

Abstract
We are interested in developing oncolytic adenoviruses for the treatment of prostate cancer (PCa) bone metastases. A key limitation of Adenovirus 5 (Ad5) is that upon systemic administration, it produces major liver and systemic toxicities. To address this issue, a chimaeric Ad5/48 adenovirus mHAd.sTβRFc was created. Seven hypervariable regions of Ad5 hexon present in Ad5-based Ad.sTβRFc expressing soluble transforming growth factor beta receptorII-Fc fusion protein (sTGβRIIFc), were replaced by those of Ad48. mHAd.sTβRFc, like Ad.sTβRFc, was replication competent in the human PCa cells, and produced high levels of sTGβRIIFc expression. Compared to Ad.sTβRFc, the systemic delivery of mHAd.sTβRFc in nude mice resulted in much reduced systemic toxicity, and reduced liver sequestration. Ad.sTβRFc produced significant liver necrosis, and increases in alanine transaminase, aspartate transaminase, lactate dehydrogenase, tumor necrosis factor-α, and interleukin-6 levels, while mHAd.sTβRFc produced much reduced responses of these markers. Intravenous delivery of Ad.sTβRFc or mHAd.sTβRFc (5 × 1010 viral particles/mouse) in nude mice bearing PC-3-luc PCa bone metastases produced inhibition of bone metastases. Moreover, a larger dose of the mHAd.sTβRFc (4 × 1011 viral particles /mouse) was also effective in inhibiting bone metastases. Thus, mHAd.sTβRFc could be developed for the treatment of PCa bone metastases.
Introduction
In the United States, prostate cancer (PCa) is the second leading cause of cancer-related deaths among men. During the advanced stages of PCa, a majority of the patients develop bone metastases and suffer from skeletal-related events resulting in morbidity and mortality.1 Androgen-deprivation therapy and chemotherapy are usually insufficient for patients with metastatic castration-resistant PCa.2,3 Bisphosphonates, such as zoledronic acid can bind with bone mineral, and inhibit bone resorption in order to relieve pain and tumor-induced hypercalcemia.4 Denosumab, a human monoclonal antibody against receptor activator of nuclear factor kappa-B ligand (RANKL), can improve bone density and suppress bone turnover by inhibiting osteoclast-mediated bone destruction.5,6 In spite of these new modalities of treatment, skeletal-related events continue to occur, albeit at a reduced rate, and it is not clear if they can help castration-resistant PCa patients live longer. Towards that end, there is an urgent need to develop novel therapies for bone metastases of PCa, with the hope of improving patients' overall survival.7
In recent years adenoviruses have emerged as promising vectors for cancer gene therapy.8,9,10,11,12,13,14,15 However, their clinical application in targeting bone metastasis is not yet described.16 To target PCa bone metastases, we wish to develop oncolytic adenoviruses that will kill PCa cells, and will simultaneously inhibit signaling pathways that promote bone metastasis. We have previously studied Ad.sTβRFc, an Adenovirus 5 (Ad5)-based oncolytic virus expressing soluble transforming growth factor beta receptorII-Fc fusion protein (sTGβRIIFc) that can inhibit TGFβ signaling;17 aberrant TGFβ signaling is known to promote bone metastases in PCa.17,18,19
For targeting bone metastases, the prefered route to deliver adenoviral vectors would be via systemic administration. A key limitation in the use of Ad5-based adenoviruses is that, upon systemic administration, a majority of the virus is taken up by the liver, producing severe hepatic damage, innate immune response, and systemic toxicity.20,21,22,23,24,25,26,27,28 Upon systemic delivery of Ad5 in mice, the viral hexon protein can bind with blood coagulation Factor X (FX), and Ad5-FX complex is taken up by the liver via heparin sulfate proteoglycan present on the hepatocytes.29,30,31,32,33 However, Ad48 hexon has poor binding affinity for FX, and therefore, Ad48 and chimaeric Ad5/48 hexon adenoviruses have reduced hepatic uptake.29,30,31,32,33 With the goal of developing oncolytic adenoviruses which upon systemic delivery will bypass the hepatic uptake, we have now created a chimaeric oncolytic adenovirus, mHAd.sTβRFc, in which seven hypervariable regions of Ad.sTβRFc were substituted with the corresponding sequence of Ad48. The goals of this study were to examine: (i) if the mHAd.sTβRFc is replication competent in PCa cells, and produces sTGFRIIFc protein, (ii) if upon systemic delivery, mHAd.sTβRFc will have reduced hepatic uptake, producing minimum hepatic and systemic toxicity, and (iii) if mHAd.sTβRFc will be effective in inhibiting the skeletal metastases, and the tumor-induced bone destruction in a PCa bone metastasis model in mice. The results indicate that, mHAd.sTβRFc exhibits reduced toxicity in mice, and is effective in inhibiting the bone metastases.
Results
Construction of hexon-chimaeric oncolytic adenovirus mHAd.sTβRFc, and mHAd.sTβRFc replication, virus-induced cytotoxicity and sTGFβRIIFc protein expression in PCa cell lines
A hexon-chimaeric mHAd.sTβRFc, in which the seven hypervariable regions of Ad5 were substituted with the corresponding sequence of Ad48, was constructed using dl01/07 backbone (Figure 1a). dl01/07, an adenoviral mutant, has two deletions in E1A region, one deletion is 4–25 amino acids (dl01), and the second deletion is 111–123 amino acids (dl07).34 The resultant E1A proteins cannot bind with p300/CBP or pRb proteins. Therefore, in primary cells, dl01/07 is not effective for S-phase induction, and the adenovirus can not replicate. However, cancer cells are able to progress to S phase, thus permitting virus replication in these cells.34 The replication potential of mHAd.sTβRFc and the vector-induced sTGFβRIIFc expression was examined in two human PCa cell lines, PC-3 and DU-145, and a mouse PCa cell line TRAMP-C2. In PC-3 and DU-145 cell lines, viral titers of mHAd.sTβRFc and Ad.sTβRFc were about 5,000-times higher than that of a replication-deficient adenovirus Ad(E1-).Null, but in TRAMP-C2, much reduced viral replication was detected (Figure 1b). Both mHAd.sTβRFc and Ad.sTβRFc produced a similar dose-dependent cytotoxicity in PC-3 cells (Figure 1c). On the basis of IC50 values (viral dose required to kill 50% of the cells), mHAd.sTβRFc and Ad.sTβRFc were about 60- and 100-fold, respectively, more toxic than Ad(E1-).Null. Similar cytotoxicity results were also observed in DU-145 cells (data not shown). In TRAMP-C2 cells, minimum cell cytotoxicity was produced by all viruses (data not shown). Infection of the three PCa cell lines by mHAd.sTβRFc and Ad.sTβRFc produced sTGFβRIIFc protein which could be detected in both the media, and the cell lysates (Figure 1d); mHAd.sTβRFc and Ad.sTβRFc produced similar levels of sTGFβRIIFc protein (1–10 µg/ml) in each cell type (Figure 1e). These results suggest that mHAd.sTβRFc and Ad.sTβRFc induce similar viral replication, cytotoxicity and sTGFβRIIFc expression in human prostate tumor cells.
Figure 1.

