

Abstract
In fetal valve maturation the mechanisms by which the relatively homogeneous proteoglycan-rich extracellular matrix (ECM) of endocardial cushions is replaced by a specialized and stratified ECM found in mature valves are not understood. Therefore, we reasoned that uncovering proteases critical for ‘remodeling’ the proteoglycan rich (extracellular matrix) ECM may elucidate novel mechanisms of valve development. We have determined that mice deficient in ADAMTS5, (A Disintegrin-like And Metalloprotease domain with ThromboSpondin-type 1 motifs) which we demonstrated is expressed predominantly by valvular endocardium during cardiac valve maturation, exhibited enlarged valves. ADAMTS5 deficient valves contained a reduction in cleavage of its substrate versican, a critical cardiac proteoglycan. In vivo reduction of versican, in Adamts5−/− mice, achieved through Vcan heterozygosity, substantially rescued the valve anomalies. An increase in BMP2 immunolocalization, Sox9 expression and mesenchymal cell proliferation were observed in Adamts5−/− valve mesenchyme and correlated with expansion of the spongiosa (proteoglycan-rich) region in Adamts5−/− valve cusps. Furthermore, these data suggest that ECM remodeling via ADAMTS5 is required for endocardium to mesenchymal signaling in late fetal valve development. Although adult Adamts5−/− mice are viable they do not recover from developmental valve anomalies and have myxomatous cardiac valves with 100% penetrance. Since the accumulation of proteoglycans is a hallmark of myxomatous valve disease, based on these data we hypothesize that a lack of versican cleavage during fetal valve development may be a potential etiology of adult myxomatous valve disease.
Keywords: ADAMTS5, versican, extracellular matrix, myxomatous valves, endocardial cushions
Introduction
Adult cardiac valve disease is a public heath problem of growing concern affecting up to 5% of individuals, with 25% of the aged population eventually developing aortic valve sclerosis, a diagnostic indicator of valve disease (Hinton and Yutzey, 2010; Hoffman and Kaplan, 2002). The fact that most diseased valves have an underlying malformation (Roberts et al., 2005) suggests the possibility that adult disease could originate, at least in part, through subtle, subclinical anomalies of valve development. A hallmark of cardiac valve developmental anomalies and adult disease is accumulation of proteoglycans and loss of extracellular matrix (ECM) stratification (Gupta et al., 2009a; Hinton et al., 2006; Weismann and Gelb, 2007). In early valve development the provisional ECM is proteoglycan-rich but during valve maturation the is replace by stratified ECM, which includes collagen and elastin. This specialized, ECM is required to effectively regulate unidirectional blood flow in the rapidly growing fetus and in the adult (Combs and Yutzey, 2009; Hinton et al., 2006). The process by which the initial ECM is remodeled during valve maturation remains poorly understood since gene deletions in mice often result in embryonic death or a subclinical or incompletely penetrant valve phenotype. These studies address the overarching hypothesis that proteoglycan accumulation in myxomatous valves results from reduced ECM cleavage and directly contributes to anomalous valve maturation or homeostasis and is not simply a secondary manifestation in disease.
Initially endocardial cushions, the precursors to adult valves, are rather large and block shaped with a proteoglycan-rich ECM (Cooley et al., 2008; Kern et al., 2007; Wirrig et al., 2007). This provisional ECM is required for the epithelial to mesenchymal transformation (EMT) (Runyan and Markwald, 1983) that contributes to the cellularization of the cushions and migration of extra-cardiac cells (Hutson and Kirby, 2003). The endocardial cushion ECM also permits the proliferation of mesenchyme during early stages of valve development (Hinton et al., 2006; Lincoln et al., 2004; Rabkin-Aikawa et al., 2005). Versican is a large aggregating proteoglycan of ECM (Cooley et al., 2008; Henderson and Copp, 1998; Ito et al., 1995; Kern et al., 2007; Kern et al., 2006; Yamamura et al., 1997; Zako et al., 1995) that is abundant during the formation of endocardial cushions, and is essential for their formation, but is dramatically reduced with restricted expression in mature valves. There are 4 splice variants of versican, each containing an N-terminal domain that binds hyaluronan and a C terminal domain that interacts with other ECM molecules (Margolis and Margolis, 1994)-(Aspberg et al., 1999; Aspberg et al., 1997; Wu et al., 2005). The variants differ in the presence/absence of internal glycosaminoglycan (GAGα or GAGβ) domains that are susceptible to cleavage by a subset of ADAMTS proteases referred to as proteoglycanases (ADAMTS1,4,5,9,20). ADAMTS5 cleavage of aggrecan contributes to the progression of osteoarthritis disease (Bondeson et al., 2008; Huang and Wu, 2010; Yatabe et al., 2009) and recent work demonstrated its requirement for versican turnover during limb morphogenesis (Enomoto et al., 2010; Longpre et al., 2009; McCulloch et al., 2009b).
Our previous studies established that versican is cleaved during cardiac development and that versican cleavage fragments are localized subjacent to the endocardium of maturing valves (Kern et al., 2006). Examination of mice heterozygous for a LacZ insertion in Adamts9 revealed a constellation of adult cardiovascular phenotypes including enlarged adult cardiac valves with increased proteoglycans. However, the early embryonic death of the Adamts9 null mice precluded analysis of its role in valve development (Kern et al., 2010). Here, we tested the specific hypothesis that the endocardial expression of ADAMTS5 in developing cardiac valves was essential for versican cleavage during the fetal sculpting phase of valve development (Longpre et al., 2009; McCulloch et al., 2009b). The experimental findings validated the hypothesis and revealed a novel role for ADAMTS5 in differentiation of valvular mesenchyme and stratification of ECM. Since an increase in proteoglycans, including versican, is associated with myxomatous valve disease (Kenagy et al., 2009; Kern et al. 2010; Seidelmann et al., 2008; Theocharis, 2008), the data suggest further investigation of altered ADAMTS5 gene expression and versican proteolysis as contributing factors in congenital valve malformations and adult valve disease.
Methods
Gene-targeted mice
All mouse experiments were done under protocols approved by the Cleveland Clinic Foundation s Institutional Animal Care and Use Committee (IACUC) and the Medical University of South Carolina IACUC. The Adamts5−/− mice used in this study were the Adamts5tm1Dgen/J gene-targeted allele available from Jackson laboratories (Bar Harbor, ME), and were bred into C57Bl/6 (> 10 generations) and maintained as previously described (Jungers et al., 2005; McCulloch et al., 2009b). The Vcanhdf lacZ insertion allele was provided by Roche™ (Mjaatvedt et al., 1998) and also maintained on a pure C57Bl/6 background. Genotyping of Adamts5 mice was by PCR as previously published (McCulloch et al., 2009b). β-gal staining was done as previously described (Jungers et al., 2005; McCulloch et al., 2009b). To mark cardiac neural crest cells, double transgenic Wnt1-Cre (C57Bl/6) mice, provided by Dr. Andrew McMahon, Harvard University, Cambridge, Mass and TgR(ROSA26)26SOR mice were used as described previously (Danielian et al., 1998; Soriano, 1999). Adamts5 expression was evaluated by β-gal staining of Adamts5tm1Dgen/J mice generated by an inactivating insertion of a dicistronic IRES-lacZ-Pgk-Neomycin cassette into exon 2 (McCulloch et al., 2009a).
Antibodies
ADAMTS5 immunolocalization was determined using an ADAMTS5 polyclonal antibody (Abcam, Cambridge, MA). Rabbit polyclonal antibodies against the GAGβ domain of versican were generated by S. Hoffman (Medical University of South Carolina) (Kern et al., 2007). Rabbit polyclonal antibodies against the C-terminal sequence of the neo-epitope of versican V0/V1 generated by proteolytic cleavage (anti-DPEAAE) (Sandy et al., 2001) were purchased from Affinity BioReagents (Golden, CO). Mouse monoclonal anti-α sarcomeric actin and anti-α-smooth muscle actin were obtained from Sigma Chemical Co. (St. Louis, MO). Rabbit polyclonal antibodies against BMP2 were purchased from Abcam (Cambridge, MA). Anti-Sox9 was purchased from Santa Cruz (Santa Cruz, California). PhosphoHistone H3 was purchased from Cell Signaling, (Carlsbad, CA). Biglycan antibody (LF-159) was a generous gift from Dr. L. Fisher at the National Institute of Health. Fluor-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
Histology and Immunohistochemistry
Standard histological procedures were used (Kern et al., 2007); for the ADAMTS5, GAGβ, Sox9 and DPEAAE staining, E17.5 hearts were fixed in 4% paraformaldehyde. For ADAMTS5 expression proteinase K digestion was required with post-fixation of antigen-antibody complex to inhibit diffusion of fluorescent signal. Amsterdam fixed tissue (35% methanol; 35% acetone; 5% acetone; 25% water) was used for DPEAAE staining at E15.5 and BMP2. Note the Amsterdam fixative for DPEAAE revealed increased amount of staining relative to paraformaldehyde fixed tissues at these fetal time points. Modified Movat staining was performed as previously described (Kern et al.). An Olympus BX40 microscope was used for bright-field microscopy and the DP2-BSW (v1.4, build 2743) software (Olympus Corporation, Center Valley, PA) was used for valve cusp and leaflet measurements. For E17.5 Wnt-1Cre-lacZ CNC ‘marked’ sections with overlay immunolocalization, the transmission mode was used on the Leica SP2/SP5 (Bannockburn, IL) confocal microscope to detect β-gal stained cells. In the same image, emission of the fluorescent-conjugated secondary antibody was detected by sequential scan. Using Adobe Photoshop™ software, the image from the transmission mode showing β-gal stained cells was inverted and placed in the blue channel to visualize CNC ‘lineage-marked’ cells with αSMA, cleaved versican DPEAAE and ADAMTS5 immunostaining.
