [DOI] [PubMed] [Google Scholar]; (c) Shawe TT, Meyers AI. J. Org. Chem. 1991;56:2751. [Google Scholar]; (d) Stead D, Carbone G, O'Brien P, Campos KR, Coldham I, Sanderson A. J. Am. Chem. Soc. 2010;132:7260. doi: 10.1021/ja102043e. [DOI] [PubMed] [Google Scholar]; (e) Beng TK, Tyree WS, Parker T, Su C, Williard PG, Gawley RE. J. Am. Chem. Soc. 2012;134:16845. doi: 10.1021/ja307796e. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Iula DM, Gawley RE. J. Org. Chem. 2000;65:6196. doi: 10.1021/jo000786j. [DOI] [PubMed] [Google Scholar]; (g) Low E, Gawley RE. J. Am. Chem. Soc. 2000;122:9562. [Google Scholar]; (h) Gawley RE, Hart GC, Bartolotti LJ. J. Org. Chem. 1989;54:175. [Google Scholar]; (i) Gawley RE, Zhang Q. J. Am. Chem. Soc. 1993;115:7515. [Google Scholar]; (j) Gawley RE, Zhang Q. J. Org. Chem. 1995;60:5763. [Google Scholar]
- 6.(a) Sheikh NS, Leonori D, Barker G, Firth JD, Campos KR, Meijer AJHM, O'Brien P, Coldham I. J. Am. Chem. Soc. 2012;134:5300. doi: 10.1021/ja211398b. [DOI] [PubMed] [Google Scholar]; (b) Xiao D, Lavey BJ, Palani A, Wang C, Aslanian RG, Kozlowski JA, Shih N-Y, McPhail AT, Randolph GP, Lachowicz JE, Duffy RA. Tetrahedron Lett. 2005;46:7653. [Google Scholar]
- 7.Beng TK, Woo JS, Gawley RE. J. Am. Chem. Soc. 2012;134:14764. doi: 10.1021/ja306276w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Seel S, Thaler T, Takatsu K, Zhang C, Zipse H, Straub BF, Mayer P, Knochel P. J. Am. Chem. Soc. 2013;133:4774. doi: 10.1021/ja201008e. [DOI] [PubMed] [Google Scholar]; (b) Millet A, Larini P, Clot E, Baudoin O. Chem. Sci. 2013;4:2241. [Google Scholar]
- 9.Xiao D, Wang C, Palani A, Tsui H-C, Reichard G, Paliwal S, Shih N-Y, Aslanian R, Duffy R, Lachowicz J, Varty G, Morgan C, Liu F, Nomeir A. Bioorg. Med. Chem. Lett. 2010;20:6313. doi: 10.1016/j.bmcl.2010.08.059. [DOI] [PubMed] [Google Scholar]
- 10.Occhiato EG, Trabocchi A, Guarna A. J. Org. Chem. 2001;66:2459. doi: 10.1021/jo001807c. [DOI] [PubMed] [Google Scholar]
- 11.Shono T, Matsumura Y, Tsubata K, Sugihara Y, Yamane S, Kanazawa T, Aoki T. J. Am. Chem. Soc. 1982;104:6697. [Google Scholar]
- 12.C3-Alkenylation, arylation, trifluoromethylation and sulfonylation reactions have been achieved under transition metal-catalyzed and photoredox conditions. For a recent review, see: Gigant N, Chausset-Boissarie L, Gillaizeau I. Chem. -Eur. J. 2014;20:7548. doi: 10.1002/chem.201402070.
- 13.Seebach D. Angew. Chem. Int. Ed. 1979;18:239. [Google Scholar]
- 14.For a review on carbolithiations see: Minko Y, Marek I. Advances in Carbolithiation. In: Luisi R, Capriati V, editors. Lithium Compounds in Organic Synthesis: From Fundamentals to Applications. Weinheim, Germany: Wiley-VCH; 2014. p. 329. For an acyclic prior example with ureas, see: Tait M, Donnard M, Minassi A, Lefranc J, Bechi B, Carbone G, O'Brien P, Clayden J. Org. Lett. 2013;15:34. doi: 10.1021/ol3029324.
- 15.Young DW, Comins DL. Org. Lett. 2005;7:5661. doi: 10.1021/ol052313a. [DOI] [PubMed] [Google Scholar]
- 16.Lepifre F, Cottineau B, Mousset D, Bouyssou P, Coudert G. Tetrahedron Lett. 2004;45:483. [Google Scholar]
- 17.Williams DR, Reeves JT, Nag PP, Pitcock WH, Jr, Baik M-H. J. Am. Chem. Soc. 2006;128:12339. doi: 10.1021/ja063243l. [DOI] [PubMed] [Google Scholar]
- 18.Ortho-lithiation predominates when sec-BuLi/TMEDA is employed. See the paragraph before the conclusion in the following: Beng TK, Fox N. Tetrahedron Lett. 2015;56:119.
- 19.CYLview, 1.0b; Legault CY. Université de Sherbrooke; 2009. ( http://www.cylview.org).
- 20.1H-NMR analysis of the crude reaction mixture showed significant dehalogenation.
- 21.Beng TK, Wilkerson-Hill SM, Sarpong R. Org. Lett. 2014;16:916. doi: 10.1021/ol403671s. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Relative configuration of 9mC was determined by NOE analysis.
- 23.See the Supporting Information for details of computations.
- 24.(a) Momose T, Toyooka N. J. Org. Chem. 1994;59:943. [Google Scholar]; (b) Beak P, Lee WK. J. Org. Chem. 1993;58:1109. [Google Scholar]
- 25.Fürst A, Plattner PA. Helv. Chim. Acta. 1949;32:275. doi: 10.1002/hlca.19490320139. [DOI] [PubMed] [Google Scholar]
- 26.Pedrosa R, Andres C, Iglesias JM, Perez-Encabo A. J. Am. Chem. Soc. 2001;123:1817. doi: 10.1021/ja002864q. [DOI] [PubMed] [Google Scholar]
- 27.(a) Peoples PR, Grutzner JB. J. Am. Chem. Soc. 1980;102:4709. [Google Scholar]; (b) Ahlbrecht H, Harbach J, Hoffmann RW, Ruhland T. Liebigs Ann. 1995:211. [Google Scholar]; (c) Fraenkel G, Martin KV. J. Am. Chem. Soc. 1995;117:10336. [Google Scholar]
- 28.Wilkinson T, Stehle N, Beak P. Org. Lett. 2000;2:155. doi: 10.1021/ol9912534. [DOI] [PubMed] [Google Scholar]
- 29.For a discussion of distortion and a correlation to reaction rate, see: Fernández I, Bickelhaupt FM. Chem. Soc. Rev. 2014;43:4953. doi: 10.1039/c4cs00055b.
- 30.For a recent application of the distortion model in explaining aryne regioselectivities, see: Medina JM, Mackey JL, Garg NK, Houk KN. J. Am. Chem. Soc. 2014;136:15798. doi: 10.1021/ja5099935.
- 31.A table describing the correlation between the Li-C2-C7 bond angle and the experimental yield is in the Supporting Information.
- 32.During the review of this manuscript, a related carbolithiation of α-aryl ene-piperidyl ureas was reported: Tait MB, Butterworth S, Clayden J. Org. Lett. 2015;17:1236. doi: 10.1021/acs.orglett.5b00199.
- 33.CCDC 1048615 (11), and 1048616 (12) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.