

Abstract
Regulated transgene expression may improve the safety and efficacy of hematopoietic stem cell (HSC) gene therapy. Clinical trials for X-linked chronic granulomatous disease (X-CGD) employing gammaretroviral vectors were limited by insertional oncogenesis or lack of persistent engraftment. Our novel strategy, based on regulated lentiviral vectors (LV), targets gp91phox expression to the differentiated myeloid compartment while sparing HSC, to reduce the risk of genotoxicity and potential perturbation of reactive oxygen species levels. Targeting was obtained by a myeloid-specific promoter (MSP) and posttranscriptional, microRNA-mediated regulation. We optimized both components in human bone marrow (BM) HSC and their differentiated progeny in vitro and in a xenotransplantation model, and generated therapeutic gp91phox expressing LVs for CGD gene therapy. All vectors restored gp91phox expression and function in human X-CGD myeloid cell lines, primary monocytes, and differentiated myeloid cells. While unregulated LVs ectopically expressed gp91phox in CD34+ cells, transcriptionally and posttranscriptionally regulated LVs substantially reduced this off-target expression. X-CGD mice transplanted with transduced HSC restored gp91phox expression, and MSP-driven vectors maintained regulation during BM development. Combining transcriptional (SP146.gp91-driven) and posttranscriptional (miR-126-restricted) targeting, we achieved high levels of myeloid-specific transgene expression, entirely sparing the CD34+ HSC compartment. This dual-targeted LV construct represents a promising candidate for further clinical development.
Introduction
Chronic granulomatous disease (CGD) is caused by defects in genes encoding the subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, responsible for the respiratory burst.1,2 The oxidase catalyzes the production of reactive oxygen species, which are critical to the killing of phagocytosed pathogens. Accordingly, CGD patients are affected by severe, life-threatening bacterial and fungal infections as well as extensive tissue granuloma formation. X-linked CGD (X-CGD) is due to mutations in the CYBB gene which encodes for the gp91phox subunit.3,4 To date, CGD patients are treated with antimicrobial and antifungal prophylaxis, but mortality remains high (3% per year).5 Hematopoietic stem cell transplantation (HSCT) represents a definitive treatment for patients with a suitable human leukocyte antigen-matched donor.6,7 Despite recent improvements in HSCT protocols,8,9,10 treating CGD patients in whom conventional treatment has failed and lack an human leukocyte antigen-matched donor remains challenging and is still associated with substantial complications. Gene transfer into hematopoietic stem/progenitor cells (HSPC) represents a promising approach, especially for this patient group. Gene therapy (GT) trials for X-CGD conducted so far have resulted in low-level engraftment or transient clinical benefit.11,12,13,14,15 Lack of long-term efficacy has been attributed to a progressive decrease in transduced cell engraftment or methylation of the viral promoter leading to silencing of transgene expression. These findings led to the hypothesis that ectopic gp91phox expression in HSPC could cause the production of reactive oxygen species that may damage DNA, alter cell growth, or induce apoptosis.16,17 A subtle competitive disadvantage of HSPC engineered with gp91phox expressing vector in a host with a highly activated bone marrow (BM) environment due to recurrent infections might have favored the loss of gene-modified cells. Alternatively, immune-mediated mechanisms against gp91phox expressing cells could have contributed to the lack of long-term persistence. In contrast, most patients treated with HSPC transduced with a spleen focus forming virus-based retroviral vector (SFFV-RV) developed myelodysplasia with monosomy 7 caused by insertional activation of the EVI1-MDS1 proto-oncogene.8,18 The frequency of this adverse event underlines the fact that only gp91phox-transduced cells with a gain-of-function event could persist in patients treated with GT protocols employing LTR-driven RV.
Overall, these findings support the need for safer and more effective gene correction strategies for X-CGD. Self-inactivating lentiviral vectors (SIN-LVs) with an internal promoter are the most promising candidates for this approach due to their higher proficiency in transducing HSC and their improved safety profile compared with retroviral vectors.19 To restore physiological expression of gp91phox, SIN-LVs were designed to allow transcriptional targeting of gp91phox to myeloid cells using different promoters, including a fusion cathepsin G and c-fes promoter,20 a human micro-RNA 223 promoter,21 an A2UCOE insulator element in combination with the MRP8 myeloid specific promoter,22 and a minimal gp91phox synthetic promoter combined with various transcription factor-binding sites.23 These vectors showed good specificity in differentiated cells, but a variable degree of leakiness in murine progenitors, and no information was provided on the specificity in human repopulating HSPC.
To further restrict transgene expression from human HSPC and, at the same time, allow robust expression in differentiated mature cells, we exploited posttranscriptional regulation by microRNA (miRNA),24,25,26,27 inserting target sequences for HSPC-specific miRNA into the LV cassette. Using this approach, we obtained a 5–30-fold downregulation of the transgene in mouse and human HSPC, while expression remained largely unaffected in differentiated cells which do not express these miRNA.27 This strategy allowed us to overcome HSPC toxicity by galactocerebrosidase and conditional suicide genes achieving a stable BM graft in mice.27 However, the optimal number and combination of miRNA target sequences remained to be determined.