Quantification of Immunofluorescence and Valve Anomalies
Three-dimensional reconstructions: Amira™ 5.3.3 (Visage Imaging, Andover, MA) was used to generate reconstructions. 100–140 5μm-thick paraffin sections were used to generate each E17.5 construction. Volumetric measurements derived from an average of three reconstructions each of Adamts5−/− and WT valves. Data from crosses of Adamts5−/− mice and Vcanhdf/+ were derived from eleven independent internally controlled litters dissected at E17.5. Volumes for valve reconstructions are the average of 3 independent reconstructions each for the Adamts5+/+;Vcan+/+ and Adamts5−/−;Vcan+/+ and 2 from independent litters for Adamts5−/−Vcan+/hdf.
Quantification of valve thickness (not 3d)
The widest portion of the cusps or leaflets of valves were used over a minimum depth of 50 μm using an Olympus BX40 microscope with DP2-BSW (v1.4, build 2743) software. Three independent measurements were taken per cusp, of four different sections. The values were averaged. A minimum number of N=4 animals were used per genotype for statistical analysis.
Quantification of IHC data
To quantify expression from immunohistochemical data, digital images of Adamts5−/− and WT heart sections were acquired at identical confocal settings using the Leica TCS SP5 AOBS Confocal Microscope System (Leica Microsystems Inc., Exton, PA). Using Amira™ software the positive pixels were measured and normalized to total tissue area. A minimum of three separate experiments with three different litters of matched Adamts5−/− were used in the analysis. To quantify the pixel intensity of pHH3 and Sox9, the total number of positive nuclear pixels above a background threshold were counted and normalized to total number of mesenchymal nuclei in the image. For BMP2 quantification the total pixel area of BMP2 positive pixels over background threshold was quantified to the total pixels per cusp area (minus hinge region) of the PA valve and graphed.
Quantification of nuclei
The total nuclei in the valve cusps were counted in sections spanning a depth of at least 50 μm and the number of nuclei/valve area μm2 was determined using Amira™ in 6 Adamts5−/− hearts and 4 WT hearts.
Statistical significance was determined using the Student t-test (2 tailed, type 2), with significance (P<0.05). Statistical data are presented as the mean ± one standard deviation (SD) from the mean.
Results
Adamts5 is expressed in the endocardium and a subset of mesenchymal cells of maturing and adult murine cardiac valves
Adamts5 expression was evaluated by β-gal staining of Adamts5−/− mice generated by an inactivating insertion of a dicistronic IRES-lacZ-Pgk-Neomycin cassette into exon 2. The β-gal staining was shown previously to overlap with the in situ localization of Adamts5 RNA (McCulloch et al., 2009a). ADAMTS5 was detected in the myocardium underlying the OFT cushions at E11.5 (Fig. 1A, open arrow). β-gal indicative of Adamts5 RNA expression was first detected in endocardial cushions of the OFT in a subset of embryos at E12.5 in the endocardium (Fig. 1B, arrow). Immunolocalization of ADAMTS5 in developing valves was first detected at E13.5 in the ECM substratum of the endocardium (Fig. E, arrow) and strong expression was also observed in the underlying myocardium (Fig. 1E, open arrow). The detection of ADAMTS5 using the anti-ADAMTS5 antibody required protease K digestion as well as post-fixation of the ADAMTS5-antibody complex to improve our threshold of detection. At E14.5, ADAMTS5 was detected in the ECM and associated with the endocardium (Fig. 1F–H, solid arrows) of the PV and AV cusps as well as the developing arterial wall (Fig. 1F, I (E15.5) open arrow). There was also expression of ADAMTS5 associated with the endocardium of the developing ventricles (1I, arrowhead). Using a neoepitope antibody that recognizes the ADAMTS cleavage site in versican (Glu 441-Ala442, V1 sequence enumeration) but not intact versican (Sandy et al., 2001), versican fragments were detected in developing valve cusps, the arterial wall and subjacent to the ventricular endocardium at E15.5 (Fig. 1J), all regions where ADAMTS5 was also expressed at this stage. At E17.5, Adamts5 was detected using LacZ expression in the valvular endocardium (Fig. 1K–M) and most readily detected on the ventricular aspect of the OFT valve cusps (Fig. 1K–M). Mesenchymal cells of the semilunar valves also expressed Adamts5 in the hinge region of the maturing cusps at E14.5–17.5 (Fig. 1G, L asterisks). Cleaved versican (anti-DPEAAE immunoreactivity) colocalized with ADAMTS5 expression in maturing SLV cusps, and was most prominent in the subendocardial regions at E17.5 (Fig. 1O). Expression of Adamts5 was maintained in the adult murine cardiac valves detected by β-gal staining (Fig. 1P, R, T) and immunolocalization (Fig. 1Q, S). The detection of ADAMTS5 in valvular mesenchyme and differentiated VIC gradually increased from E15.5 to adult in valve cusps. In the adult ADAMTS5 is localized within VIC and ECM of the distal cusp and the annulus (Fig. S, ‘A’). These expression patterns suggested that ADAMTS5 and/or proteolytically cleaved versican had potential roles in valve maturation and homeostasis. It is important to note that ADAMTS5 is a secreted protein and is active extracellularly. Although it is initially expressed by the endocardium, it is associated with the ECM of mesenchyme subjacent to the endocardium. Therefore we do not expect protein expression and beta-gal as well as the DPEAAE cleaved versican fragments to always overlap. In addition other ECM proteases including ADAMTS1 {Kern, 2006 #2489} and ADAMTS9 {Kern, 2010 #6621} are present in the cushions and likely produce DPEAAE versican cleavage fragment in the developing valve cusps. Since the ADAMTS5 protein is very difficult to detect by IHC, (it requires protease digestion of the tissue and post-fixation of the ADAMTS5-antibody complex) the enzymatic beta gal reaction facilitated detection of gene expression prior to our ability to detect the protein at E12.5 and was used to readily and clearly detect cells that express ADAMTS5 during valve maturation. We acknowledge that there might be differential stability of transcripts and proteins expressed between the wild type ADAMTS5 allele and the LacZ inserted allele, with LacZ products likely more stable. However, this would only impact interpretation of the shutting down of gene expression and not the initiation of expression at E12.5 which we have demonstrated. The presence of DPEAAE fragments supports this E12.5 expression of ADAMTS5, although there is still potential of other ADAMTS molecules cleaving versican at this early timepoint.
Figure 1. Adamts5 is expressed during cardiac valve maturation in regions where versican cleavage fragments are detected.

The images show β-galactosidase staining indicative of Adamts5 RNA expression, using mice homozygous for the lacZ insertion in Adamts5 (B, F–H, K–M, P, R, T), immunolocalization of ADAMTS5 (A, C–E, I, N, Q, S), and immunodetection of cleaved versican using an anti-DPEAAE441 neo-epitope antibody (J, O). ADAMTS5 was strongly expressed in the myocardium surrounding the developing OFT cushions at E11.5 (A, open arrow). β-galactosidase staining of Adamts5 expression was first detected in developing valves at E12.5 in the endocardium (B, solid arrow) but not detected by immunolocalization until E13.5 (E, solid arrow). ADAMTS5 immunolocalization was most intense surrounding developing PV cushions at E12.5 and E13.5 (C, E open arrows) and less intense but evident in the myocardium surrounding the developing AV (D, open arrow). At E14.5 β-galactosidase staining was found in the endocardium of the PV and AV (F–H, solid arrow) and in mesenchymal cells of the hinge region (G, H; asterisk). β-gal staining was evident in the walls of the great arteries at E14.5 (F, open arrow). Immunolocalization of ADAMTS5 was observed at E15.5 subjacent to the endocardial endothelium (I, solid arrow), in the wall of the aortic artery (I, open arrow) and subjacent to endocardial lining of ventricular trabeculae (I, arrowhead). By E17.5 β-gal staining was present in the endocardium (K–M, arrow) as well as in the hinge region of the maturing cusps (L, asterisk). ADAMTS5 was immunolocalized at E17.5 subjacent to the endocardium (N, arrow). The N-terminal cleavage fragment of versican, DPEAAE, was found subjacent to the endocardium at E15.5 (J, solid arrow) and in the PA wall (J, open arrow). At E17.5 the cleaved versican DPEEAE fragment was localized subjacent to the endocardium (O, solid arrow), and in the hinge region (O, asterisk). In adult murine cardiac valves β-galactosidase staining is evident in the PV (P), the AV (R) and MV (T). Immunolocalization of ADAMTS5 in adult cardiac valves (Q, S) revealed expression throughout the cusps as well as in the annulus (S). PA-pulmonary artery; Ao-aorta; VS-valvulosinus. Green (A, C–E, I, N, Q, S)-ADAMTS5; green (J, O)-anti-DPEAAE cleaved versican; red (A, I, J, N, O)-α smooth muscle actin (αSMA); blue (A, I, J)-Cardiac neural crest (Wnt-1Cre: Rosa-LacZ “marked” cardiac neural crest (CNC). Scale bars: A, B, D, E, H, L, N–T)=50 μm; G, M =100 μm; F, K = 150 μm; C, I, J=200 μm.*-DPEEAE expression is more readily detected in Amsterdam fixative compared to paraformaldehyde (see methods section).
