

Abstract
Mapping the precise position of endonucleolytic cleavage sites is a fundamental experimental technique used to describe the function of a homing endonuclease. However, these proteins are often recalcitrant to cloning and over-expression in biological systems because of toxicity induced by spurious DNA cleavage events. In this chapter we outline the steps to successfully express a homing endonuclease in vitro and use this product in nucleotide-resolution cleavage assays.
Keywords: Selfish DNA, Intron, Intein, Mobile DNA, Mobile genetic element
1 Introduction
Homing endonucleases (HEs) are site-specific DNA endonucleases that promote the horizontal transfer of the gene encoding them (i.e., homing endonuclease gene or HEG) and flanking DNA. These HEGs are considered selfish/parasitic elements and can be found as free-standing genes or embedded within intervening sequences such as introns and inteins. Typical HEs recognize relatively large sequences (14–40 bp) as compared to restriction endonucleases (4–8 bp). These recognition sequences usually contain a distinguishing characteristic that enables the HE differentiate between a genome containing the HEG and one that lacks the sequence [1–3]. The associated intervening sequence of intron-and intein-encoded HEGs lies within the HE recognition sequence thereby disrupting it and preventing self-cleavage. However, the same uninterrupted sequence in another genome is sensitive to the HE. A HE-induced DNA break initiates a gene conversion event whereby the HEG is copied into the new location (intron/intein homing). The process for free-standing HEGs is similar, but the mode of protection can vary; the recognition site typically contains sequence changes that prevent its cleavage (intronless homing) [4], but it has also been observed that an intron, which does not encode the HEG [5] or other genetic element positioned at this location, can prevent cleavage (collaborative homing) [6].
One of the key pieces of information describing the function of an HE is the exact position where the nuclease cuts DNA. The location of the cleavage site (CS) is important for biotechnological application and can also be informative as to how the genome is protected from cleavage. For example, is the CS near an intervening sequence, or are there sequence differences between sensitive and resistant sites? Cleavage information also provides insight into the enzymatic properties of the nuclease, such as whether both strands are cut and the nature of the extensions if there is a double-strand break. The experimental design to map CSs is conceptually straightforward; one simply mixes the endonuclease with potential DNA targets and looks for changes in mobility of the target DNA by gel electrophoresis. The exact cleavage point is mapped by comparing cleavage product migration to a DNA sequencing ladder.
The toxicity associated with HEs is a less tractable problem. Efforts to alleviate the toxicity of certain proteins by tuning their over-expression in vivo have been a long-standing course of investigation [7–9]. Various strategies to resolve this problem have been employed, including the use of tightly controlled araPBAD [10] and rhaPBAD [11] inducible promoters, antisense promoters [12, 13], the generation of new hybrid promoters [14], introduction of RNA polymerase (RNAP) by bacteriophage infection [15], and control of plasmid copy number [16, 17]. One way to overcome this hurdle is to produce the endonuclease in vitro using commercially available cell-free in vitro transcription/translation systems. This method is fast and simple, and since no living cells are involved, toxicity is not an issue [18, 19]. Cell-free protein synthesis has been used to produce several HEGs including I-TevI [20, 21], I-TevII [22], SegF [4], Hef [23], SegA [24], I-DmoI [25], F-CphI [5], MobA [6], MobE [23], I-TslI [3], and I-Ssp6803I [26]. Such studies employing in vitro-synthesized HEs have contributed greatly to our understanding of the structure–function and evolution of these remarkable proteins.
Coupling in vitro-synthesized HEs with radiolabeled DNA targets in endonuclease cleavage assays can provide a vast amount of information about the enzyme. Although such data are generally qualitative in nature, results are produced quickly by simple standard protocols and can be extremely valuable in determining a path that the research will take. In this chapter, we focus on producing a homing endonuclease in vitro using a cell-free system and mapping its cleavage site at nucleotide resolution with single-strand, end-radiolabeled DNA targets.
2 Materials
2.1 Kinasing Reagents
T4 polynucleotide kinase (10 Units/mL).