Construction of hexon-chimaeric adenovirus mHAd.sTβRFc, mHAd.sTβRFc replication, mHAd.sTβRFc-induced cytotoxicity, and expression of sTGFβRIIFc protein in PCa cells. (a) Construction of mHAd.sTβRFc. The Ad48 hexon sequence in pcDNA3-hxn between ApaI and HpaI restriction sites was exchanged for a corresponding Ad5 hexon sequence in PTG01/07 with two deletions in the E1A region.33 The sTGFβRIIFc gene was cloned in the E3 region, but the adenoviral death protein sequence was left intact. (b) mHAd.sTβRFc replication in PCa cells. (c) mHAd.sTβRFc-induced cytotoxicity in PC-3 cells. (d) mHAd.sTβRFc-mediated expression of sTGFβRIIFc protein in PCa cells by western blot. (e) mHAd.sTβRFc-mediated expression of sTGFβRIIFc protein in PCa cells by ELISA.
Systemic administration of mHAd.sTβRFc in nude mice exhibits reduced systemic toxicity, reduced hepatotoxicity, reduced uptake in the liver and spleen, and attenuated innate immune response
To investigate the toxicity of mHAd.sTβRFc and Ad.sTβRFc, viral vectors were injected into nude mice via tail vein with different doses (low dose (LD) = 2.5 × 1010 viral particles (VPs)/mouse, medium dose (MD) = 1.0 × 1011 VPs/mouse, and a high dose (HD) = 2.0 × 1011 VPs/mouse). Mice were monitored for 3 days for their body weight and signs of morbidity. The HD of Ad.sTβRFc resulted in the death of animals within 24 hours, and, hence, could not be further analyzed. By day 3, only the MD of Ad.sTβRFc produced significant body weight loss (P < 0.01) (Figure 2a). On day 3, livers in all the Ad.sTβRFc groups had a pale/yellow appearance, while livers from the mHAd.sTβRFc and the buffer-treated groups had the normal bright red appearance (Figure 2b, top panel). Both the LD and the MD of Ad.sTβRFc produced liver necrosis, while no distinct morphological changes were observed in any of the mHAd.sTβRFc-treated groups (Figure 2b, second panel from top). Serum alanine transaminase and aspartate transaminase levels in each of the Ad.sTβRFc groups were significantly higher than those in the corresponding mHAd.sTβRFc groups (LD: P < 0.05; MD: P < 0.001) (Figure 2c,d). These results indicate that systemic administration of mHAd.sTβRFc could produce much reduced hepatic damage compared to Ad.sTβRFc.
Figure 2.

Systemic administration of mHAd.sTβRFc produces reduced systemic toxicity, reduced hepatic toxicity, and reduced liver and spleen uptake. (a) Analysis of mice body weight 3 days after intravenous injection of viruses. Average body weight per group (n = 4) is plotted as the mean ± SEM. (b) Gross liver morphology (upper panel), H&E staining showing microscopic morphology of liver (second panel from top), and immunohistochemistry staining of sTGFβRIIFc expression (third panel from top) in liver 3 days after intravenous injection of viruses, immunohistochemistry staining of sTGFβRIIFc expression in spleen 3 days after intravenous injection of viruses (lowest panel) (Scale bar = 300 µm). (c) Serum alanine transaminase levels 3 days after intravenous injection of viruses (n = 4). (d) Serum aspartate transaminase levels 3 days after intravenous injection of viruses (n = 4). (e) Viral genomic copies in liver (n = 4) P value comparisons are shown for a, c, d, and e (*P < 0.05, **P < 0.01, ***P < 0.001), ++P < 0.01 for Ad.sTβRFc (MD) versus mHAd.sTβRFc (HD).
To investigate, if the adenovirus-induced hepato-toxicity is associated with the increased viral uptake in the liver, adenoviral uptake in the liver was examined by quantifying viral genomic DNA in the liver, and the adenovirus-mediated sTGFβRIIFc expression. Viral DNA copy number in the mHAd.sTβRFc groups was significantly lower than in the Ad.sTβRFc groups (LD: P < 0.05, MD: P < 0.01; Ad.sTβRFc MD versus mHAd.sTβRFc HD: P < 0.01) (Figure 2e). In mice that received Ad.sTβRFc, a large number of liver cells produced sTGFβRIIFc (brown stained cells); while the comparable viral dose of mHAd.sTβRFc produced sTGFβRIIFc in much fewer liver cells (Figure 2b, third panel from top), suggesting that hexon modification resulted in the reduced hepatic uptake of mHAd.sTβRFc. Furthermore, serum sTGFβRIIFc protein levels in mHAd.sTβRFc groups were also lower than those in Ad.sTβRFc groups, when compared with the similar viral dose (data not shown). Ad.sTβRFc also produced a dose-dependent increase in the sTGFβRIIFc expressing spleen cells, and the corresponding dose of mHAd.sTβRFc produced much fewer sTGFβRIIFc-expressing cells (Figure 2b, lowest panel). These results indicated that unlike Ad.sTβRFc, the mHAd.sTβRFc is not as readily targeted to the spleen, and the findings were consistent with the previous observations that Ad5-FX complexes can also be taken up by the spleen.35
To examine the effects of mHAd.sTβRFc and Ad.sTβRFc on the liver Kupffer cell necrosis and the innate immune responses, the serum lactate dehydrogenase (LDH), tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) levels were measured at 1 hour and at 48 hours after intravenous administration in nude mice. One hour after Ad.sTβRFc injection, both LD and MD produced significant increase in the LDH activity (LD, P < 0.01; MD, P < 0.001) (Figure 3a); however, at 1 hour, only MD and HD of mHAd.sTβRFc produced significant increases in LDH (P < 0.001) (Figure 3a). Since, a rapid rise in serum LDH is a marker of early Kupffer cell necrosis,27 the findings suggested that the LD and the MD of Ad.sTβRFc, and the MD and HD of mHAd.sTβRFc can induce rapid Kupffer cell necrosis. While the serum LDH levels remained much higher in Ad.sTβRFc-treated mice (LD, P < 0.001; MD, P < 0.001) (Figure 3b), at 48 hours the serum LDH in any of the mHAd.sTβRFc-treated groups were not significantly higher (Figure 3b). The high serum LDH levels at 48 hours likely reflect a more significant tissue damage (e.g., liver damage) by Ad.sTβRFc, while it was significantly attenuated in mHAd.sTβRFc-treated mice.
Figure 3.

Effects of systemic administration of mHAd.sTβRFc and Ad.sTβRFc on LDH and cytokine levels. (a) Serum LDH levels 1 hour after intravenous injection of viruses (n = 4). (b) Serum LDH levels 48 hours after intravenous injection of viruses (n = 4). (c) Serum TNF-α levels 1 hour after intravenous injection of viruses (n = 4). (d) Serum TNF-α levels 48 hours after intravenous injection of viruses (n = 4). (e) Serum IL-6 levels 1 hour after intravenous injection of viruses (n = 4). (f) Serum IL-6 levels 48 hours after intravenous injection of viruses (n = 4). The detection limits of TNF-α, and IL-6 were 6.2 and 3.1 pg/ml, respectively. The IL-6 levels shown in each of the groups are above the detectable limits. The TNF- α levels in 1 hour samples had detectable levels of TNF-α, however, in the 48 hours samples, some of the treatment groups (LTD and HTD of mHAd.sTβRFc) had barely detectable levels of TNF-α. P value comparisons with buffer group are shown for all panels (* represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001).