ADAMTS5 deficiency results in myxomatous cardiac valves with the most dramatic anomalies in the pulmonary valve cusps
Previous investigators had reported that adult mice containing an in-frame deletion of the region encoding the catalytic domain of Adamts5−/− were externally normal and that analysis of internal organs did not reveal any abnormalities (Rogerson et al., 2008). In fact our analysis of E17.5 hearts revealed no obvious cardiac abnormalities at E17.5. (Supplemental Fig. 2 A, B). Upon more detailed examination we discovered that the pulmonary valve cusps were grossly enlarged in late fetal development and in the adult. At E14.5 subtle differences in the hinge region and valve outline (Fig. 2A,B) were evident in Adamts5−/− mice. Three-dimensional (3D) reconstructions revealed increased volumes and highlighted these changes in valve shape (Fig. 2C,D; Supplemental Table 1). During fetal development the severity of the abnormalities increased and by E17.5 the PV of Adamts5−/− mice were grossly enlarged and malformed (Fig. 2E,F). 3D reconstructions of E17.5 PV cusps showed an average of 2.4 fold increase in Adamts5−/− cusp volume compared to WT littermates (Fig. 2G,H). Reconstructions highlighted the “balloon shaped” PV at E17.5 in the Adamts5−/− mice, which resembled the endocardial cushions earlier in development (E14.5) and prior to maturation (Fig. 2 A–D). The penetrance of the PV phenotype in Adamts5−/− mice was 100% (n=53; Adamts5−/− and n=56 WT littermates). Although not as dramatic as the PV, the aortic valve (AV) cusps were also larger in the Adamts5−/− with an average of 1.5 fold greater volume at E17.5. (Supplemental Table 1; Supplemental Fig. 2). The mitral leaflets at E17.5 also showed a statistically significant increase in leaflet thickness in the Adamts5−/− mice compared to WT littermates (Supplemental Fig. 2). Adult Adamts5−/− mice are viable with a normal lifespan however examination of the PV, AV and MV from 6 month old Adamts5−/− mice revealed significantly larger, myxomatous PA (*P<0.02), AV (*P<0.04) and MV (*P<0.04) compared to WT (Fig. 3) (n=9; Adamts5−/−; n=6; WT). Therefore the Adamts5−/− mice did not recover from the developmental valve anomalies. ADAMTS5 expression was maintained in the endocardial and VIC in all adult cardiac valves in regions associated with fibrous ECM layers (Fig. 1), notably the hinge region and subjacent to the endocardium.
Figure 2. Adamts5 null mice have enlarged and malformed pulmonary valves.
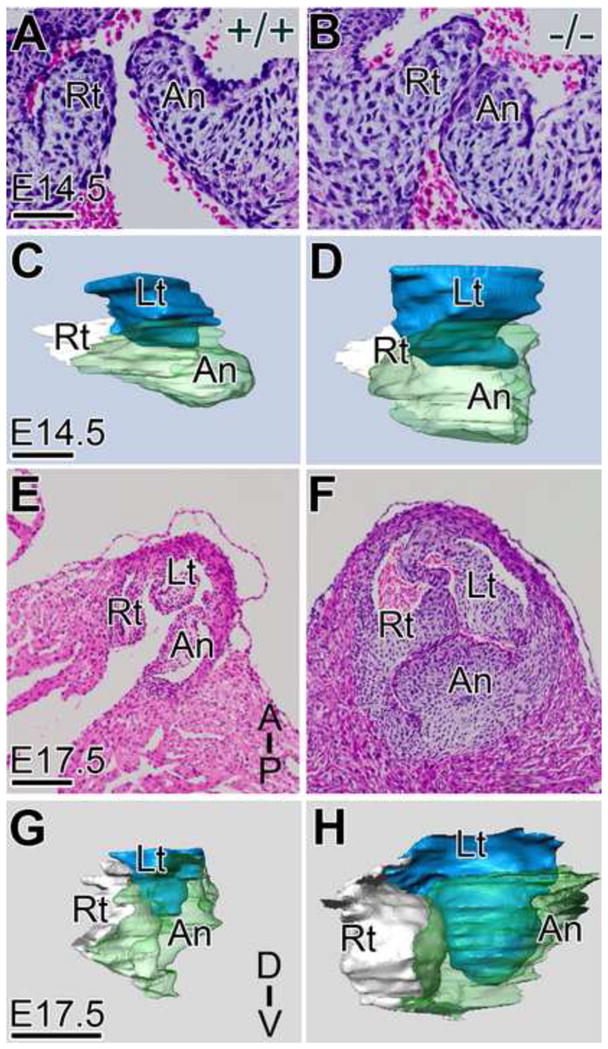
H & E stained frontal sections of the PV from WT (A, E) and Adamts5−/− (B, F) hearts at E14.5 (A, B) and E17.5 (E, F) same magnification are shown. Three-dimensional reconstructions of WT (C, G) and Adamts5−/− mice (D, H) were generated from histological sections of E14.5 and E17.5 PV. Lt- left cusp of the PV (blue); Rt- right cusp of the PV (white); An-anterior cusp of the PV (green); D-dorsal; V-ventral; Scale bars: A = 50 μm; C=75 μm; E =150 μm; G = 200 μm. Corresponding quantitative data from reconstructions are listed in Supplemental Table I.
Figure 3. Adult Adamts5−/− mice have myxomatous pulmonary, aortic and mitral valves.

Movat pentachrome stained sections of adult WT (A, D, G) and Adamts5−/− valves (D, E, H). Increased blue staining in the cusps and leaflets of Adamts5−/− is indicative of proteoglycans (B, E, H), verified as versican by IHC (not shown). Quantification of the thickness of the Adamts5−/− versus WT adult PV (A, B), AV (D, E), and MV (G, H) valves is shown in C (*P< 0.02), F (*P< 0.04), and I (*P<0.04) respectively. PV- n=6 Adamts5−/− ; n= 6, WT; AV- n=6 Adamts5−/− ; n=3 WT; MV- n=5 Adamts5−/− ; n=4 WT. PA-pulmonary artery; Ao-aorta; MV-mitral valve; Lt-left cusp; An-anterior cusp. Black/green bars in (A, B, D, E, G, H) indicate regions of proteoglycan staining in the free edge where measurements were taken. Scale bars: A, B, D, E, G, H = 200 μm.
Increased intercellular space and increased mesenchymal cell number contributed to the enlarged cardiac valves in ADAMTS5 deficient mice
In our previous work, we determined that the subendocardial mesenchyme subjacent to the endocardium is condensed concomitant with detection of the versican cleaved fragments (Kern et al., 2006). High magnification of histological sections from developing PV from Adamts5−/− mice beginning at E12.5, when ADAMTS5 was initially detected (Fig. 1B) revealed a decrease in compaction of subendocardial mesenchyme Fig. 4A, B (blue/red bars). At E14.5 there is an increase in subendocardial mesenchyme accumulation and compaction in the WT (Fig. 4C, blue bar) not evident in the cusp from Adamts5−/− mice (Fig. 4D, red bar). By E17.5 there was a dramatic increase in intercellular mesenchymal space throughout the Adamts5−/− cusps compared to WT (*P<0.0001). These data were quantified in Fig. 4G, and reported as cell density. There were also significantly more mesenchymal cells within the cusps of Adamts5−/− deficient hearts (Fig. 4H, *P<0.03). We further investigated whether an increase in cell proliferation or reduced apoptosis contributed to the increased mesenchymal cell number in PV cusps in Adamts5−/− mice. Using a TUNEL assay at E14.5 and E17.5 there was minimal apoptosis in valvular mesenchyme and no difference between Adamts5−/− and WT littermates (data not shown). Using phospho-Histone H3 (pHH3) as a marker of cell proliferation, we identified a statistically significant increase in proliferating mesenchymal cells in the maturing PV cusps of Adamts5−/− mice at both E14.5 and E17.5 (Fig. 5, *P<0.01, *P<0.03, respectively); however at postnatal day 8 endocardial and mesenchymal PV cell proliferation did not differ significantly from WT littermates (Fig. 5F,G). These findings implicated that both increased extracellular matrix accumulation resulting in increased mesenchymal space as well as increased mesenchymal cell proliferation contributed to the grossly enlarged PV in Adamts5−/− mice by E17.5.
Figure 4. An increase in mesenchymal cell number with increased intercellular space contributes to the enlarged valve cusps in Adamts5−/− mice.

H & E stained sections of maturing PV cusps of E12.5, E14.5 and E17.5 WT (A, C, E) and Adamts5−/− (B, D, F) embryos are shown. Blue bars in A, C, and E represent the normal progression of subendocardial mesenchyme compaction evident in WT embryo cusps but absent in the Adamts5−/− cusps (highlighted by the red bars shown in panels B, D, and F). In panel G the number of total cells (E17.5) per relative cusp area in μm2 is represented by the mean ± SD; * denotes P<0.0001. Total nuclei were counted in the semi-lunar cusps from WT and Adamts5 null embryos at E17.5 spanning a 40μm depth. The mean of the total nuclei/cusp is graphed in H; error bars represent +/− SD of the mean; * denotes P<0.03. Scale bar: A= 30 μm.
Figure 5. Mesenchymal cell proliferation is increased in pulmonary valve cusps from Adamts5−/− mice at E14.5 and E17.5.
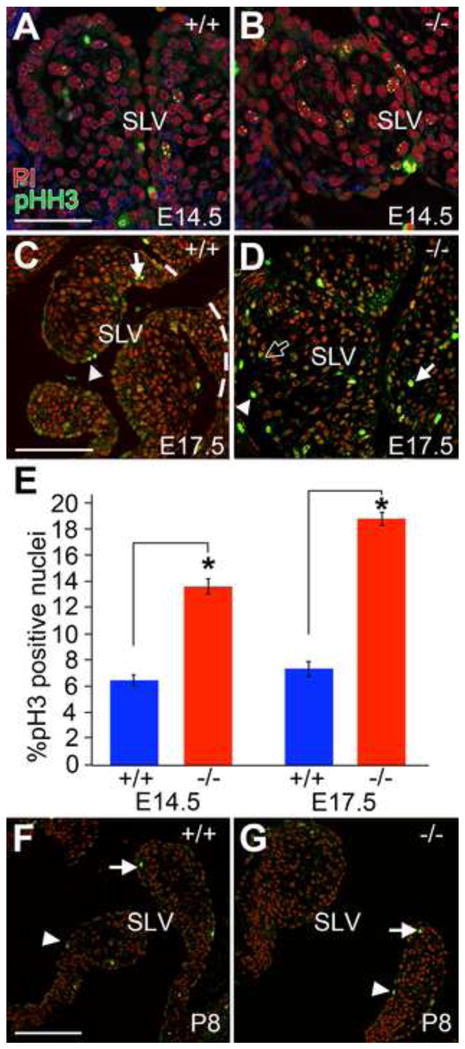
Proliferation of SLV leaflets using anti-phosphoHistoneH3 (pHH3-green) in WT (A, C, F) and ADAMTS5 null (B, D, G) embryos was evaluated in frontal sections. Mesenchymal cells positive for pHH3 (arrows) and endothelial cells (arrowheads) are shown in developing SLV leaflets (demarcated in C by dashed lines) (A, B, E14.5; C, D, E17.5; F, G, P8). Solid arrows and arrowheads denote examples of strong staining of nuclei in mesenchymal and endocardial cells respectively. The open arrow shows an example of punctate staining of mesenchymal nuclei. Red - propidium iodide (PI) staining of nuclei. Panel E shows mean ± SD of the percent positive mesenchymal pHH3 nuclei from Adamts5−/− and WT from E14.5 (* denotes P<0.01) and E17.5 (* denotes P<0.03); n=6 Adamts5−/− and n=6 WT. pHH3 at P8 was not significantly different between Adamts5−/− and WT. SLV-semilunar valve; P8-postnatal day 8. Scale bars: A=25 μm, C,F=50 μm.