10× Kinase buffer A (500 mM Tris–HCl pH 7.6, 100 mM MgCl2, 50 mM DTT, 1 mM spermidine).
γ-32 P-ATP (3,000 Ci/mmol; 10 uCi/μL).
5 % Trichloroacetic acid.
Glass microfiber filter (Whatman) 2.3 cm grade GF/B.
Scintillation cocktail.
2.2 Polymerase Chain Reaction Reagents
Taq DNA polymerase (5 Units/mL).
10× PCR buffer (100 mM Tris–HCl pH 8.8, 500 mM KCl, 0.8 % v/v, Nonidet P40).
dNTP Mix (2 mM each dATP, dCTP, dGTP, and dTTP).
25 mM MgCl2.
2.3 Sequencing Reagents
2 N NaOH.
5 mM EDTA.
Sequenase v2.0 (Affymetrix/USB).
5× Sequenase buffer (200 mM Tris–HCl, pH 7.5, 100 mM MgCl2, 250 mM NaCl).
Modified labeling mix (7.5 μM each dNTP).
ddATP, ddCTP, ddGTP, and ddTTP mixes (Each mix contains: 80 μM each dNTP, 8 μM specific ddNTP, 50 mM NaCl).
0.1 M DTT.
Sequencing loading dye (95 % formamide, 20 mM EDTA, 0.05 % bromophenol blue, 0.05 % xylene cyanol FF).
2.4 In Vitro Transcription/Translation Reagents
TNT T7 Quick Coupled Transcription/Translation System (Promega) see Note 1 for discussion of different cell-free protein expression systems.
1 mM methionine.
35S-methionine (>1,000 Ci/mmol).
Nuclease-free water.
2.5 Endonuclease Assay Reagents
10× ECA buffer (0.5 M Tris–HCl pH 7.5, 0.5 M NaCl).
0.25 mg/mL Poly (dI-dC) (Sigma).
0.1 M MgCl2.
Phenol, saturated, pH 6.6/7.9.
2.6 Denaturing Polyacrylamide Gel Electrophoresis (PAGE) Reagents
40 % (19:1 acrylamide–bis acrylamide) polyacrylamide (see Note 2).
10× Tris-borate EDTA (TBE) buffer (890 mM Tris, 890 mM Boric Acid, 20 mM EDTA, pH 8.3).
Ultrapure urea.
10 % ammonium per sulfate (APS) stored at 4°C.
N,N,N,N′-tetramethyl-ethylenediamine (TEMED) stored at 4°C.
2.7 Molecular Biology
Plasmid miniprep kit.
PCR purification kit.
2.8 Equipment
Thermal cycler.
Spectrophotometer.
High-voltage power supply.
Phosphor imager.
Model S2 Sequencing Gel Electrophoresis Apparatus (or equivalent) (see Note 3).
Glass plates (30 cm × 40 cm) for Gel Electrophoresis Apparatus.
0.4 mm spacers and combs to fit Gel Electrophoresis Apparatus.
Whatman filter paper (30 cm × 40 cm).
Gel dryer.
Vacuum apparatus to fit glass microfiber filters for scintillation counting.
Scintillation counter.
3 Methods
The overall strategy is straightforward. A primer is designed to incorporate a T7 promoter upstream of a HEG (Fig. 1a, see Note 4 for discussion of optimal parameters) for PCR amplification. This PCR product is mixed with a coupled in vitro T7 RNA polymerase transcription/translation system and the T7 promoter directs transcription of the PCR amplified HEG. The RNA is then translated by ribosomes in the cell-free system producing the homing endonuclease (Fig. 1b). This in vitro-synthesized HE is mixed directly with the PCR-generated putative target DNA that has been radiolabeled on the 5′ end of a single strand (Fig. 1c). Double-strand cleavage by the HE results in a total of four DNA strands, only one of which is physically connected to the radioactive atom (Fig. 1d). The products are then separated by denaturing gel electrophoresis alongside a DNA sequencing ladder containing DNA molecules with the identical 5′ radiolabeled end as the substrate DNA (Fig. 2a).