Serum TNF-α (an early alarm response cytokine) levels were significantly increased in both the Ad.sTβRFc-treated groups (LD: P < 0.05; MD: P < 0.01) at 1 hour following intravenous administration, and were sustained till 48 hours (P < 0.001). However, none of mHAd.sTβRFc groups demonstrated any significant changes in TNF-α levels at either 1 hour (a small increase in TNF-α levels was observed at 1 hour that did not reach significance) (Figure 3c), or at 48 hours (Figure 3d). Serum IL-6 (an NF-kB-dependent proinflammatory cytokine) level was significantly increased at 1 hour following the Ad.sTβRFc administration (MD) (P < 0.001) (Figure 3e), which was again sustained till 48 hours (P < 0.05) (Figure 3f). However, in mHAd.sTβRFc-treated mice, a significant increase in IL-6 level was observed only in the HD group at 1 hour (P < 0.01) (Figure 3e), and was reduced to basal levels by 48 hours (Figure 3f). Thus, compared to Ad.sTβRFc, mHAd.sTβRFc elicits a weaker and transient innate immune response, which is resolved by 48 hours postviral injections.
Systemic administration of mHAd.sTβRFc produces viral replication and sTGFβRIIFc protein in the skeletal tumors, and inhibits bone metastases
Next, we investigated if following the systemic delivery of mHAd.sTβRFc, the virus can be taken up by the skeletal tumors, and can inhibit bone metastasis in an animal model of PCa. PC-3-luc cells were inoculated in the left heart ventricle of male nude mice to produce skeletal metastases. Viral vectors were administered intravenously, and 3 days later, hind limbs and liver samples were collected, and subjected to immunohistochemistry of the adenoviral hexon, and sTGFβRIIFc proteins. mHAd.sTβRFc and Ad.sTβRFc produced nearly equal expression of the hexon, and sTGFβRIIFc proteins (as indicated by brown stained cells) in the skeletal tumors (Figure 4a, panels 1 and 3 from the top). However, mHAd.sTβRFc produced much lower hexon and sTGFβRIIFc expression in the liver (Figure 4a, panels 2 and 4 from the top) indicating that the chimaeric hexon modification in mHAd.sTβRFc did not affect the viral uptake and its replication in the skeletal tumors, while its uptake/replication in the liver was reduced.
Figure 4.

mHAd.sTβRFc-mediated expression of sTGFβRIIFc protein in skeletal tumors and effects of adenoviral vectors on the skeletal tumor progression by BLI analyses. (a) Three days after intravenous injection of the viruses, hexon or sTGFβRIIFc were detected in the skeletal tumors and in the liver, by immunohistochemistry staining using antihexon antibody or antihuman IgG Fcγ antibody, respectively. Brown cells indicate hexon or sTGFβRIIFc expression. Scale bar equals 300 µm. (b) Representative whole body dorsal and ventral BLI images on day 9, day 30 and day 44 in buffer and each treatment group are shown. Regions of interest are pointed out with red circles. (c) To measure bone metastases, BLI signals in the hind limbs were quantified in buffer and each treatment group, and are shown. (d) Fold-increases of BLI signal intensity from days 9 to 44 were calculated and are shown. P value comparisons with buffer group are shown for c and d (* represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001).
To determine the effectiveness of mHAd.sTβRFc to inhibit the established PCa bone metastases, PC-3-luc cells were injected into the left heart ventricle of male nude mice; and the bioluminescence imaging (BLI) data observed on day 9 were used to create experimental groups. On day 10, mice were intravenously administered with either buffer or viral vectors; and another injection of either buffer or viral vectors was given on day 13. This dose scheduling has been previously found to be effective in this animal model.17 Two treatment groups received a low therapeutic dose (LTD, two doses, 2.5 × 1010 VPs/mouse each, a total of 5 × 1010 VPs/mouse) of mHAd.sTβRFc or Ad.sTβRFc, and a third treatment group received a high therapeutic dose (HTD, two doses, 2.0 × 1011 VPs/mouse each, a total of 4.0 × 1011 VPs/mouse) of mHAd.sTβRFc. The HD of Ad.sTβRFc resulted in the animal deaths within 24 hours (as described earlier, Figure 2a), and hence the effect of HTD of Ad.sTβRFc could not be examined. Mice were imaged once a week, and signal intensity of combined dorsal and ventral hind limbs was quantified. Figure 4b shows BLI of a representative mouse from each group on days 9, 30, and 44. In the buffer-treated group, there was a time-dependent increase in BLI signal (Figure 4c), and all the three treatment groups, exhibited significant reductions in the BLI signal (P < 0.001). Analysis of the fold-induction of BLI signal from day 9 to day 44, showed that the LTD of mHAd.sTβRFc and Ad.sTβRFc exhibited significant reductions in the BLI fold increases (P < 0.05). HTD of the mHAd.sTβRFc also inhibited the tumor growth (P < 0.01) (Figure 4d). However, there were no significant differences in the BLI signals among the three treatment groups, indicating that the mHAd.sTβRFc and Ad.sTβRFc were equally effective in inhibiting the skeletal tumor growth.
Bone metastases were further examined by radiographic analyses on day 16, and once a week thereafter. Figure 5a shows representative X-ray images on days 16, 38, and 51 from each group. Osteolytic lesions on the skeletal tumors are marked by yellow arrows. To quantify tumor size, X-ray lesions were measured in both hind limbs of each mouse. An increase in tumor area was observed in the buffer group, while there was significant inhibition of the tumor progression in each of the treatment groups (P < 0.001) (Figure 5b). However, suppression of tumor growth by the HTD of mHAd.sTβRFc produced antitumor response significantly better than the LTD of Ad.sTβRFc (P < 0.05) or mHAd.sTβRFc (P < 0.01) (Figure 5b). On day 51, a significant inhibition of tumor size was observed in mice treated with mHAd.sTβRFc (HTD) (P < 0.001), and this inhibitory effect was better than that observed in Ad.sTβRFc (LTD) (P < 0.01) or mHAd.sTβRFc (LTD) (P < 0.01) treatment groups (Figure 5c). More importantly, only the mHAd.sTβRFc (HTD) treatment resulted in significantly higher number of skeletal tumor-free mice (P < 0.01) (Figure 5d). These results suggest that both mHAd.sTβRFc and Ad.sTβRFc are equally effective in inhibiting the skeletal tumor growth, and a higher dose of mHAd.sTβRFc, is more effective in inhibiting bone metastases.
Figure 5.

Effect of adenoviral vectors on the skeletal tumor progression by radiography analyses. (a) Representative radiographs of mouse hind limbs on days 16, 38, and 51 in the buffer and each treatment group are shown. Yellow arrows indicate the sites of osteolytic lesions. (b) Lesion sizes in each of the hind limb bones during the duration of the study were calculated using Image J software. Results shown are the average lesion area in the hind limbs in buffer group and each of the treatment groups. (c) Lesion sizes in the hind limb bones on day 51 were calculated and are shown. (d) Bone metastasis free incidences (mice without X-ray positive lesions) on day 51 are shown. P value comparisons with buffer group are shown for b, c, and d (* represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001).
The effect of vectors' treatment on the tumor burden at the terminal time point (day 53), was examined by histomorphometric analyses of the hind limb bones. Figure 6a shows a representative bone sample from the buffer group and each of the treatment groups. The tumor area in the tibia and femur, outlined with yellow line in Figure 6a, were measured. The LTD of Ad.sTβRFc and the mHAd.sTβRFc caused a significant reduction in the tumor burden (P < 0.05); the HTD of mHAd.sTβRFc was also effective in reducing the tumor burden (P < 0.01) (Figure 6b). Only mHAd.sTβRFc (HTD) treatment resulted in a significant increase in the tumor-free bones by histomorphometric analyses (P < 0.05) (Figure 6c). On day 53, serum sTGFβRIIFc were detected in all the treatment groups (Figure 6d). Serum sTGFβRIIFc protein levels in mHAd.sTβRFc groups were substantially lower than those in the Ad.sTβRFc groups (Ad.sTβRFc (LTD) vs mHAd.sTβRFc (LTD), P < 0.001). However, the HTD of mHAd.sTβRFc produced similar levels of sTGFβRIIFc as the LTD of Ad.sTβRFc. This is not surprising considering that upon systemic delivery, greater amounts of Ad.sTβRFc are taken up by the liver as compared to mHAd.sTβRFc (as shown earlier in Figure 2b). Since, following the intravenous delivery of Ad.sTβRFc or mHAd.sTβRFc, liver is likely the major site for sTGFβRIIFc protein production and its subsequent release in the blood, greater amounts of sTGFβRIIFc are detected in Ad.sTβRFc-treated mice.