The ADAMTS5 substrate versican accumulates in Adamts5−/− cardiac valves concomitant with a reduction of versican cleavage fragments
We hypothesized that the accumulation of ADAMTS5 substrates would be the primary and initial cause underlying the myxomatous valve phenotype in the Adamts5−/− valves. In addition this hypothesis is consistent with the observation of increased intercellular space in Adamts5−/− valve cusps. Therefore we analyzed whether processing of the ADAMTS5 substrate versican (Longpre et al., 2009; McCulloch et al., 2009b) was altered in the enlarged valves of Adamts5−/− mice beginning at E12.5 at the onset of detection of ADAMTS5 expression. Intact versican was detected (using anti-GAGβ, versican antibody (Kern et al., 2007)) prominently in the endocardial cushions of WT PV cusps at E12.5. However, compacted subendocardial mesenchymal cells at E12.5 show a loss of versican staining (Fig. 6A, bar). In Adamts5−/− valves there is a loss of compaction at E12.5 (Fig. 6B, bar) and increased versican. We initially detected Adamts5−/− expression in the endocardium of developing cushions at E12.5. At this time point in WT valve cusps detection of the versican cleaved fragment DPEAAE was evident (Fig. 6C) but not in the Adamts5−/− cusps (Fig. 6D). By E14.5 the increase in intact versican and reduction in subendocardial compacted mesenchyme was more prominent. A statistically significant increase in intact versican (using anti-GAGβ, versican antibody (Kern et al., 2007)) (Fig. 6E, F, I, J; *P<0.003, O) concomitant with marked decrease in anti–DPEAAE immunostaining (versican N-terminal ADAMTS cleavage fragment (Sandy et al., 2001)) (Fig. 6G, H, K, L *P<0.03, P) was observed in Adamts5−/− mice compared to WT littermates in E14.5, E17.5. At postnatal day 1 (P1), increased Alcian blue staining was evident in the Movat pentachrome stained sections of PV from Adamts5−/− mice (Fig. 6M, N blue) and is associated with versican accumulation (Fig. 6Q, R). In adult valves an increase in versican was also observed in the myxomatous valves of ADAMTS5 deficient mice (Fig. 6S, T) and a reduction of cleaved DPEAAE immunodetection (Fig. 6U, V). These data suggested that loss of ADAMTS5 resulted in decreased proteolytic processing of versican during semilunar valve maturation, accumulation of uncleaved versican, and resulted in the myxomatous valve phenotype in Adamts5 deficient mice. We conclude that the dramatic increase in versican accumulation also contributes to the increase in the spongiosa or proteoglycan-containing layer in the valves of Adamts5−/− mice compared to WT.
Figure 6. Versican processing is decreased in semilunar valves from ADAMTS5 deficient mice and contributes to their myxomatous phenotype.

Histological sections of PV cusps from WT (A, C, E, G, I, K, M, Q, S, U) and Adamts5−/− (B, D, F, H, J, L, N, R, T, V) are shown. PV cusps were immunostained for intact versican (GAGβ, green) in WT (A, E, I, Q, S) and Adamts5−/− littermates (B, F, J, R, T). Immunolocalization of cleaved versican (anti-DPEAAE, green) from WT (C, G, K, U) and an Adamts5−/− littermates (D, H, L, V). Quantification (mean ± SD) of intact versican (GAGβ, O; * denotes P<0.003) immunolocalization and cleaved versican (anti-DPEAAE, P; * denotes P<0.03) was performed using Amira™ software to calculate positive pixels from digital confocal images obtained at identical gain settings. Data was obtained from 6 pairs of Adamts5−/− and WT littermate embryos at E14.5 and E17.5. Movat pentachrome stained sections from the PV of a P1 WT (M) and Adamts5−/− (N) highlight ECM, i.e. proteoglycans, blue; fibrous layer red. Sister sections from M, N were stained for versican (GAGβ, green and red-propidium iodide) in Q, R respectively. PA-pulmonary artey; RV right ventricle; An-Anterior cusp; Rt-right cusp; S-spongiosa. Scale bars: A, C, E, K, U = 50μm; M, Q = 100μm; G, I, S = 50 μm.
The other aggregating proteoglycan substrate of ADAMTS5, aggrecan, has not been detected in mammalian cardiac valves (Angel et al., 2011) and data not shown). However, biglycan, a small leucine rich proteoglycan (SLRP) has been reported as an in vitro substrate of ADAMTS5 (Melching et al., 2006), and its accumulation has been associated with human valve disease (Derbali et al.; Gupta et al., 2009b). Therefore we tested if accumulation of biglycan contributed to the myxomatous valve phenotype in Adamts5−/− mice in late fetal development and/or in the adult (Supplemental Fig. 1). Biglycan was detected in the PV of both Adamts5−/− and WT at E17.5 notably in the attachment point of the PV cusps (Supplemental Fig. 1A, B, arrow). However there was not an accumulation of biglycan at E17.5 (Supplemental Fig. 1A–D) in the Adamts5−/− PV. In the adult Adamts5−/− mice biglycan was misexpressed in the PV, although the extent of expression was not increased compared to WT littermates (Supplemental Fig. 1G–J) when normalized to valve area. This is in contrast to the ADAMTS5 substrate versican that showed dramatic accumulation by E17.5 and in the adult (Fig. 6).
BMP2 localization and Sox9 expression are not down-regulated in Adamts5−/− valve cusps compared to WT littermates
It is well established that the TGFβ superfamily is involved in valve formation, maturation and disease by mediating changes in the ECM (Carta et al., 2006; Carta et al., 2009; Combs and Yutzey, 2009; Galvin et al., 2000; Jian et al., 2003; Kim et al., 2008; Ng et al., 2004; Snarr et al., 2008). We examined expression and localization of BMP2 during fetal valve maturation. Using digital images from confocal microscopy, the percent positive pixel area of BMP2 immunolocalization in the PV cusps from Adamts5 deficient mice was compared to WT littermates at E15.5, E17.5 and P1. At E15.5 BMP2 is localized throughout the developing cusps in the WT and Adamts5−/− PV (Fig. 7A, B). We determined that both expression domain and intensity of BMP2 was not down-regulated and restricted in the ECM of valve cusps of Adamts5 deficient mice compared to WT littermates during valve maturation (Fig. 7). At E17.5 in WT mice the BMP2 was restricted to the spongiosa layer and the expression intensity was reduced (Fig. 7C, C′) while in the Adamts5−/− PV there was increased intensity as well as a lack of restriction of BMP2 (Fig. 7D, D′, *P<0.001). The BMP2 expression differences in the Adamts5−/− PV continued until after birth in postnatal day 1 (P1) (Fig. 7E, F). Therefore mesenchyme in the valve cusps from ADAMTS5 deficient mice has both an increased amount of BMP2 as well as an increased domain of expression compared to WT littermates.
Figure 7. Pulmonary valve cusps from Adamts5−/− mice do not exhibit BMP2 down-regulation and restricted expression domain compared to WT littermates.

Immunohistochemistry of BMP2 (green) is shown in the PV of WT (A, C, C′, E) and Adamts5−/− (B, D, D′, F) at E15.5 (A, B), E17.5 (C, C′, D, D′) and P1 (E, F). Colormaps depict positive pixels for BMP2 staining at E17.5 (C′, D′). The graph in G depicts mean fold increase above WT of positive BMP2 pixel area/valve cusp area at E17.5 (* denotes P<0.001); n=6 WT; n=6 Adamts5−/−; bars = +/− SD of the mean. Green (A, B, C, D) -BMP2; red (A, B, C, D) - propidium iodide; An- anterior cusp of the PV; Rt- right cusp; Lt-left cusp; PA-pulmonary artery; S-spongiosa, (versican containing) layer; P1-postnatal day 1. Scale bar A=50μm.
Since BMP2 has been shown to increase expression of the transcription factor Sox9, a SRY transcription factor that is highly expressed in undifferentiated valve mesenchyme, we also examined the expression pattern and levels of Sox9 in the ADAMTS5 null mice (Fig. 8). Sox9 is highly expressed in endocardial mesenchyme throughout the cushion at E12.5 in both Adamts5−/− and WT littermates (Fig. 8A, B). At E14.5 prior to significant stratification of ECM, there were slight differences in the Sox9 staining in the Adamts5 deficient mice evident at the ventricular aspect of the cusps and subjacent to the endocardium (Fig. 8C, D). Colormaps were utilized to identify the pixel intensities of Sox9 staining in individual nuclei to highlight the differences between WT and Adamts5−/− PV (Fig. 8C′, D′, E and F). Quantification of Sox9 staining at E17.5 when the WT has decreased expression (Fig. 8E) compared to earlier in development, showed significant differences compared to Adamts5−/− (Fig. 8F, G, *P<0.003). These studies revealed that both the expression intensity and domain of Sox9 was not developmentally down-regulated in the cusps of ADAMTS5 null mice at E17.5. Sox9 expression in WT mice at E17.5 was restricted to mesenchymal cells (Fig. 8E) in the arterial region of the cusps while ADAMTS5 deficient mice showed little or no mesenchymal restriction of Sox9 (Fig. 8F) and were reminiscent of valve mesenchyme earlier in development (E14.5 (Fig. 8C, C′) and E12, A,B?). Collectively these data show that loss of versican processing by ADAMTS5 in the endocardium affects BMP2 and Sox9 expression in underlying mesenchyme during late fetal stages of PV maturation.