Fig. 1.
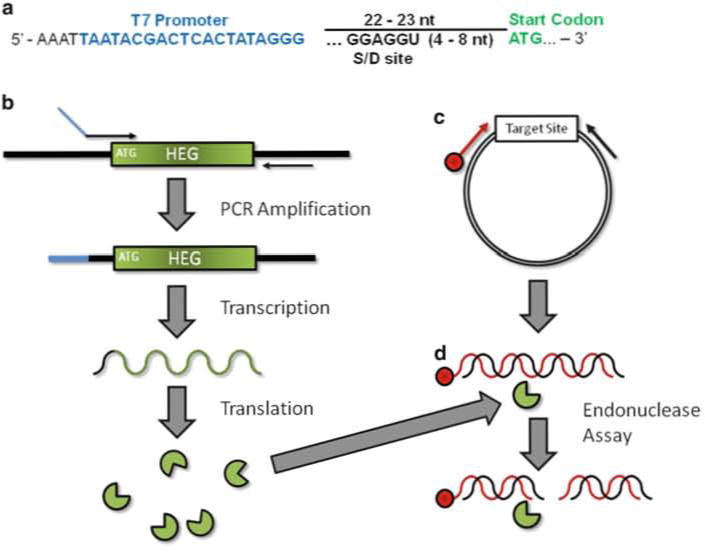
Schematic of in vitro homing endonuclease expression and endonuclease assay. (a) Upstream primer design for amplification of a HEG. T7 promoter (blue) should be placed ~22–23 bp upstream of the HEG initiation codon (green). Sequence at the 5′ end in black is thought to stabilize RNAP–promoter interaction. If using a prokaryotic system a ribosome binding site/Shine–Delgarno sequence (S-D) should be incorporated. (b) In vitro homing endonuclease production. PCR amplification results in the incorporation of a T7 promoter (blue) upstream of the HEG. This product is used to direct protein synthesis in the coupled in vitro transcription/translation reaction. (c) Target site DNA is amplified from a plasmid with a 5′-end labeled (red) and unlabeled primer resulting in a duplex DNA molecule labeled on one strand. The same labeled primer and plasmid DNA is used to generate a DNA sequencing ladder. The resulting ladder and the PCR product are labeled at the exact same position. (d) Endonuclease Assay. The HE (from b) is mixed with precursor dsDNA labeled on the 5′-end. Cleavage of both strands results in two dsDNA products. Of the resulting four individual strands, only one is covalently linked to the label and therefore visible by phosphor imaging
Fig. 2.
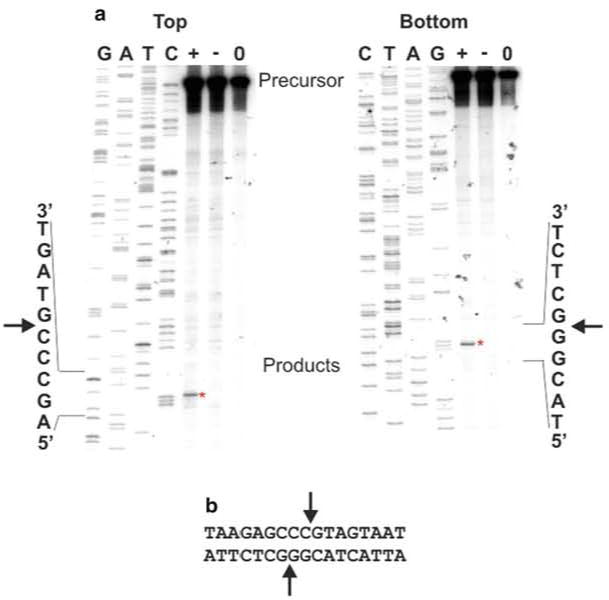
Cleavage site mapping using in vitro-synthesized protein. (a) Precursor/substrate DNA was amplified, and each strand was individually 5′-end-labeled with 32P in different PCRs. Sequencing ladders were generated using the corresponding labeled primer. The ddNTP used in the sequencing reactions is indicated above each lane. The sequence of the target DNA immediately flanking the cut site (represented by an arrowhead) is indicated next to each autoradiogram image. Precursor/substrate DNA was incubated with in vitro-synthesized protein (+), a mock unprogrammed in vitro synthesis reaction (−) or DNA only (0) and the reactions were separated on a denaturing polyacrylamide gel by electrophoresis. The regions where the precursors and products (asterisk) migrate are indicated. (b) Summary of the cleavage reaction. The DNA sequence for a portion of the region is shown below the images. The cut sites on each strand are indicated by arrows and result in 2 nt 3′ extensions
3.1 Generation of Singly 5′-End Labeled Target DNA Substrate
Kinase primer by mixing 9.5 μL H2O, 1.5 μL 10× buffer A, 1.0 μL 6 μM oligo, 2.0 μL γ-32 P-ATP, and 1.0 μL T4 PNK.