Figure 6.

Effect of adenoviral vectors on the skeletal tumor progression by histomorphometric analyses, and microCT analyses. (a) Representative longitudinal, midsagittal H&E-stained sections of tibia and femur from each group on day 53 are shown. Scale bar equals 1 mm. (b) Tumor areas outlined with yellow in panel a were used to measure tumor burden in each treatment group. The average tumor sizes in the hind limbs in each of the treatment groups are shown. Eight bone samples were used in each group. (c) Bone metastasis free incidences (mice without histomorphometric positive lesions) on day 53 are shown (n = 8). (d) Virus-mediated expression of sTGFβRIIFc (measured by ELISA) in mouse serum 53 days after intravenous injection of viruses. (e) Representative microCT slices near the growth plate (top panel), and 1.45 mm distal of the growth plate (middle panel), and 3D reconstructions of the tibia bones form the various treatment groups (lower panel). Arrows indicate the site of bone destruction. Solid line (in the lower panel) indicates the growth plate and dashed line indicates 1.45 mm downstream of the growth plate. P value comparisons with buffer group are shown for b and c (* represents P < 0.05, ** represents P < 0.01); for panel d, student t-tests were performed among the treatment groups shown (*** represents P < 0.001).
Bone samples randomly selected from each group were subjected to synchrotron microcomputed tomography (microCT) analyses. Normal bone showed typical scan images near the growth plate and 1.45 mm distal to the growth plate, and the normal reconstructed 3D image of the scanned region (Figure 6e). However, the buffer-treated group had massive tumor-induced trabecular and cortical bone destructions, and the osteolytic lesions are visible in the 3D reconstructed image (marked as yellow arrows). However, in the treatment groups, normal bone mass and architecture were observed (Figure 6e).
Systemic administration of mHAd.sTβRFc inhibits osteoclast activity and hypercalcemia, and increases animal survival
To examine the effect of mHAd.sTβRFc treatment on the tumor-induced osteolytic bone destruction, we performed the tartrate resistant acid phosphatase (TRAP) staining for osteoclasts on median sagittal sections of the hind limb bones. Figure 7a shows a representative TRAP staining along the bone/tumor interface, and arrows point to the activated mature osteoclasts with multiple nuclei. All the treatment groups had a significant reduction in the osteoclast number compared to the buffer group (Ad.sTβRFc (LTD) (P < 0.01), mHAd.sTβRFc (LTD) (P < 0.01), mHAd.sTβRFc (HTD) (P < 0.001) (Figure 7b). The treatment groups also showed significant inhibition of the serum TRACP 5b, a secreted marker of osteoclast activity compared to the buffer group (Ad.sTβRFc (LTD) (P < 0.05), mHAd.sTβRFc (LTD) (P < 0.01), mHAd.sTβRFc (HTD) (P < 0.01) (Figure 7c). However, there were no statistical differences in the osteoclast numbers or in the TRACP 5b levels among the three treatment groups (Figure 7b,c). Since osteolytic bone destruction can result in hypercalcemia, the serum calcium levels were measured. Ad.sTβRFc (LTD), mHAd.sTβRFc (LTD), and mHAd.sTβRFc (HTD) treatments, all lowered calcium levels (P < 0.01). The calcium levels in the vector treatment groups were statistically similar to the age matched normal mice (Figure 7d). The animal body weights were also monitored once a week, as an indicator of cancer cachexia. In the buffer group, the mice began to lose body weight from day 44 onwards. By day 53, there was a significant reduction in the body weight of mice in the buffer group compared to the normal mice (P < 0.001) (Figure 7e). However, there was no significant difference between the body weights of the Ad.sTβRFc, and the mHAd.sTβRFc treatment groups compared to the normal mice. Using the criteria of more than 10% body weight loss from day 44 to 53 as a predictor of poor survival, all the treatment groups had significant survival advantage over the buffer group (data not shown).
Figure 7.

Effect of adenoviral vectors on the skeletal tumor progression: Osteoclast numbers, mice serum TRACP5b levels, serum calcium, and mice body weight analysis. (a) A representative TRAP staining of bone (arrows indicate multinucleated TRAP positive osteoclasts). Scale bar = 500 µm. (b) Osteoclast (OC) number per mm calculated at the tumor–bone interface in each group. Eight bone samples were used in each group. (c) Serum TRACP 5b concentration on day 53 in Units/liter. (d) Serum samples collected on day 53 were used to measure calcium levels. (e) Mice body weight analysis on day 53. Average body weight per group throughout the experiment is plotted as the mean ± SEM. P value comparisons with buffer group are shown for b, c, and d (* represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001), for e (***P < 0.001, represents buffer versus all treatments groups, and normal mice).
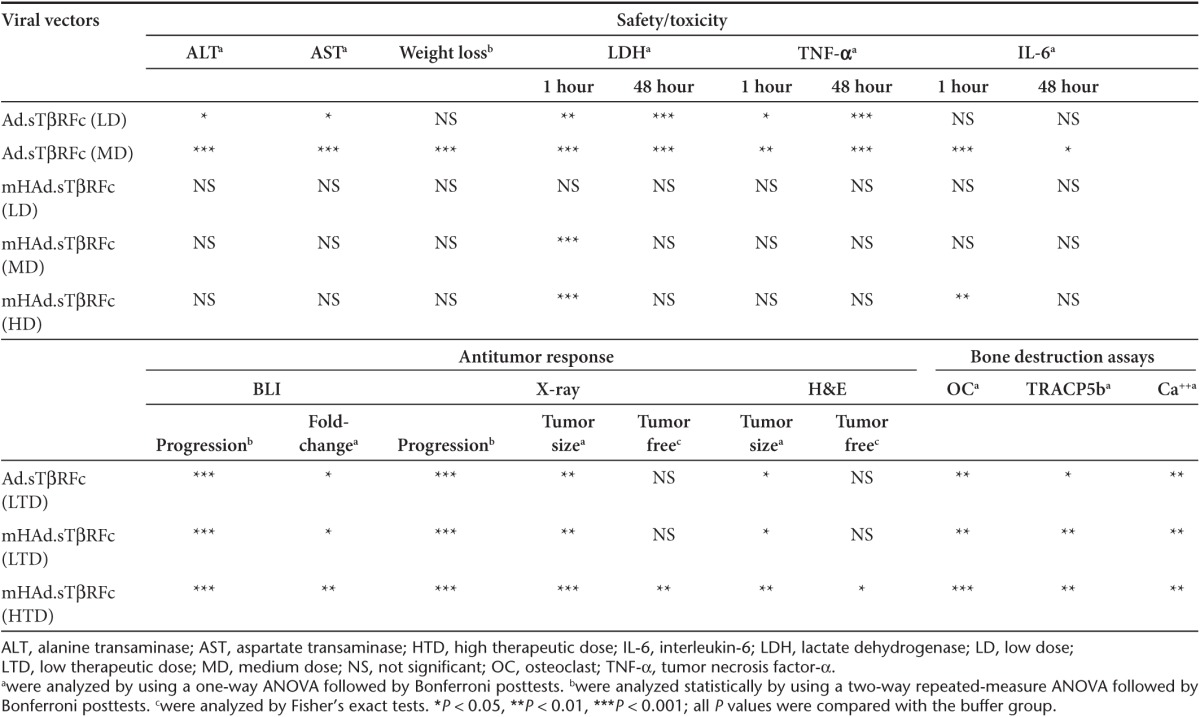
In conclusion, systemic delivery of mHAd.sTβRFc in nude mice, when compared with Ad.sTβRFc, produces reduced liver sequestration, reduced hepatotoxicity, reduced spleen uptake, attenuated innate immune response, and reduced systemic toxicity. However, in the PC-3-luc bone metastasis model of PCa, mHAd.sTβRFc is equally effective as Ad.sTβRFc in inhibiting the bone metastasis and bone destruction, as evaluated by multiple assays. In addition, a higher dose of mHAd.sTβRFc further improves the therapeutic effect in the PCa bone metastasis model in several assays (Table 1).