Figure 8. In late fetal stage valves from Adamts5−/− mice Sox9 expression intensity and domain is not decreased like WT littermates.

Sox9 expression obtained by immunolocalization is shown in the PV of WT (A, C, C′, E) and Adamts5−/− (B, D, D′, F) at E12.5 (A, B), E14.5 (C, C′, D, D′) outlined in white and E17.5 (E, F) outlined in white. Colormaps depict pixel intensity of Sox9 staining at E14.5 (C′, D′) and E17.5 (E, F). The graph in G shows mean positive Sox9 pixels per nuclei in PV cusps at E17.5 (* denotes P<0.003); n=6 WT; n=6; bars = +/− SD of the mean. Green (A, B, C, D)-SOX9; red (A, B, C, D)- propidium iodide; An-anterior cusp of the PA valve; Rt-right cusp; S-spongiosa. Scale bar in A=150μm, C=50μm.
Versican heterozygousity rescues the Adamts5−/− cardiac valve morphology and BMP2/Sox9 signaling, implicating failure to cleave versican is a primary mechanism that contributes to myxomatous valve formation
To test the hypothesis that the clearance of versican was a significant role of ADAMTS5 in developing valves we reduced versican (Vcan) in the Adamts5−/− mice using the Vcanhdf mutant. The Vcanhdf allele mouse results in inactivation of the Vcan gene (Mjaatvedt et al., 1998). Although Vcanhdf/hdf mice die during mid-gestation, the Vcan+/hdf heterozygous mice are viable, albeit with reduced amount of versican in the (atrioventricular) AVC (Wirrig et al., 2007), and as we have determined, in the PV (Fig. 9A, D). When the Vcan+/hdf were interbred with the Adamts5−/− there was significant rescue of the PV phenotype at E17.5 in the Adamts5−/−;Vcan+/hdf (rescue; n=11) mice (Fig. 9C, G, K, O) when compared quantitatively with the appropriate littermate controls Adamts5−/−; Vcan+/+ (n=11, *P<0.003) (Fig. 9B, F, J, N), Adamts5+/+; Vcan+/+ (n=6, *P<0.008) (Fig. 9A, E, I, M). The AV and MV of the Adamts5−/−;Vcan+/hdf mice also had statistically significant rescue of the enlarged valve phenotype compared to Adamts5−/−;Vcan+/+ mice. While both leaflets of the MV at E17.5 showed phenotypic rescue in the Adamts5−/−;Vcan+/hdf compared to Adamts5−/−;Vcan+/+, only the left cusp of the AV showed statistically significant rescue, the right cusp did not (Supplemental Fig. 2). Taken together, these data showed that versican heterozygousity significantly rescued the valvular anomalies in Adamts5−/− mice, but did not completely restore normal architecture.
Figure 9. Genetic reduction of versican in Adamts5−/− mice significantly rescues the phenotype of developing pulmonary valves.
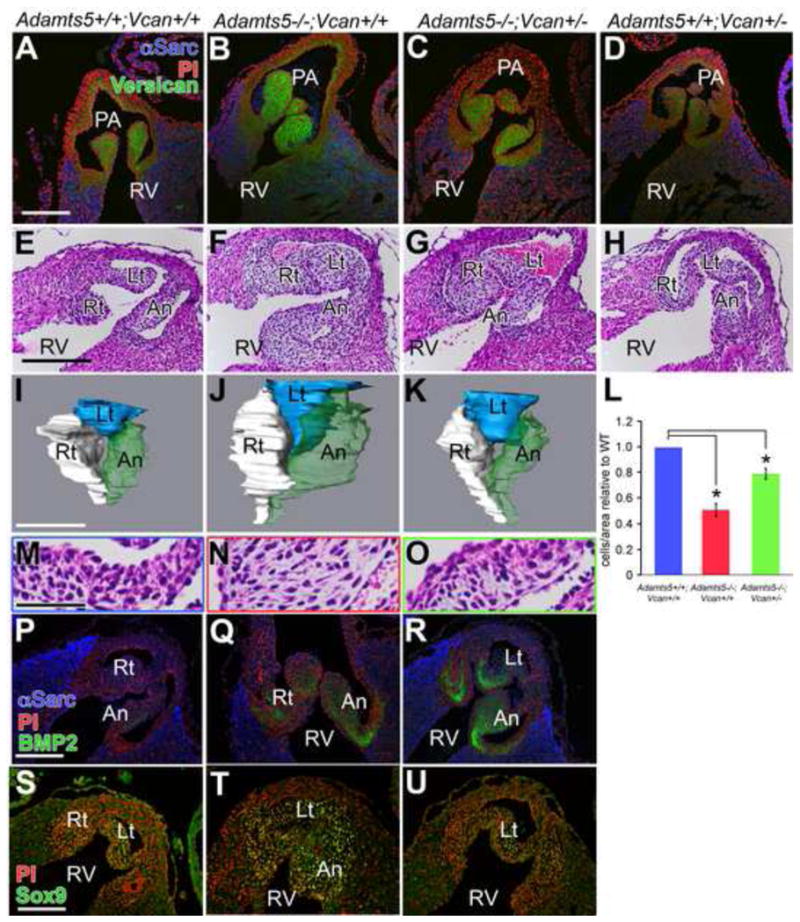
Frontal sections of E17.5 hearts from the indicated genotypes were immunostained for intact versican (GAGβ - green; propidium iodide - red; α-sarcomeric actin - blue) (A, B, C, D) or with H & E to outline their morphology (E, F, G, H). Three-dimensional reconstructions of the PV from Adamts5+/+; Vcan+/+ (I); Adamts5−/−; Vcan+/+ (J) and Adamts5−/−; Vcan+/− mice (K) were generated using Amira™ reconstruction software to outline their shape, and obtain volume measurements of the PV at E17.5. Reconstruction volumes listed (Supplemental Table 1, also includes aortic valve data). Sections of PV cusps of Adamts5+/+,Vcan+/+ (M); Adamts5−/−; Vcan+/+ (N) or Adamts5−/−; Vcan+/− mice (O) were H & E stained. Mesenchymal cell compaction (cells/unit area) was measured using Amira™ software (L) and is expressed as mean ±SD of the mean. * denotes a significant difference in the compaction of the Adamts5−/−; Vcan+/− PV compared to Adamts5−/−; Vcan+/+ mice (P<0.003) and of Adamts5−/−; Vcan+/− PV compared to Adamts5+/+; Vcan+/+ PV (P<0.008). BMP2 immunolocalization (P, Q, R) of the indicated genotypes. Sox9 localization depicted in panels S, T, U. PA- pulmonary artery; RV-right ventricle; Rt-right cusp of the PV; An-anterior cusp of the PV; Lt-left cusp of the PV. Scale bars: A = 150 μm, E, P, S = 100 μm; I = 200 μm; M = 30 μm.
We hypothesized that morphological rescue of the Adamts5−/−;Vcan+/hdf indicated that BMP2 and Sox9 expression would also be down-regulated in the Adamts5−/−;Vcan+/hdf valves compared to Adamts5−/−. Both BMP2 and Sox9 were down-regulated compared to Adamts5−/−. These data demonstrate a direct correlation between the expansion of the spongiosa region and versican accumulation in the Adamts5−/− with BMP2/Sox9 expression in myxomatous valves, suggesting that this signaling pathway contributes to myxomatous valve formation during development.
Discussion
These data show for the first time that loss of an ECM protease results in abnormal valve development that contributes to myxomatous degeneration in the adult. Thus, these data provide a novel example of how valve malformations during development can result in adult valve disease. The expression of ADAMTS5 in the valvular endocardium suggests that underlying mesenchyme are dependent on versican proteolysis to remodel the subendocardial ECM for mesenchymal differentiation during valve maturation. Furthermore loss of ADAMTS5 could not be compensated by ADAMTS1 or ADAMTS9 that we have previously shown to be present during cardiac valve development (Kern et al., 2006; Kern et al., 2010).