Incubate at 37°C for 30 min.
Place reactions on ice or store at −20°C.
PCR amplify the target DNA by adding 56.0 μL H2O, 10.0 μL 10× PCR buffer, 10.0 μL dNTP mix, 6.0 μL 25 mM MgCl2, 1.0 μL 6 μM unlabeled paired oligonucleotide, 1.0 μL template DNA (1:100 dilution of a miniprep), and 1.0 μL Taq DNA polymerase to the labeled primer.
Incubate for the following cycle: 95°C for 10 min (hot start), followed by 25 cycles of 95°C for 30 s, 50°C for 30 s, 72°C for 30 s. Do a final extension at 72°C for 5 min (see Note 5).
The PCR product should be purified with a commercially available PCR cleanup kit and eluted in 30–50 μL H2O.
Reactions should be placed on ice or stored at −20°C.
Check incorporation of label by placing a glass microfiber filter onto the vacuum apparatus and wet filter with 5 % TCA.
Add 1 μL of purified PCR product and wash for 1 min with 5 % TCA followed by 95 % ethanol to help dry the filter.
Place filter in scintillation vial with scintillation cocktail and read on 32P channel. This allows calculation of the amount of radiolabel incorporated into the DNA.
Measure total label by placing 1 μL of purified PCR product on glass filter and add directly (no washing) to scintillation vial with cocktail (see Note 6).
3.2 Generation of DNA Sequencing Ladder
Kinase primer by mixing 1.5 μL H2O, 0.5 μL 10× Buffer A, 1.0 μL 2 μM oligo, 2.0 μL γ-32 P-ATP, and 0.2 μL T4 PNK.
Incubate at 37°C for 30 min.
Place reactions on ice or store at −20°C.
Prepare plasmid DNA from 1.5 to 3.0 mL culture using a commercially available miniprep kit.
Elute DNA in 100 μL H2O.
Denature plasmid DNA for sequencing by mixing 15.0 μL plasmid DNA, 1.75 μL 2 N NaOH, and 0.7 μL 5 mM EDTA.
Incubate at 37°C for 30 min.
Precipitate by adding 1.7 μL 3 M sodium acetate and 50.0 μL 100 % ethanol and vortex.
Incubate for at least 30 min at −80°C or > 2 h at −20°C.
Centrifuge at high speed for 10 min.
Remove the supernatant.
Dry and resuspend the pellet in 5.4 μL H2O.
Anneal primer to plasmid DNA by mixing 5.4 μL denatured plasmid DNA, 2.6 μL kinased primer, and 2.0 μL 5× Sequenase buffer.
Heat to >90°C and cool slowly to 37°C in a heat block with heat turned off.
Spin briefly and place on ice.
Aliquot 2.5 μL of each ddNTP mix into four separate tubes labeled G, A, T, and C that have been pre-warmed to 37°C.
For the extension and termination reactions add 1.0 μL 0.1 M DTT, 2.0 μL modified labeling mix and 0.5 μL H2O to denatured DNA mix.