Table 1. Safety/toxicity studies and antitumor responses of mHAd.sTβRFc and Ad.sTβRFc.
Discussion
We are reporting here the construction of mHAd.sTβRFc, a hexon chimaeric Ad5/Ad48 oncolytic adenovirus expressing sTGFβRIIFc. It is important that the mHAd.sTβRFc in which seven hypervariable regions of Ad5 hexon were replaced with the corresponding regions of Ad48, retained its capacity to bind and replicate within the PCa cells, and produced sTGFβRIIFc protein. Thus, in the prostate tumor cells studied here, the mHAd.sTβRFc retains its tropism for the tumor cells.
We have shown that systemic delivery of mHAd.sTβRFc virus has much reduced liver sequestration (measured by viral genomic DNA, hexon and sTGFβRIIFc expression in the liver). This is consistent with the model that following systemic delivery of Ad5 in mice, the adenovirus hexon binds with FX in the blood, and the Ad5-FX complex is taken up by the liver. In contrast, the hepatic uptake of Ad5/48 hexon, because of diminished capacity to bind with FX, was significantly reduced. More importantly, compared to Ad.sTβRFc, much reduced hepatotoxicity (measured by the liver necrosis, and the serum alanine transaminase and aspartate transaminase levels), and perhaps other tissue(s)' damage (measured by serum LDH), are observed following mHAd.sTβRFc treatment of nude mice. It is quite intriguing that the reduced liver tropism of mHAd.sTβRFc was accompanied by the reduced tropism to the spleen, and produced a highly attenuated innate immune response (measured by serum pro-inflammatory cytokines TNF-α, IL-6), suggesting the role of Ad5-FX complex in producing liver toxicity, as well as inducing innate immune responses. In a recent very elegant study, the role of FX in activating the innate immunity in group C viruses, (which includes the Ad5 virus) has been well documented.23
It is worth noting that, at an early time point (1 hour), relatively high doses (MD and HD) of mHAd.sTβRFc also induced cell necrosis (measured by serum LDH), and innate immune response (IL-6 induction). This early induction of toxic response by mHAd.sTβRFc is not unexpected. The research conducted in many laboratories has indicated multiple biochemical events which can be potentially triggered within minutes following systemic delivery of adenoviruses into mice. These events are: viral sequestration and destruction by the liver Kupffer cells, Kupffer cells activation and/or necrosis, early liver toxicity, endothelial cell activation, virus uptake in the spleen, and the activation of the innate immune response.20,21,22,23,24,26,27,35–38 Some, or all of these steps can potentially be activated independent of the hexon, e.g., the uptake of adenovirus by Kupffer cells via the scavenger receptor is well described.20,21,22 It should be noted that the systemic delivery of a nonreplicating (E1 minus) Ad5/48 (seven hypervariable regions of Ad48 inserted in Ad5 backbone), has been shown to produce inflammatory responses and toxicity in MF1 immune-competent mice.39 In our studies, while the Ad.sTβRFc-mediated induction of LDH and IL-6 remained significantly elevated at 48 hours, mHAd.sTβRFc effects on LDH and IL-6 levels were no longer significantly increased at 48 hours. In addition, in nude mice, the maximum tolerable dose of mHAd.sTβRFc was much higher than that of Ad.sTβRFc. This could perhaps be due to the combination of reduced hepatotoxicity, attenuated innate immune responses, and decreased systemic toxicity elicited by the mHAd.sTβRFc.
The systemic administration of mHAd.sTβRFc, just like Ad.sTβRFc, resulted in its uptake in the skeletal tumors, and resulted in viral replication, and sTGFβRIIFc expression in the tumors. Thus, in the PC-3-luc prostate tumor model, the modified hexon also retains its tropism for the human prostate tumor cells in vivo. Systemic administration of mHAd.sTβRFc produced antitumor responses (detected by radiography, BLI, and histomorphometric analyses), inhibited bone destruction (measured by osteoclast activity, bone architect, and hypercalcemia) and increased the overall animal survival. In this regard, mHAd.sTβRFc and Ad.sTβRFc were equally effective. These results confirm that the two features—viral replication, and simultaneous sTGFβRIIFc expression targeting TGFβ signaling at the tumor site, which are critical for the antitumor effects,17 are preserved in Ad5/48 hexon-based mHAdsTβRFc virus. Moreover, because of reduced toxicity of mHAd.sTβRFc, a higher dose of mHAd.sTβRFc also resulted in antitumor responses (Table 1).
Systemic delivery of oncolytic adenoviruses in clinical trials in cancer patients have resulted in the elevation of liver enzymes and inflammatory cytokines, and the adenoviruses were detected in the liver tissues upon the biopsies.40,41,42,43 Therefore, because of the reduced toxicity and the efficacy of mHAd.sTβRFc, it could be used as a novel approach for treating advanced stage PCa patients affected with bone metastases. However, before testing mHAd.sTβRFc in the human clinical trials, we realize that a few other critical issues regarding mHAd.sTβRFc biology and host interactions should be addressed. It is known that following systemic delivery, Ad5 can also bind with other blood components including erythrocytes, platelets, IgM antibodies, and the components of the complement system.21,44 Therefore, we need to evaluate the binding of mHAd.sTβRFc with these cellular and noncellular components, and if necessary to include additional modifications in mHAd.sTβRFc to alter the virus tropism.
Another in vivo barrier in the use of adenovirus is the humoral immune response directed against the virus, which will preclude the multiple systemic administration of the adenoviruses, and even the initial viral dose might be ineffective in patients harboring preexisting neutralizing antibodies.26 In that regard, it is interesting to note that in a vaccine trial, the Ad5/48 chimaeric hexon adenovirus has been shown to evade the pre-existing Ad5 neutralizing antibodies in mice and rhesus monkeys.30 However, following systemic delivery, whether mHAd.sTβRFc can circumvent the Ad5 neutralizing antibodies remains to be examined. Human adenoviruses in general (including mHAd.sTβRFc), have poor replication potential in the mouse prostate tumor cells, and hence are difficult to evaluate in the immune-competent syngeneic host. However, efforts are underway in our laboratory to develop an immune competent mouse model of PCa bone metastasis that can be used to evaluate the antitumor responses.