Our studies indicate that by late fetal development the ADAMTS5 deficient mice have grossly myxomatous PV. At this time point we hypothesize that multiple intersecting signaling pathways including BMP2/Sox9 as well as factors considered secondary to the direct role of ADAMTS5 contribute to the cardiac valve malformations. To begin to elucidate the initial mechanism or underlying etiology of the myxomatous phenotype in the ADAMTS5 deficient valves our analysis focused on the proteoglycan substrates that are directly cleaved by ADAMTS5. (Longpre et al., 2009; McCulloch et al., 2009b). Our data strongly implicated versican, a critical component of the endocardial ECM (Mjaatvedt et al., 2001; Mjaatvedt et al., 1998) that is cleaved during murine cardiac development (Kern et al., 2006) as a critical substrate of ADAMTS5 in developing cardiac valves. In fact we observed reduced versican cleaved fragments in ADAMTS5 deficient valve cusps, with concomitant accumulation of intact versican, and increased histochemical staining for proteoglycans. Although the ADAMTS5 protein was not detectable at E12.5, the loss of versican cleaved fragments in the subjacent ECM was evident in the ADAMTS5 deficient endocardial cushions. β-gal staining indicated that the Adamts5 gene was expressed by the overlying endocardium prior to this time point. As development progressed, intact versican accumulated in the ADAMTS5 deficient valve cusps, leading us to hypothesize that versican cleavage was needed for its clearance during valve maturation. However, determining a primary defect in the ADAMTS5 deficient valves is complicated by the fact that in addition to a loss of versican cleavage fragments we observed accumulation of the ‘intact’ substrate versican. Although the loss of versican cleavage fragments was observed in close proximity to where the ADAMTS5 proteinase was localized in the ECM, the accumulation of versican was observed throughout the valve cusps by E17.5 greatly augmenting and impacting valve mesenchyme that do not express or normally require ADAMTS5 activity. To further investigate versican as a key ADAMTS5 substrate in this context, in vivo reduction of Vcan in the Adamts5−/− mice showed significant restoration of normal valve architecture and strongly suggests versican was a definitive substrate of ADAMTS5 in the maturing valves. However, the fact that the ADAMTS5 valve phenotype was not completely rescued on Vcan+/hdf background leaves open the possibility that cleaved versican fragments as well as the lack of proteolysis of other ADAMTS5 substrates may also play a role in valve development. Aggrecan, the other aggregating proteoglycan and well-characterized substrate of ADAMTS5 is not present in mammalian valves. Biglycan has been reported as an in vitro substrate of ADAMTS5 (Melching et al., 2006) however it at the time point when the Adamts5−/− myxomatous valve phenotype is dramatic (E17.5) there is not accumulation of biglycan in the Adamts5−/− PV. Although ADAMTS5 is a member of the subfamily of proteases that cleaves proteoglycans (Apte, 2004; Apte, 2009) it is conceivable that additional unknown substrates may also play a role in the valve phenotype observed in the ADAMTS5 deficient heart valves. Collectively, these data indicated that clearance of versican is likely a predominant mechanism underlying the Adamts5−/− myxomatous valve phenotype.
Our morphometric data revealed that the PV cusps in ADAMTS5 deficient mice were more significantly affected than other valves. During development the surrounding tissue of PV cusps and AV cusps are significantly different. The PV cusps mature surrounded by a transient myocardial cuff, a subset of myocardium during fetal stages that we have shown exhibits the strongest expression of ADAMTS5 in the heart. Therefore the morphological differences of the valve cusp phenotype in the ADAMTS5 deficient mice may be due to a dependence of the surrounding myocardial ADAMTS5 expression, a hypothesis that will require further experimentation. In addition, previous reports show differential activation of VICs in the PV compared to the AV (Aikawa et al., 2006; Merryman et al., 2007) suggesting differences in genetic profiles may contribute to the differential phenotype observed in the maturing cusps of ADAMTS5 deficient embryos. Since changes in ECM are often induced by altered mechanical strain, differences in underlying genetic programs among the cardiac valves cannot be viewed in isolation of the differences in biomechanical forces. In fact, transplant studies have shown that lower pressure in the PV compared to the AV produces thicker AV in humans irrespective of the origin of the valve tissue (Rabkin-Aikawa et al., 2004).
The increased mesenchymal cell proliferation in the ADAMTS5 deficient mice is highly relevant to the pathology of myxomatous valve degeneration (Akhtar et al., 1999; Aupperle et al., 2009; Gupta et al., 2009a; Morales et al., 1992) since activation of VIC (Aikawa et al., 2007; Hinton et al., 2008; Rabkin-Aikawa et al.) is prevalent in myxomatous valves of patients and mouse models of disease. Additional reports demonstrate a versican-rich ECM promotes mesenchymal cell proliferation (Shepard et al., 2007; Shibata et al., 2003; Snow et al., 2005). Versican accumulation is also associated with intimal hyperplasia where vascular smooth muscle cells (VSMC) proliferate (Wight, 2008); conversely, as the intimal lesion regresses, versican is cleaved concomitant with quiescence of VSMC (Kenagy et al., 2005; Kenagy et al., 2006). A loss of intact versican due to over-expression of ADAMTS1 in the developing myocardium results in a lack of myocardial growth (Stankunas et al., 2008). However, the increase in ADAMTS1 also results in an over-production of versican fragments (Stankunas et al., 2008) that may have alternative bioactivity and suppress proliferation of cardiomyocytes. The fact that ADAMTS5 is more readily detected on the ventricular face and hinge regions of the SLV cusps may indicate that versican proteolysis is required in the specialized ECM of cardiac valves that is subjected to severe tension or shear stress. The aorta, a tissue that withstands severe biomechanical force, exhibits expression of multiple ADAMTS proteases (Jonsson-Rylander et al., 2005; Kenagy et al., 2005; Sandy, 2001; Sandy et al., 2001) and is where versican proteolysis was initially discovered (Sandy, 2001; Sandy et al., 2001). In fact, differential blood flow increases activity of ADAMTS proteases and versican cleavage in the descending aorta (Kenagy et al., 2005; Kenagy et al., 2009). These data may also indicate a role for versican proteolysis in elastogenesis either directly in elastin fiber assembly or indirectly in distribution of other fibrous matrix components and growth factor sequestration. We hypothesize that ADAMTS5 proteolysis of versican is essential for fibrous ECM formation and elastogenesis in the ventricular aspect of the semi-lunar valves to provide appropriate strength and stretch for mature cardiac valves. Further investigation will be required to determine a role if any, for versican cleaved fragments in the maturation of the cardiovascular ECM.
ADAMTS5 represents one of the few endocardially-expressed genes that influences underlying mesenchyme during valve maturation. Initially the loss of versican cleavage at E12.5 resulted in a decrease in mesenchymal compaction subjacent to the endocardium. How the developing valves sense the increased shear stress of blood flow in the growing fetus and respond in the form of ECM production is a critical and unresolved question, but there is an emerging role for the transcription factor NFATc1 (Combs and Yutzey, 2009; de la Pompa et al., 1998; Jang et al., 2010; Ranger et al., 1998). We hypothesize that subendocardial ECM remodeling by ADAMTS5 may in turn promote HB-EGF signaling and dysregulation results in an increase in mesenchymal cell proliferation and lack of BMP down-regulation (Jackson et al., 2003). Increased BMP2 also correlates with hyperplastic valves in other mouse models (Clark et al., 1999; Delot, 2003; Delot et al., 2003; Kim et al., 2001; Shelton and Yutzey, 2007). BMP2 activates Sox9, a SRY-related transcription factor (Akiyama et al., 2002),(Lincoln et al., 2006) and studies by Lincoln et. al, (Peacock et al., 2010) show that loss of Sox9 in developing cardiac valves correlates with decreased proteoglycan staining which is consistent with findings presented here. These data suggest that Sox9 is critical for maintaining a proteoglycan compartment, i.e. the spongiosa. Here we demonstrated that BMP2 and Sox9 were restricted to the spongiosa in normal valves by late fetal development but in the ADAMTS5 deficient mice BMP2 and Sox9 were expressed throughout the valve mesenchyme. To date membrane receptors that directly bind versican have not been identified although it has been extensively investigated. However, persistence of a proteoglycan-rich ECM has been shown to facilitate growth factor binding, prevent growth factor degradation as well as stabilize growth factor receptor interactions (Kantola et al., 2008; Lee et al., 2006).
In addition, versican binds to other matrix components including hyaluronan (N-terminal domain), fibronectin and fibulin-1 (C-terminus domain) (Ohno-Jinno et al., 2008), therefore we speculate that changes in versican proteolysis may mediate changes in growth factor signaling and binding through its ECM binding partners. Furthermore, it is likely that other substrates of ADAMTS5 have not been elucidated in the context of valve maturation. How additional i.e. yet to be discovered, ADAMTS5 substrates may affect other intersecting signaling pathways and/or augment the changes in BMP2/Sox9 that we have observed in the ADAMTS5 deficient valves remains to be investigated. Our ongoing investigation into the role ECM remodeling in valve development has brought to the forefront challenges in terminology when referring to primary and secondary defects that contribute to the myxomatous valve phenotype of the ADAMTS5 deficient mice. Analysis of the ADAMTS5 deficient valves has revealed changes in versican cleavage fragment levels, accumulation of the ‘intact’ substrate, and strongly suggests that other ECM proteins that directly bind versican may also be altered in the ADAMTS5 valve cusps. In addition versican directly affects growth factor binding sequestration in the ECM. At what point is the accumulation of the ADAMTS5 substrate versican considered a primary defect in the context of ECM remodeling? Data outlined here suggests that further investigation will be required to determine which factors are primary or secondary and to what extent versican binding ECM partners contribute to the phenotype and their temporal role in progression of valve phenotype in the ADAMTS5 deficient mouse model. Our approach here was to define the initial observable defects in the ADAMTS5 deficient mice that were in close proximity to ADAMTS5 expression, and correlated with known ADAMTS5 ECM substrates. Progression of these subtle malformations was followed to late fetal development where there was a dramatic myxomatous valve phenotype. This work also suggests that analysis of the entire ECM proteomic profile during normal development and in disease may be required to understand how ECM remodeling events impact not only ECM organization but also cell behavior, cell signaling and cross-talk of pathway components during valve development and disease states.
The studies and genetic models used here to experimentally alter versican abundance and cleavage during valve maturation identify versican as a novel modulator of endothelial-mesenchymal signaling that is critical for cardiac valve maturation. Finally, while previous studies have demonstrated that increased versican is a hallmark of myxomatous valve degeneration and disease (Akhtar et al., 1999; Aupperle et al., 2009; Gupta et al., 2009a; Morales et al., 1992), the present work as well as published findings on ADAMTS9 (Kern et al., 2010) suggest that the myxomatous phenotype may result from loss of versican-degrading protease activity, including ADAMTS5. These studies suggest mutations affecting the expression or function of ADAMTS5 may have the potential to cause myxomatous valve disease. The ADAMTS5 deficient mice presented here will provide a viable model with full penetrance of myxomatous degeneration that will aid in testing future therapeutic interventions for the correction of developmental abnormalities and adult myxomatous valve disease. Collectively these data demonstrate for the first time the mechanism of versican cleavage and ADAMTS5 as important mediators of ECM stratification required for valve maturation in the four-chambered heart.