Begin extension reaction by adding 2.5 μL of a 1:8 dilution of Sequenase (in the provided dilution buffer).
Incubate for 4 min at 37°C.
Aliquot 3.5 μL of this extension mix to the pre-aliquotted ddNTP termination mix.
Incubate at 37°C for 4 min.
Add 5 μL sequencing loading dye.
Place reactions on ice or store at −20°C.
3.3 Amplification of HEG for In Vitro Expression
PCR amplify the HEG by mixing 68.0 μL H2O, 10.0 μL 10× PCR buffer, 10.0 μL dNTP mix, 6.0 μL 25 mM MgCl2, 2.0 μL 20 μM upstream (T7 promoter-containing) primer (see Note 4), 2.0 μL 20 μM downstream primer, 1.0 μL template DNA, and 1.0 μL Taq DNA polymerase.
Incubate for the following cycle 95°C for 10 min (hot start), followed by 25 cycles of: 95°C for 30 s, 50°C for 30 s, 72°C for 1 min (see Notes 5 and 7).
Purify the PCR product with a commercially available PCR cleanup kit and elute in 30–50 μL H2O.
Quantify DNA on a spectrophotometer.
3.4 Generation of In Vitro-Synthesized Homing Endonuclease
To produce the homing endonuclease mix 40 μL TNT T7 Quick Master Mix, 1 μL 1 mM methionine, 2.5–5 μL PCR-generated DNA template (~100–800 ng), and bring the total volume up to 50 μL with H2O (see Note 8).
In parallel produce a mock in vitro synthesis control (see Note 9) by mixing 40 μL TNT T7 Quick Master Mix, 1 μL 1 mM methionine, and 9 μL H2O.
Incubate the reactions at 30°C for 60–90 min.
The protein products are ready to use and should be kept on ice or frozen at −70°C in aliquots to prevent numerous freeze– thaw cycles for storage.
3.5 Endonucleolytic Cleavage Assay (EC Assay)
Thaw all reagents at 25°C and place on ice before mixing.
Gently Mix 2 μL 10× ECA buffer, 2 μL 0.25 mg/mL Poly (dI-dC), 2 μL 0.1 M MgCl2, ~105 cpm target DNA, 2 μL in vitro-synthesized protein, and H2O to 20 μL on ice.
Briefly centrifuge to move all liquid to the bottom of the tube.
Incubate reactions at 30°C for 30 min (see Note 10).
Stop reactions on ice.
3.6 Phenol Extraction
Add an equal volume (20 μL) of phenol.
Vortex and centrifuge for 2 min.
Transfer aqueous phase to a fresh 1.7 mL tube.
Add 5 μL sequencing loading buffer to each reaction.
3.7 Denaturing Polyacrylamide Gel
To make an 8 % polyacrylamide/7 M urea denaturing gel mix 38 mL H2O, 20 mL 40 % polyacrylamide, 10 mL 10× TBE buffer, and 36.7 g urea in a 250 mL beaker with a stir bar (see Note 11).
Gently heat and stir the mixture to dissolve the urea (see Note 12). While the solution is mixing proceed to step 3.
Clean glass plates, spacers, and combs with H2O and 95 % ethanol. Assemble glass plates and spacers. Tape the bottom and sides of the gel to prevent leaking.
Add 0.5 mL 10 % APS and 20 μL TEMED to the dissolved gel solution, mix well, and slowly pour the gel solution into one corner of the opening between the two plates with this “sandwich” held at a ~45° angle, until the gel is filled.
Lay the gel flat, place the comb between the glass plates, and clamp each side of the gel with three clamps per side. Allow the gel to polymerize completely (typically at least an hour).
Before running the gel, remove the comb and tape carefully. Place the gel into the electrophoresis apparatus and fill the upper and lower tanks with 1× TBE, ensuring that the glass plates are submerged in buffer.
3.8 Electrophoresis
Pre-run the gel at 60 W for at least 30 min (see Note 13).