Materials and Methods
Cell lines and viruses. Two human prostate tumor cell lines PC-3 and DU-145, and a mouse prostate tumor cell line TRAMP-C2 were purchased from ATCC (Manassas, VA). A PC-3-luc cell line was generated by stable transfection of a retrovirus expressing luciferase gene into PC-3 cells as previously described.45 All prostate tumor cell lines were cultured in RPMI-1640 media containing 10% fetal calf serum. All media components were purchased from Invitrogen (Grand Island, NY). Adenoviral vectors are: Ad.sTβRFc, an oncolytic Ad5-expressing sTGβRIIFc gene,34 mHAd.sTβRFc that was created by genetically replacing seven hypervariable regions of Ad.sTβRFc hexon by the corresponding sequence of Ad48 hexon (see Figure 1a), and Ad(E1-).Null, a nonreplicating adenovirus without any foreign gene.34 All adenoviral vectors were amplified in HEK293 cells and purified as described earlier.46
Adenoviral replication and cytotoxicity assays. Cells were plated in six-well dishes (5 × 105 cells/well). The next day, cells were incubated with adenoviral vectors (2.5 × 104 VPs/cell) for 3 hours. After washing the cells three times with media, either the crude viral lysates were collected at 3 hours samples, or incubations were continued at 37°C for 48 hours. Various aliquots of 3 and 48 hours crude viral lysates were used to infect HEK293 cells according to Adeno-X Rapid Titer Kit protocol (Clontech, Mountain view, CA) as described earlier.47 Hexon-expressing positive brown cells were photographed, and counted under the microscope to quantify viral replication. Viral titers were represented as the viral burst size (an increase in positive hexon expressing cells from 3 to 48 hours). To examine viral-induced cytotoxicity, cells were plated in 96-well plates (103 cells/well) as previously described.17 The next day, cells were infected with various doses of adenoviral vectors, and the incubations were continued for 7 days. Cells were washed, fixed and stained with sulforhodamine B (Sigma-Aldrich, St Louis, MO), and the absorbance at 564 nm (A564) was measured as previously described.46 Untreated control cells were considered to have 100% survival.
sTGFβRIIFc expression in the PCa cells. Cells were plated in six-well dishes (5 × 105 cells/well). The next day, cells were incubated with adenoviral vectors (2.5 × 103 VPs/cell) for 24 hours. The media were changed to serum free media, and the incubations continued for another 24 hours. Both media and cell lysates were subjected to western blot analyses for sTGFβRIIFc expression as previously described.17 Blots were probed with antibodies against TGFβRII (H-567, Santa Cruz Biotechnology, Santa Cruz, CA), human IgG, Fcγ fragment (Jackson Immunoresearch, West Grove, PA), or actin protein (A2066, Sigma-Aldrich, St Louis, MO). sTGβRIIFc levels in the media were also examined by ELISA using antihuman IgG, Fcγ fragment antibodies, biotinylated anti-TGFβRII antibodies (BAF241, R&D systems, Minneapolis, MN), as previously described.17
Animal studies. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at NorthShore University HealthSystem.
Liver toxicity studies. Six-week-old male nude mice were injected with low dose (LD = 2.5 × 1010 VPs/mouse), medium dose (MD = 1.0 × 1011 VPs/mouse) or high dose (HD = 2 × 1011 VPs/mouse) of Ad.sTβRFc or mHAd.sTβRFc via tail vein. The high dose of Ad.sTβRFc (2.0 × 1011 VPs/mouse) led to the deaths of all the mice within 24 hours. Each group contains four mice. Three days after virus injection, their body weights were measured before they were euthanized. The livers were photographed. The liver samples were collected and processed for hematoxylin and eosin (H&E) staining, immunohistochemistry staining by antihuman IgG, Fcγ fragment antibody, and viral genome copy measurement by quantitative PCR (qPCR). Mice spleens were also processed for immunohistochemistry staining by antihuman IgG, Fcγ antibody. Mouse blood was centrifuged at 10K rpm for 5 minutes, and serum samples were used to determine the sTGFβRIIFc levels by ELISA as described earlier,17 and the alanine transaminase and aspartate transaminase levels by the commercially available kits (Cayman Chemical, Ann Arbor, MI).33
Innate immune response studies and serum LDH assay. Six-week-old male nude mice were injected with low dose (LD = 2.5 × 1010 VPs/mouse) or medium dose (MD = 1.0 × 1011 VPs/mouse) of Ad.sTβRFc or mHAd.sTβRFc via tail vein. An additional high dose (HD = 2.0 × 1011 VPs/mouse) of mHAd.sTβRFc was also administered. Each group contains four mice. One hour after injection, mice were anesthetized with Ketamine (25 mg/ml)/Xylazine (2 mg/ml) cocktail and 300 µl blood were withdrawn via the heart. Forty eight hours after virus injection, mice were anesthetized by isoflurane and killed after collecting blood samples. Mice sera were used to determine LDH levels (ab102526, Abcam, Cambridge, UK), IL-6 levels (M6000B, R&D systems, Minneapolis, MN), and TNF-α levels (R&D systems, Minneapolis, MN) according to the suggested protocols.
Viral uptake in skeletal tumors. PC-3-luc cells (2.0 × 105/mouse) were injected into the left heart ventricle of four week old male nude mice (Nu/Nu) (Charles River Laboratories, Wilmington, MA) on day 0, as described earlier.17 Mice with X-ray positive skeletal lesions (on 46 days post tumor cells injection) were injected with Ad.sTβRFc or mHAd.sTβRFc (1.0 × 1011 VPs/mouse) via tail vein. Each group contains three mice. Three days later, mice were killed and the liver and hind limbs were harvested, processed, and stained with antihexon antibody (MAB8044, Millipore, Billerica, MA) or antihuman IgG, Fcγ fragment antibody as described previously.48 Serum samples were used to measure sTGFβRIIFc levels by ELISA as previously described.17,25
Bone metastasis model and bioluminescence imaging. To establish bone metastasis, PC-3-luc cells (2.0 × 105/mouse) were injected into the left heart ventricle of 4-week-old male nude mice (Nu/Nu) (Charles River laboratories) on day 0, as described earlier.17 On day 9, mice were subjected to BLI in dorsal and ventral positions using Xenogen IVIS Spectrum imaging equipment (Caliper life sciences, Hopkinton, MA) after being injected intraperitoneally with 100 μl (150 mg/kg) of the D-luciferin solution (Gold BioTechnology, St Louis, MO). During image acquisition, mice were kept under anesthesia with 1.5–2.0% isoflurane. Signal intensity was quantified as the total flux (photons/seconds) within regions of interest in both left and right hind limbs using Living Image software 3.0 (Caliper Life Sciences, Hopkinton, MA) as described.17,25 We obtained 42 mice that had the regions of interest flux within the range of 2.0 × 105–1.0 × 106 photons/second. They were divided into four groups, with statistically indistinguishable BLI signals amongst each group (an average of about 5.0 × 105 photons/second per group). For treatment groups, various viral vectors were administered via tail vein on days 10 and 13. These groups were: Ad.sTβRFc group (LTD, total 5.0 × 1010 VPs/mouse, n = 11), mHAd.sTβRFc group (LTD, total 5.0 × 1010 VPs/mouse, n = 11), and a mHAd.sTβRFc group (HTD, total 4.0 × 1011 VPs/mouse, n = 10). The control group of mice (n = 10) was administered with the buffer alone. BLI was conducted weekly for the duration of the study. All of the mice were euthanized after blood was collected on day 53.