Supplementary Material
Highlights.
ADAMTS5 deficient mice develop enlarged cardiac valves by late fetal stages.
In vivo reduction of the ADAMTS5 proteoglycan substrate versican rescues the valve phenotype in Adamts5−/− mice.
Endothelial expression of ADAMTS5 is required for underlying mesenchymal cell differentiation in maturing cardiac valves.
BMP2 and Sox9 expression is not down-regulation and compartmentalized in Adamts5−/− cardiac valve cusps.
Acknowledgments
The authors would like to thank Aimee Phelps for her technical expertise with histology.
Sources of funding: This project was supported by the American Heart Association: Beginning Grant in Aid-076236U (CBK), Scientist Development Grant 10SDG2610168 (CBK), Grant-in-Aid 0655530U (AW), NIH RR016434 (CBK, AW), HL084285 (AW), AR49930 (SSA) and AR53890 (SSA) and The Foundation for Children, 2010-162 (DM).
Non-standard abbreviations and Acronyms
- ECM
extracellular matrix
- ADAMTS
A Disintegrin-like and Metalloprotease domain with ThromboSpondin type motifs
- EMT
epithelial to mesenchymal transformation
- WT
wild type
- OFT
outflow tract
- αSMA
smooth muscle alpha-actin
- VIC
valvular interstitial cells
- AVC
atrioventricular canal
- SLV
semilunar valves
- PA
pulmonary artery
- PV
pulmonary valve
- AV
aortic valve
- MV
mitral valve
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–86. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–52. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- Akhtar S, Meek KM, James V. Ultrastructure abnormalities in proteoglycans, collagen fibrils, and elastic fibers in normal and myxomatous mitral valve chordae tendineae. Cardiovasc Pathol. 1999;8:191–201. doi: 10.1016/s1054-8807(99)00004-6. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–28. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel PM, Nusinow D, Brown CB, Violette K, Barnett JV, Zhang B, Baldwin HS, Caprioli RM. Networked-based characterization of extracellular matrix proteins from adult mouse pulmonary and aortic valves. J Proteome Res. 2011;10:812–23. doi: 10.1021/pr1009806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte SS. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: the ADAMTS family. Int J Biochem Cell Biol. 2004;36:981–5. doi: 10.1016/j.biocel.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily-functions and mechanisms. J Biol Chem. 2009 doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspberg A, Adam S, Kostka G, Timpl R, Heinegard D. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. Journal of Biological Chemistry. 1999;274:20444–9. doi: 10.1074/jbc.274.29.20444. [DOI] [PubMed] [Google Scholar]
- Aspberg A, Miura R, Bourdoulous S, Shimonaka M, Heinegard D, Schachner M, Ruoslahti E, Yamaguchi Y. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10116–21. doi: 10.1073/pnas.94.19.10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aupperle H, Marz I, Thielebein J, Kiefer B, Kappe A, Schoon HA. Immunohistochemical characterization of the extracellular matrix in normal mitral valves and in chronic valve disease (endocardiosis) in dogs. Res Vet Sci. 2009;87:277–83. doi: 10.1016/j.rvsc.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Bondeson J, Wainwright S, Hughes C, Caterson B. The regulation of the ADAMTS4 and ADAMTS5 aggrecanases in osteoarthritis: a review. Clin Exp Rheumatol. 2008;26:139–45. [PubMed] [Google Scholar]
- Carta L, Pereira L, Arteaga-Solis E, Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, Sukoyan M, Kerkis A, Hazeki N, Keene DR, Sakai LY, Ramirez F. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281:8016–23. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, Ramirez F. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 2009;284:5630–6. doi: 10.1074/jbc.M806962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T, Conway S, Scott I, Labosky P, Winnier G, Bundy J, Hogan B, Greenspan D. The mammalian tolloid-like 1 gene, Tll1, is necessary for normal septation and positioning of the heart. Deveopment. 1999;126:2631–2642. doi: 10.1242/dev.126.12.2631. [DOI] [PubMed] [Google Scholar]
- Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105:408–21. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley MA, Kern CB, Fresco VM, Wessels A, Thompson RP, McQuinn TC, Twal WO, Mjaatvedt CH, Drake CJ, Argraves WS. Fibulin-1 is required for morphogenesis of neural crest-derived structures. Dev Biol. 2008;319:336–45. doi: 10.1016/j.ydbio.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–6. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum [see comments] Nature. 1998;392:182–6. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- Delot EC. Control of endocardial cushion and cardiac valve maturation by BMP signaling pathways. Mol Genet Metab. 2003;80:27–35. doi: 10.1016/j.ymgme.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Delot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development. 2003;130:209–220. doi: 10.1242/dev.00181. [DOI] [PubMed] [Google Scholar]
- Derbali H, Bosse Y, Cote N, Pibarot P, Audet A, Pepin A, Arsenault B, Couture C, Despres JP, Mathieu P. Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via Toll-like receptor 2. Am J Pathol. 176:2638–45. doi: 10.2353/ajpath.2010.090541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Nelson CM, Somerville RP, Mielke K, Dixon LJ, Powell K, Apte SS. Cooperation of two ADAMTS metalloproteases in closure of the mouse palate identifies a requirement for versican proteolysis in regulating palatal mesenchyme proliferation. Development. 2010;137:4029–38. doi: 10.1242/dev.050591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MA, Jr, Falb D, Huszar D. A role for smad6 in development and homeostasis of the cardiovascular system. Nature Genetics. 2000;24:171–4. doi: 10.1038/72835. [DOI] [PubMed] [Google Scholar]
- Gupta V, Barzilla JE, Mendez JS, Stephens EH, Lee EL, Collard CD, Laucirica R, Weigel PH, Grande-Allen KJ. Abundance and location of proteoglycans and hyaluronan within normal and myxomatous mitral valves. Cardiovasc Pathol. 2009a;18:191–7. doi: 10.1016/j.carpath.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Tseng H, Lawrence BD, Grande-Allen KJ. Effect of cyclic mechanical strain on glycosaminoglycan and proteoglycan synthesis by heart valve cells. Acta Biomater. 2009b;5:531–40. doi: 10.1016/j.actbio.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson DJ, Copp AJ. Versican expression is associated with chamber specification, septation, and valvulogenesis in the developing mouse heart. Circulation Research. 1998;83:523–32. doi: 10.1161/01.res.83.5.523. [DOI] [PubMed] [Google Scholar]
- Hinton RB, Jr, Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, Yutzey KE. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart Circ Physiol. 2008;294:H2480–8. doi: 10.1152/ajpheart.91431.2007. [DOI] [PubMed] [Google Scholar]
- Hinton RB, Jr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–8. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- Hinton RB, Yutzey KE. Heart Valve Structure and Function in Development and Disease. Annu Rev Physiol. 2010 doi: 10.1146/annurev-physiol-012110-142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- Huang K, Wu LD. Suppression of aggrecanase: a novel protective mechanism of dehydroepiandrosterone in osteoarthritis? Mol Biol Rep. 2010;37:1241–5. doi: 10.1007/s11033-009-9495-5. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res C Embryo Today. 2003;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- Ito K, Shinomura T, Zako M, Ujita M, Kimata K. Multiple forms of mouse PG-M, a large chondroitin sulfate proteoglycan generated by alternative splicing. J Biol Chem. 1995;270:958–65. doi: 10.1074/jbc.270.2.958. [DOI] [PubMed] [Google Scholar]
- Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, Lee DC. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. Embo J. 2003;22:2704–16. doi: 10.1093/emboj/cdg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang GH, Park IS, Yang JH, Bischoff J, Lee YM. Differential functions of genes regulated by VEGF-NFATc1 signaling pathway in the migration of pulmonary valve endothelial cells. FEBS Lett. 2010;584:141–6. doi: 10.1016/j.febslet.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian B, Narula N, Li QY, Mohler ER, 3rd, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–65. doi: 10.1016/s0003-4975(02)04312-6. discussion 465–6. [DOI] [PubMed] [Google Scholar]
- Jonsson-Rylander AC, Nilsson T, Fritsche-Danielson R, Hammarstrom A, Behrendt M, Andersson JO, Lindgren K, Andersson AK, Wallbrandt P, Rosengren B, Brodin P, Thelin A, Westin A, Hurt-Camejo E, Lee-Sogaard CH. Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler Thromb Vasc Biol. 2005;25:180–5. doi: 10.1161/01.ATV.0000150045.27127.37. [DOI] [PubMed] [Google Scholar]