Heat the samples at 95°C for 5 min, place on ice for 1–2 min, centrifuge for 30 s, and place the tubes back on ice.
Prior to loading, rinse the wells with buffer using a syringe, being careful not to damage the wells.
Load 1–10 μL into each well (see Note 14).
Run gel for 2.5 h at a constant 60 W (see Note 11).
When the electrophoresis is complete, carefully disassemble the gel sandwich by prying the glass plates apart with a spatula. If the gel remains attached to only one plate, press a large filter paper on top of the gel, flip so that the paper is on the bottom, and transfer the gel to the paper. If the gel remains attached to both plates, gently vibrate the glass plate to help the gel fall onto one plate.
Place on gel dryer and cover with plastic wrap. Dry under vacuum with heat until completely dry (~30–60 min) (see Note 15).
Expose to film or phosphor imaging screen overnight (see Note 16).
Acknowledgments
We would like to thank Caren J. Stark, Matthew Stanger, Dorie Smith, and Carol Lyn Piazza for critical reading of the manuscript. Research in the Belfort Lab is supported by NIH grants GM39422 and GM44844.
Footnotes
Cell-free protein synthesis systems are mainly derived from E. coli, rabbit reticulocyte lysates and wheat germ extracts. The protocol described here uses a rabbit reticulocyte cell-free system, but can easily be adapted for other cell-free protein synthesis systems. In fact, cell-free systems derived from wheat germ and E. coli have also been successfully used in our hands [20, 21, 26]. The manufacturers provide detailed protocols for using their systems and typically no modification is necessary. The E. coli system generally provides the highest yield, but our personal experience suggests that this system can be contaminated by DNA exonucleases and is not the best choice when studying homing endonucleases. We have not experienced any nonspecific nuclease problems with either of the eukaryotic systems. Additionally, eukaryotic systems have better RNA unwinding activities and should be used when translational start signals are sequestered in stem-loop structures.
Acrylamide is a neurotoxin. Use protective equipment when handling.
Our preferred electrophoresis apparatus is a Model S2 Sequencing Gel Electrophoresis Apparatus that fits 30 cm × 40 cm gel sequencing plates. This apparatus is equipped with an aluminum plate that disperses heat generated during the electrophoresis and minimizes gel “smiling.” It also has a convenient built-in drain for the top buffer chamber.
The upstream primer needs to incorporate a T7 promoter at a sufficient distance upstream of the translational start of the HEG (~22 bp Fig.1a). The sequence between the T7 promoter and translational start can anneal to the template though this is not necessary. If using a prokaryotic cell-free extract, production of the HE will require the inclusion of a ribosome binding or Shine–Delgarno (S-D) site (5′-GGAGGU-3′) between 4 and 8 nt upstream of the translational start [27]. A naturally occurring site can be used, but if no site exists upstream of the HEG, one can be incorporated into the upstream primer.
The annealing temperature, extension time and number of cycles should be determined empirically for each set of oligonucleotides and template. As the optimal PCR conditions will be dependent on the particular template and primers, the conditions should first be established using unlabeled primers. In our experience, for cleavage site mapping target DNA PCR products between 0.2 and 0.4 kb work best and therefore only a short extension time is necessary.
The yield of labeled DNA ranges from 103 to 105 cpm/μL depending on the individual oligonucleotide used.
To ensure that the HEG is wild type, the template should be genomic DNA since clones of HEGs are often mutated. However, if a stable, wild-type clone of the HEG exists that is not conducive to over-expression that would work also.
The Master Mix should be thawed, distributed into 20 μL or 40 μL aliquots and frozen at −70°C. When handling the Master Mix, it should be placed on ice when not used in incubation. Also, protein synthesis can be monitored by the incorporation of radioactive methionine. Simply substitute 2 μL 35S-methionine for the unlabeled methionine. The technical manual for this system is quite detailed and no modifications are necessary.
A mock synthesis reaction, where the cell-free system lacks a PCR product and is therefore not programmed to produce an HEG should be performed in parallel. This allows non-HE derived cleavages of the target to be “subtracted” from the result.