X-ray radiography imaging. Mice were subjected to X-ray radiography in the prone position using Faxitron (Faxitron X-ray Corporation, Wheeling, IL) on day 16 and once a week until day 51 as described.17 Bone lesions were quantified in femur and tibia of both hind limbs using Image J software (NIH, Bethesda, MD) as described earlier.17,25
Bone histology and histomorphometric analysis. On day 53, mice were euthanized, and hind limbs were harvested, processed and stained with hematoxylin and eosin (H&E) as previously described.17 Tumor burden per tibia/femur was quantified on H&E-stained sections using NIS-Elements BR 3.10 Software (Nikon, Melville, NY) as previously described.17,25 Multinucleated TRAP positive osteoclasts (OCs) at the bone–tumor interface were measured as previously described.25
Synchrotron microCT. Synchrotron microCT, was performed using station 2-BM of the Advanced Photon Source (APS) at Argonne National Laboratory (Argonne, IL) using the dedicated microCT instrument.49 The following conditions were used for data collection: 15 keV, 0.12º rotation increment, 180º rotation range, (2K)2 reconstructions with 2.9 µm isotropic volume elements (voxels). 2D slices were stacked in order to produce a 3D volume. The image was down sampled by including every 20th slice for 3D rendering. An isosurface was then generated to identify the edges of the bone for visualization. 3D images of bone sections spanning 3.0 mm below the growth plate regions were constructed using MATLAB R2011a (The MathWorks, Natick, MA).
Quantification of calcium, TRACP 5b, and sTGFβRIIFc in serum. At terminal time point (day 53); the mice blood was collected via cardiac puncture. Mice sera were obtained by centrifuging blood at 10K rpm for 5 minutes. Calcium concentrations were measured using QuantiChrom calcium assay kit (BioAssay Systems, Hayward, CA). Serum concentrations of osteoclast derived TRACP 5b were measured by using a solid phase immunofixed enzyme activity, MouseTRAP kit, according to the manufacturer's instructions (Immunodiagnosticsystems, Phoenix, AZ). Serum sTGFβRIIFc levels were determined by ELISA as described earlier.17,25
Statistical analysis. Data were presented as mean ± SEM and statistically analyzed using GraphPad Prism software version 5 (GraphPad Software, San Diego, CA). Safety/toxicity endpoints and antitumor responses of mHAd.sTβRFc and Ad.sTβRFc with various doses were compared using one-way ANOVA followed by Bonferroni post hoc tests adjusting for multiplicity. Longitudinal data were analyzed using a two-way repeated measure ANOVA followed by Bonferroni post hoc tests for all the data of over time course. The Student's t-tests were performed to analyze viral genomic copy number, and sTGFβRIIFc levels in the blood. A Fisher's exact test was used for the bone metastasis incidence data in the X-ray and histomorphometric analyses. A Kaplan–Meier survival test was used for the survival data. Differences were considered significant at two sided P < 0.05.
Acknowledgments
The work was funded in part by the National Institutes of Health grant # R01CA12738 (P.S.), and philanthropic support through John and Carol Walter Center for Urological Health, NorthShore University HealthSystem. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We are thankful to the Kovler Family Foundation, and Richard Hulina, Jimmie Alford and Maree Bullock, and an anonymous donor for their generous philanthropic support. We are thankful to Janardan Khandekar, Theodore Mazzone, Bruce Brockstein and Michael Caplan for their continuous support. This work is dedicated to the fond memory of Jimmie Alford.
References
- Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001;27:165–176. doi: 10.1053/ctrv.2000.0210. [DOI] [PubMed] [Google Scholar]
- Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannata DH, Kirschenbaum A, Levine AC. Androgen deprivation therapy as primary treatment for prostate cancer. J Clin Endocrinol Metab. 2012;97:360–365. doi: 10.1210/jc.2011-2353. [DOI] [PubMed] [Google Scholar]
- Coleman R. The use of bisphosphonates in cancer treatment. Ann N Y Acad Sci. 2011;1218:3–14. doi: 10.1111/j.1749-6632.2010.05766.x. [DOI] [PubMed] [Google Scholar]
- Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. 2012;379:39–46. doi: 10.1016/S0140-6736(11)61226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fizazi K, Carducci M, Smith M, Damião R, Brown J, Karsh L, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377:813–822. doi: 10.1016/S0140-6736(10)62344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturge J, Caley MP, Waxman J. Bone metastasis in prostate cancer: emerging therapeutic strategies. Nat Rev Clin Oncol. 2011;8:357–368. doi: 10.1038/nrclinonc.2011.67. [DOI] [PubMed] [Google Scholar]
- Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- Seth P.ed) (1999Adenoviruses: Basic Biology to Gene Therapy R G Landes Company; Austin; pp. 1–314. [Google Scholar]
- Toth K, Dhar D, Wold WS. Oncolytic (replication-competent) adenoviruses as anticancer agents. Expert Opin Biol Ther. 2010;10:353–368. doi: 10.1517/14712590903559822. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Curiel DT. Current issues and future directions of oncolytic adenoviruses. Mol Ther. 2010;18:243–250. doi: 10.1038/mt.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IK, Yun CO. Recent developments in oncolytic adenovirus-based immunotherapeutic agents for use against metastatic cancers. Cancer Gene Ther. 2013;20:70–76. doi: 10.1038/cgt.2012.95. [DOI] [PubMed] [Google Scholar]
- Nakashima H, Chiocca EA. Switching a replication-defective adenoviral vector into a replication-competent, oncolytic adenovirus. J Virol. 2014;88:345–353. doi: 10.1128/JVI.02668-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JB, Johnson M, Sato M, Koh SB, Mulholland DJ, Stout D, et al. Adenovirus-mediated gene expression imaging to directly detect sentinel lymph node metastasis of prostate cancer. Nat Med. 2008;14:882–888. doi: 10.1038/nm.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vrij J, Willemsen RA, Lindholm L, Hoeben RC, Bangma CH, Barber C, et al. GIANT Consortium Adenovirus-derived vectors for prostate cancer gene therapy. Hum Gene Ther. 2010;21:795–805. doi: 10.1089/hum.2009.203. [DOI] [PubMed] [Google Scholar]
- Schenk E, Essand M, Bangma CH, Barber C, Behr JP, Briggs S, et al. GIANT FP6 Consortium Clinical adenoviral gene therapy for prostate cancer. Hum Gene Ther. 2010;21:807–813. doi: 10.1089/hum.2009.206. [DOI] [PubMed] [Google Scholar]
- Hu Z, Gupta J, Zhang Z, Gerseny H, Berg A, Chen YJ, et al. Systemic delivery of oncolytic adenoviruses targeting transforming growth factor-β inhibits established bone metastasis in a prostate cancer mouse model. Hum Gene Ther. 2012;23:871–882. doi: 10.1089/hum.2012.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juárez P, Guise TA. TGF-β in cancer and bone: implications for treatment of bone metastases. Bone. 2011;48:23–29. doi: 10.1016/j.bone.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Jones E, Pu H, Kyprianou N. Targeting TGF-beta in prostate cancer: therapeutic possibilities during tumor progression. Expert Opin Ther Targets. 2009;13:227–234. doi: 10.1517/14728220802705696. [DOI] [PubMed] [Google Scholar]