- Jungers KA, Le Goff C, Somerville RP, Apte SS. Adamts9 is widely expressed during mouse embryo development. Gene Expr Patterns. 2005;5:609–17. doi: 10.1016/j.modgep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Kantola AK, Keski-Oja J, Koli K. Fibronectin and heparin binding domains of latent TGF-beta binding protein (LTBP)-4 mediate matrix targeting and cell adhesion. Exp Cell Res. 2008;314:2488–500. doi: 10.1016/j.yexcr.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Kenagy RD, Fischer JW, Lara S, Sandy JD, Clowes AW, Wight TN. Accumulation and loss of extracellular matrix during shear stress-mediated intimal growth and regression in baboon vascular grafts. J Histochem Cytochem. 2005;53:131–40. doi: 10.1369/jhc.4A6493.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenagy RD, Min SK, Clowes AW, Sandy JD. Cell death-associated ADAMTS4 and versican degradation in vascular tissue. J Histochem Cytochem. 2009;57:889–97. doi: 10.1369/jhc.2009.953901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenagy RD, Plaas AH, Wight TN. Versican degradation and vascular disease. Trends Cardiovasc Med. 2006;16:209–15. doi: 10.1016/j.tcm.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern CB, Norris RA, Thompson RP, Argraves WS, Fairey SE, Reyes L, Hoffman S, Markwald RR, Mjaatvedt CH. Versican proteolysis mediates myocardial regression during outflow tract development. Dev Dyn. 2007;236:671–83. doi: 10.1002/dvdy.21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern CB, Twal WO, Mjaatvedt CH, Fairey SE, Toole BP, Iruela-Arispe ML, Argraves WS. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn. 2006;235:2238–47. doi: 10.1002/dvdy.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern CB, Wessels A, McGarity J, Dixon LJ, Alston E, Argraves WS, Geeting D, Nelson CM, Menick DR, Apte SS. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010;29:304–16. doi: 10.1016/j.matbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L, Kim do K, Yang WI, Shin DH, Jung IM, Park HK, Chang BC. Overexpression of transforming growth factor-beta 1 in the valvular fibrosis of chronic rheumatic heart disease. J Korean Med Sci. 2008;23:41–8. doi: 10.3346/jkms.2008.23.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RY, Robertson EJ, Solloway MJ. Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart. Dev Biol. 2001;235:449–66. doi: 10.1006/dbio.2001.0284. [DOI] [PubMed] [Google Scholar]
- Lee NV, Sato M, Annis DS, Loo JA, Wu L, Mosher DF, Iruela-Arispe ML. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006;25:5270–83. doi: 10.1038/sj.emboj.7601400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004;230:239–50. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev Biol. 2006;292:292–302. doi: 10.1016/j.ydbio.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Longpre JM, McCulloch DR, Koo BH, Alexander JP, Apte SS, Leduc R. Characterization of proADAMTS5 processing by proprotein convertases. Int J Biochem Cell Biol. 2009;41:1116–26. doi: 10.1016/j.biocel.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Margolis R, Margolis R. Aggrecan-versican-neurocan family proteoglycans. Methods Enzymol. 1994;245:105–26. doi: 10.1016/0076-6879(94)45008-0. [DOI] [PubMed] [Google Scholar]
- McCulloch DR, Le Goff C, Bhatt S, Dixon LJ, Sandy JD, Apte SS. Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues. Gene Expr Patterns. 2009a;9:314–23. doi: 10.1016/j.gep.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch DR, Nelson CM, Dixon LJ, Silver DL, Wylie JD, Lindner V, Sasaki T, Cooley MA, Argraves WS, Apte SS. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev Cell. 2009b;17:687–98. doi: 10.1016/j.devcel.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melching LI, Fisher WD, Lee ER, Mort JS, Roughley PJ. The cleavage of biglycan by aggrecanases. Osteoarthritis Cartilage. 2006;14:1147–54. doi: 10.1016/j.joca.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Merryman WD, Liao J, Parekh A, Candiello JE, Lin H, Sacks MS. Differences in tissue-remodeling potential of aortic and pulmonary heart valve interstitial cells. Tissue Eng. 2007;13:2281–9. doi: 10.1089/ten.2006.0324. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, Turner D, Markwald RR. The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol. 2001;238:97–109. doi: 10.1006/dbio.2001.0409. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Yamamura H, Capehart AA, Turner D, Markwald RR. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev Biol. 1998;202:56–66. doi: 10.1006/dbio.1998.9001. [DOI] [PubMed] [Google Scholar]
- Morales AR, Romanelli R, Boucek RJ, Tate LG, Alvarez RT, Davis JT. Myxoid heart disease: an assessment of extravalvular cardiac pathology in severe mitral valve prolapse. Hum Pathol. 1992;23:129–37. doi: 10.1016/0046-8177(92)90233-s. [DOI] [PubMed] [Google Scholar]
- Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, Dietz HC. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–92. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Jinno A, Isogai Z, Yoneda M, Kasai K, Miyaishi O, Inoue Y, Kataoka T, Zhao JS, Li H, Takeyama M, Keene DR, Sakai LY, Kimata K, Iwaki M, Zako M. Versican and fibrillin-1 form a major hyaluronan-binding complex in the ciliary body. Invest Ophthalmol Vis Sci. 2008;49:2870–7. doi: 10.1167/iovs.07-1488. [DOI] [PubMed] [Google Scholar]
- Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106:712–9. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabkin-Aikawa E, Aikawa M, Farber M, Kratz JR, Garcia-Cardena G, Kouchoukos NT, Mitchell MB, Jonas RA, Schoen FJ. Clinical pulmonary autograft valves: pathologic evidence of adaptive remodeling in the aortic site. J Thorac Cardiovasc Surg. 2004;128:552–61. doi: 10.1016/j.jtcvs.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Rabkin-Aikawa E, Mayer JE, Jr, Schoen FJ. Heart valve regeneration. Adv Biochem Eng Biotechnol. 2005;94:141–79. doi: 10.1007/b100003. [DOI] [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–90. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- Roberts WC, Ko JM, Hamilton C. Comparison of valve structure, valve weight, and severity of the valve obstruction in 1849 patients having isolated aortic valve replacement for aortic valve stenosis (with or without associated aortic regurgitation) studied at 3 different medical centers in 2 different time periods. Circulation. 2005;112:3919–29. doi: 10.1161/CIRCULATIONAHA.105.543280. [DOI] [PubMed] [Google Scholar]
- Rogerson FM, Stanton H, East CJ, Golub SB, Tutolo L, Farmer PJ, Fosang AJ. Evidence of a novel aggrecan-degrading activity in cartilage: Studies of mice deficient in both ADAMTS-4 and ADAMTS-5. Arthritis Rheum. 2008;58:1664–73. doi: 10.1002/art.23458. [DOI] [PubMed] [Google Scholar]
- Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. 1983;95:108–14. doi: 10.1016/0012-1606(83)90010-6. [DOI] [PubMed] [Google Scholar]
- Sandy JD. Proteoglycan core proteins and catabolic fragments present in tissues and fluids. Methods Mol Biol. 2001;171:335–45. doi: 10.1385/1-59259-209-0:335. [DOI] [PubMed] [Google Scholar]
- Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, Zimmermann DR, Lemire JM, Fischer JW, Wight TN, Clowes AW. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem. 2001;276:13372–8. doi: 10.1074/jbc.M009737200. [DOI] [PubMed] [Google Scholar]
- Seidelmann SB, Kuo C, Pleskac N, Molina J, Sayers S, Li R, Zhou J, Johnson P, Braun K, Chan C, Teupser D, Breslow JL, Wight TN, Tall AR, Welch CL. Athsq1 is an atherosclerosis modifier locus with dramatic effects on lesion area and prominent accumulation of versican. Arterioscler Thromb Vasc Biol. 2008;28:2180–6. doi: 10.1161/ATVBAHA.108.176800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton EL, Yutzey KE. Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev Biol. 2007;302:376–88. doi: 10.1016/j.ydbio.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard JB, Krug HA, LaFoon BA, Hoffman S, Capehart AA. Versican expression during synovial joint morphogenesis. Int J Biol Sci. 2007;3:380–4. doi: 10.7150/ijbs.3.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Fukada K, Imai H, Abe T, Yamashita Y. In situ hybridization and immunohistochemistry of versican, aggrecan and link protein, and histochemistry of hyaluronan in the developing mouse limb bud cartilage. J Anat. 2003;203:425–32. doi: 10.1046/j.1469-7580.2003.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snarr BS, Kern CB, Wessels A. Origin and fate of cardiac mesenchyme. Dev Dyn. 2008;237:2804–19. doi: 10.1002/dvdy.21725. [DOI] [PubMed] [Google Scholar]
- Snow HE, Riccio LM, Mjaatvedt CH, Hoffman S, Capehart AA. Versican expression during skeletal/joint morphogenesis and patterning of muscle and nerve in the embryonic mouse limb. Anat Rec A Discov Mol Cell Evol Biol. 2005;282A:95–105. doi: 10.1002/ar.a.20151. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, Chang CP. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharis AD. Versican in health and disease. Connect Tissue Res. 2008;49:230–4. doi: 10.1080/03008200802147571. [DOI] [PubMed] [Google Scholar]
- Weismann CG, Gelb BD. The genetics of congenital heart disease: a review of recent developments. Curr Opin Cardiol. 2007;22:200–6. doi: 10.1097/HCO.0b013e3280f629c7. [DOI] [PubMed] [Google Scholar]
- Wight TN. Arterial remodeling in vascular disease: a key role for hyaluronan and versican. Front Biosci. 2008;13:4933–7. doi: 10.2741/3052. [DOI] [PubMed] [Google Scholar]
- Wirrig EE, Snarr BS, Chintalapudi MR, O’Neal JL, Phelps AL, Barth JL, Fresco VM, Kern CB, Mjaatvedt CH, Toole BP, Hoffman S, Trusk TC, Argraves WS, Wessels A. Cartilage link protein 1 (Crtl1), an extracellular matrix component playing an important role in heart development. Dev Biol. 2007;310:291–303. doi: 10.1016/j.ydbio.2007.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YJ, La Pierre DP, Wu J, Yee AJ, Yang BB. The interaction of versican with its binding partners. Cell Res. 2005;15:483–94. doi: 10.1038/sj.cr.7290318. [DOI] [PubMed] [Google Scholar]
- Yamamura H, Zhang M, Markwald R, Mjaatvedt C. A heart segmental defect in the anterior-posterior axis of a transgenic mutant mouse. Dev Biol. 1997;186:58–72. doi: 10.1006/dbio.1997.8559. [DOI] [PubMed] [Google Scholar]
- Yatabe T, Mochizuki S, Takizawa M, Chijiiwa M, Okada A, Kimura T, Fujita Y, Matsumoto H, Toyama Y, Okada Y. Hyaluronan inhibits expression of ADAMTS4 (aggrecanase-1) in human osteoarthritic chondrocytes. Ann Rheum Dis. 2009;68:1051–8. doi: 10.1136/ard.2007.086884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zako M, Shinomura T, Ujita M, Ito K, Kimata K. Expression of PG-M(V3), an alternatively spliced form of PG-M without a chondroitin sulfate attachment in region in mouse and human tissues. J Biol Chem. 1995;270:3914–8. doi: 10.1074/jbc.270.8.3914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.