Endonuclease cleavage assay conditions including: buffer, pH, monovalent ion and divalent ion, temperature, and time should be optimized for each protein, although the conditions set out here are a good starting point. We use a lower temperature (30°C vs. 37°C) to minimize the action of nonspecific nucleases. Also, each set of reactions should include the HEG programmed in vitro synthesis reaction, a mock synthesis reaction and DNA only control (Fig. 2a).
The percentage gel and duration of run required for optimal resolution will vary depending on fragment lengths and will need to be determined empirically. This protocol is based on product fragments between 0.1 and 0.2 kb.
Make sure that the solution is not too warm or the gel may polymerize too quickly.
Wattage, pre-run and run conditions will vary depending on apparatus, gel thickness, and buffer composition. This has been optimized for 1× TBE gels cast with 0.4 mm spacers on a Model S2 apparatus. Thicker gels need to be pre-run and run at a lower wattage or voltage.
The ratio of sequencing reaction to cleavage assay reaction will have to be determined by trial and error and depends on the quality of each of the reactions.
It is imperative that the gel is completely dry; otherwise it may crack when removed from the vacuum.
For ease of interpretation, the reaction containing the in vitro-synthesized HEG, and therefore, the cleavage product to be mapped should be loaded immediately adjacent to the sequencing ladder. Ideally it should be loaded next to the ddNTP lane that corresponds to the last base contained in the cleavage product (Fig. 2a). This allows for the most accurate mapping of the cleavage site. To interpret the data, one simply has to look for the ddNTP band that aligns with the cleavage product. This corresponds to the 3′-most base contained within the cleavage product and the DNA strand is cut immediately 3′ to this base (Fig. 2a, b). Sometimes, the sequencing band that lines up with the cleavage product remains ambiguous. Since the sequencing and cleavage reactions are contained in different buffers and are not typically precipitated, slight migration differences can occur. If this is a problem, both sets of reactions could be phenol extracted, precipitated and resuspended in loading dye. Alternatively, the cleavage reactions and the sequencing reaction can be mixed before loading. In this instance, the product should produce an extra band in each of the sequencing reactions except for the ddNTP that has a band that aligns with the product (see Fig. 4.3a in [3] for an example).
Contributor Information
Richard P. Bonocora, Wadsworth Center, New York State, Department of Health, Albany, NY, USA
Marlene Belfort, Department of Biological Sciences and The RNA Institute, University at Albany, State University of New York, Albany, NY, USA.
References
- 1.Lambowitz AM, Belfort M. Introns as mobile genetic elements. Annu Rev Biochem. 1993;62:587–622. doi: 10.1146/annurev.bi.62.070193.003103. [DOI] [PubMed] [Google Scholar]
- 2.Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011;19(1):7–15. doi: 10.1016/j.str.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonocora RP, Shub DA. A likely pathway for formation of mobile group I introns. Curr Biol. 2009;19(3):223–228. doi: 10.1016/j.cub.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belle A, Landthaler M, Shub DA. Intronless homing: site-specific endonuclease SegF of bacteriophage T4 mediates localized marker exclusion analogous to homing endonucleases of group I introns. Genes Dev. 2002;16(3):351–362. doi: 10.1101/gad.960302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng Q, Bonocora RP, Shub DA. A free-standing homing endonuclease targets an intron insertion site in the psbA gene of cyanophages. Curr Biol. 2009;19(3):218–222. doi: 10.1016/j.cub.2008.11.069. [DOI] [PubMed] [Google Scholar]
- 6.Bonocora RP, et al. A homing endonuclease and the 50-nt ribosomal bypass sequence of phage T4 constitute a mobile DNA cassette. Proc Natl Acad Sci U S A. 2011;108(39):16351–16356. doi: 10.1073/pnas.1107633108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol. 1991;219(1):37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- 8.Moffatt BA, Studier FW. T7 lysozyme inhibits transcription by T7 RNA polymerase. Cell. 1987;49(2):221–227. doi: 10.1016/0092-8674(87)90563-0. [DOI] [PubMed] [Google Scholar]