- Lieber A, He CY, Meuse L, Schowalter D, Kirillova I, Winther B, et al. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J Virol. 1997;71:8798–8807. doi: 10.1128/jvi.71.11.8798-8807.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Tian J, Smith JS, Byrnes AP. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Virol. 2008;82:11705–11713. doi: 10.1128/JVI.01320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare R, Chen CY, Weaver EA, Barry MA. Advances and future challenges in adenoviral vector pharmacology and targeting. Curr Gene Ther. 2011;11:241–258. doi: 10.2174/156652311796150363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronin K, Flatt JW, Di Paolo NC, Khare R, Kalyuzhniy O, Acchione M, et al. Coagulation factor X activates innate immunity to human species C adenovirus. Science. 2012;338:795–798. doi: 10.1126/science.1226625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green NK, Herbert CW, Hale SJ, Hale AB, Mautner V, Harkins R, et al. Extended plasma circulation time and decreased toxicity of polymer-coated adenovirus. Gene Ther. 2004;11:1256–1263. doi: 10.1038/sj.gt.3302295. [DOI] [PubMed] [Google Scholar]
- Hu Z, Gerseny H, Zhang Z, Chen YJ, Berg A, Stock S, et al. Oncolytic adenovirus expressing soluble TGFbeta receptor II-Fc-mediated inhibition of established bone metastases: a safe and effective systemic therapeutic approach for breast cancer. 2011;9:1609–1618. doi: 10.1038/mt.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahi YS, Bangari DS, Mittal SK. Adenoviral vector immunity: its implications and circumvention strategies. Curr Gene Ther. 2011;11:307–320. doi: 10.2174/156652311796150372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickan E, Smith JS, Tian J, Eggerman TL, Lozier JN, Muller J, et al. Rapid Kupffer cell death after intravenous injection of adenovirus vectors. Mol Ther. 2006;13:108–117. doi: 10.1016/j.ymthe.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Engler H, Machemer T, Philopena J, Wen SF, Quijano E, Ramachandra M, et al. Acute hepatotoxicity of oncolytic adenoviruses in mouse models is associated with expression of wild-type E1a and induction of TNF-alpha. Virology. 2004;328:52–61. doi: 10.1016/j.virol.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Parker AL, Waddington SN, Nicol CG, Shayakhmetov DM, Buckley SM, Denby L, et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood. 2006;108:2554–2561. doi: 10.1182/blood-2006-04-008532. [DOI] [PubMed] [Google Scholar]
- Roberts DM, Nanda A, Havenga MJ, Abbink P, Lynch DM, Ewald BA, et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441:239–243. doi: 10.1038/nature04721. [DOI] [PubMed] [Google Scholar]
- Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL, et al. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci USA. 2008;105:5483–5488. doi: 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alba R, Bradshaw AC, Parker AL, Bhella D, Waddington SN, Nicklin SA, et al. Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: effect of mutagenesis on FX interactions and gene transfer. Blood. 2009;114:965–971. doi: 10.1182/blood-2009-03-208835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Krimmel J, Zhang Z, Hu Z, Seth P. Systemic delivery of a novel liver-detargeted oncolytic adenovirus causes reduced liver toxicity but maintains the antitumor response in a breast cancer bone metastasis model. Hum Gene Ther. 2011;22:1137–1142. doi: 10.1089/hum.2011.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P, Wang ZG, Pister A, Zafar MB, Kim S, Guise T, et al. Development of oncolytic adenovirus armed with a fusion of soluble transforming growth factor-beta receptor II and human immunoglobulin Fc for breast cancer therapy. Hum Gene Ther. 2006;17:1152–1160. doi: 10.1089/hum.2006.17.1152. [DOI] [PubMed] [Google Scholar]
- Yu D, Jin C, Ramachandran M, Xu J, Nilsson B, Korsgren O, et al. Adenovirus serotype 5 vectors with Tat-PTD modified hexon and serotype 35 fiber show greatly enhanced transduction capacity of primary cell cultures. PLoS One. 2013;8:e54952. doi: 10.1371/journal.pone.0054952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao N, Gao GP, Parr M, Johnston J, Baradet T, Wilson JM, et al. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose response of transduction in liver. Mol Ther. 2001;3:28–35. doi: 10.1006/mthe.2000.0227. [DOI] [PubMed] [Google Scholar]
- Ogawara K, Rots MG, Kok RJ, Moorlag HE, Van Loenen AM, Meijer DK, et al. A novel strategy to modify adenovirus tropism and enhance transgene delivery to activated vascular endothelial cells in vitro and in vivo. Hum Gene Ther. 2004;15:433–443. doi: 10.1089/10430340460745766. [DOI] [PubMed] [Google Scholar]
- Schiedner G, Bloch W, Hertel S, Johnston M, Molojavyi A, Dries V, et al. A hemodynamic response to intravenous adenovirus vector particles is caused by systemic Kupffer cell-mediated activation of endothelial cells. Hum Gene Ther. 2003;14:1631–1641. doi: 10.1089/104303403322542275. [DOI] [PubMed] [Google Scholar]
- Coughlan L, Bradshaw AC, Parker AL, Robinson H, White K, Custers J, et al. Ad5:Ad48 hexon hypervariable region substitutions lead to toxicity and increased inflammatory responses following intravenous delivery. Mol Ther. 2012;20:2268–2281. doi: 10.1038/mt.2012.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid T, Warren R, Kirn D. Intravascular adenoviral agents in cancer patients: lessons from clinical trials. Cancer Gene Ther. 2002;9:979–986. doi: 10.1038/sj.cgt.7700539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemunaitis J, Cunningham C, Buchanan A, Blackburn A, Edelman G, Maples P, et al. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther. 2001;8:746–759. doi: 10.1038/sj.gt.3301424. [DOI] [PubMed] [Google Scholar]
- Small EJ, Carducci MA, Burke JM, Rodriguez R, Fong L, van Ummersen L, et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther. 2006;14:107–117. doi: 10.1016/j.ymthe.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Hamid O, Varterasian ML, Wadler S, Hecht JR, Benson A, 3rd, Galanis E, et al. Phase II trial of intravenous CI-1042 in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:1498–1504. doi: 10.1200/JCO.2003.09.114. [DOI] [PubMed] [Google Scholar]
- Xu Z, Qiu Q, Tian J, Smith JS, Conenello GM, Morita T, et al. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat Med. 2013;19:452–457. doi: 10.1038/nm.3107. [DOI] [PubMed] [Google Scholar]
- Loberg RD, Day LL, Dunn R, Kalikin LM, Pienta KJ. Inhibition of decay-accelerating factor (CD55) attenuates prostate cancer growth and survival in vivo. Neoplasia. 2006;8:69–78. doi: 10.1593/neo.05679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayose D, Gudas J, Nguyen H, Srivastava S, Cowan KH, Seth P. Cytotoxic effects of adenovirus-mediated wild-type p53 protein expression in normal and tumor mammary epithelial cells. Clin Cancer Res. 1995;1:889–897. [PubMed] [Google Scholar]
- Zhang Z, Hu Z, Gupta J, Krimmel JD, Gerseny HM, Berg AF, et al. Intravenous administration of adenoviruses targeting transforming growth factor beta signaling inhibits established bone metastases in 4T1 mouse mammary tumor model in an immunocompetent syngeneic host. Cancer Gene Ther. 2012;19:630–636. doi: 10.1038/cgt.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Zhang Z, Guise T, Seth P. Systemic delivery of an oncolytic adenovirus expressing soluble transforming growth factor-β receptor II-Fc fusion protein can inhibit breast cancer bone metastasis in a mouse model. Hum Gene Ther. 2010;21:1623–1629. doi: 10.1089/hum.2010.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Carlo FD, Mancini DC, McNulty I, Tieman B, Bresnahan J, et al. A high-throughput X-ray microtomography system at the Advanced Photon Source. Rev Sci Instr. 2068 2001;72:2062. [Google Scholar]