- 9.Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189(1):113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 10.Guzman LM, et al. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177(14):4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giacalone MJ, et al. Toxic protein expression in Escherichia coli using a rhamnose-based tightly regulated and tunable promoter system. Biotechniques. 2006;40(3):355–364. doi: 10.2144/000112112. [DOI] [PubMed] [Google Scholar]
- 12.Worrall AF, Connolly BA. The chemical synthesis of a gene coding for bovine pancreatic DNase I and its cloning and expression in Escherichia coli. J Biol Chem. 1990;265(35):21889–21895. [PubMed] [Google Scholar]
- 13.O’Connor CD, Timmis KN. Highly repressible expression system for cloning genes that specify potentially toxic proteins. J Bacteriol. 1987;169(10):4457–4462. doi: 10.1128/jb.169.10.4457-4462.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1–I2 regulatory elements. Nucleic Acids Res. 1997;25(6):1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao F, Drake SK, Kompala DS. Characterization of gene expression in recombinant Escherichia coli cells infected with phage lambda. Biotechnol Prog. 1993;9(2):153–159. doi: 10.1021/bp00020a006. [DOI] [PubMed] [Google Scholar]
- 16.Bowers LM, et al. Bacterial expression system with tightly regulated gene expression and plasmid copy number. Gene. 2004;340(1):11–18. doi: 10.1016/j.gene.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Saida F, et al. Expression of highly toxic genes in E. coli: special strategies and genetic tools. Curr Protein Pept Sci. 2006;7(1):47–56. doi: 10.2174/138920306775474095. [DOI] [PubMed] [Google Scholar]
- 18.Katzen F, Chang G, Kudlicki W. The past, present and future of cell-free protein synthesis. Trends Biotechnol. 2005;23(3):150–156. doi: 10.1016/j.tibtech.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Katzen F, Peterson TC, Kudlicki W. Membrane protein expression: no cells required. Trends Biotechnol. 2009;27(8):455–460. doi: 10.1016/j.tibtech.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Bell-Pedersen D, et al. A site-specific endonuclease and co-conversion of flanking exons associated with the mobile td intron of phage T4. Gene. 1989;82(1):119–126. doi: 10.1016/0378-1119(89)90036-x. [DOI] [PubMed] [Google Scholar]
- 21.Bell-Pedersen D, et al. I-TevI, the endonuclease encoded by the mobile td intron, recognizes binding and cleavage domains on its DNA target. Proc Natl Acad Sci U S A. 1991;88(17):7719–7723. doi: 10.1073/pnas.88.17.7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell-Pedersen D, et al. Intron mobility in phage T4 is dependent upon a distinctive class of endonucleases and independent of DNA sequences encoding the intron core: mechanistic and evolutionary implications. Nucleic Acids Res. 1990;18(13):3763–3770. doi: 10.1093/nar/18.13.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandegren L, Nord D, Sjoberg BM. SegH and Hef: two novel homing endonucleases whose genes replace the mobC and mobE genes in several T4-related phages. Nucleic Acids Res. 2005;33(19):6203–6213. doi: 10.1093/nar/gki932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma M, Ellis RL, Hinton DM. Identification of a family of bacteriophage T4 genes encoding proteins similar to those present in group I introns of fungi and phage. Proc Natl Acad Sci U S A. 1992;89(14):6658–6662. doi: 10.1073/pnas.89.14.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalgaard JZ, Garrett RA, Belfort M. A site-specific endonuclease encoded by a typical archaeal intron. Proc Natl Acad Sci U S A. 1993;90(12):5414–5417. doi: 10.1073/pnas.90.12.5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonocora RP, Shub DA. A novel group I intron-encoded endonuclease specific for the anticodon region of tRNA(fMet) genes. Mol Microbiol. 2001;39(5):1299–1306. doi: 10.1111/j.1365-2958.2001.02318.x. [DOI] [PubMed] [Google Scholar]
- 27.Shine J, Delgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]