Here we show that LV encoding miRNA-mediated posttranscriptional control elements and myeloid-specific transcriptional elements allow unprecedented levels of myeloid-specific transgene expression, entirely sparing the CD34+ HSPC compartment. Therapeutic LV encoding gp91phox effectively corrected the X-CGD phenotype in gp91phox-deficient cell lines, primary cells, and gp91phox-deficient mice while maintaining strictly regulated transgene expression.
Results
Optimized detargeting of the HSPC compartment by miRNA target sequences
To optimize posttranscriptional regulation, we screened combinations of target sequences for miR-126 and miR-130a or targets for novel putative HSPC-specific miRNAs27 in human cord blood (CB) for their capacity to repress transgenes in a differentiation stage-specific manner (Supplementary Figures S1 and S2). Two targets for miR-126 (126T(2)), either alone or in combination with two miR-130a targets, performed best, resulting in regulation indices of 10.7 and 12.9, respectively, when comparing CD34+CD38− HSPC with CD13+ myeloid progeny (Supplementary Figure S2). We further characterized the regulatory potential of the 126T(2) target on CB (n = 2 additional donors) and BM (n = 4 donors) in short-term or myeloid differentiation culture using the bidirectional lentiviral reporter vector platform (BdLV)24,25,27 (Figure 1a). 126T(2) activity was similar in CB and BM, with a 10–18-fold reduction of transgene expression in CD34+CD38−CD90+ HSPC and unrestricted expression in differentiated myeloid cells, with loss of miR-126 activity occurring around the myeloblast/promyelocyte stage (Figure 1b). We then reconstituted NSG mice with human BM CD34+ cells transduced with BdLV.126T(2) or Ctrl.BdLV, and quantified transgene expression in the xenografted cells 24 weeks after transplantation (Figure 1c,d). Importantly, we found persistent downregulation of transgene expression in CD34+ HSPC, with a gradient of activity extending from HSC (25-fold downregulation) to multipotent progenitors (7–20-fold repression) and committed progenitors/precursors (3–6-fold repression). Differentiated cells (B cells and myeloid cells) reached 70–80% of transgene expression levels with respect to the Ctrl BdLV, indicating a near-complete loss of miR-126 activity, in line with the in vitro results.
Figure 1.
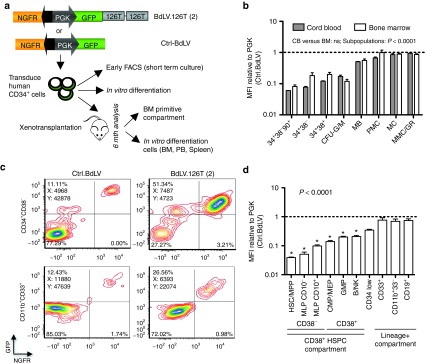
Optimized detargeting of the hematopoietic stem/progenitor cells (HSPC) compartment by miRNA target sequences. (a) Measuring miRNA activity in primary human HSPC by bidirectional reporter lentiviral vectors (BdLVs). Comparing the miR-126 reporter, BdLV.126T(2), with a control (Ctrl-)BdLV allows precise quantification of GFP downregulation without the need for matching vector copy number (see Methods section for details). The scheme shows in vitro and in vivo assays to comprehensively measure the activity of 126T(2) in human hematopoiesis. (b) Activity of the 126T(2) target in cord blood (CB) (n = 2 donors) and bone marrow (BM) (n = 4 donors), either in the HSPC compartment (CD34+CD38−CD90+, CD34+38−, and 34+38+, 2 days after transduction), or after 2 weeks in myeloid differentiating condition (G-CSF and FCS 10%). CFU-G/M, colony forming units granulocyte/monocyte, CD34hiCD13+; MB, myeloblasts, CD34+/lowCD13+CD11b−; PMC, promyelocytes, CD34−CD13+CD11b−; MC, myelocytes, CD13+CD11b+CD16−; MMC/GR, metamyelocytes/granulocytes, CD13+CD11b+CD16+. The dotted line represents the GFP mean fluorescence intensity (MFI) of the Ctrl BdLV. The differences in GFP expression between the subsets are highly significant (Two-way analysis of variance (ANOVA)), while there is no significant difference between HSPC source (CB versus BM). (c,d) Activity of 126T(2) in vivo. BM CD34+ HSPC were transduced with BdLV.126T(2) or Ctrl.BdLV, and transplanted into NSG mice (n = 4 per group). (c) Representative FACS plots show reporter expression in human CD45+ cells recovered from the BM of hematochimeric mice at 24 weeks post-transplantation. Upper row: plots gated on CD34+CD38− HSPC; lower row: FACS plots gated on CD33+CD11b+ myeloid progeny. (d) Quantification of the in vivo activity of the 126T(2) in the indicated subpopulations. The following surface marker profiles were used to define subpopulations: HSC/MPP: CD34+CD38−CD45RA−CD10−; MLP: CD34+CD38−CD45RA+CD10+ or - as indicated; CMP/MEP: CD34+CD38+CD45RA−; GMP: CD34+CD38+CD45RA+CD10−; B/NK: CD34+CD38+CD45RA+CD10+. The dotted line represents the GFP MFI of the Ctrl BdLV. Statistics were performed by one-way ANOVA, and * indicates P < 0.05 in Bonferroni's post test against CD33+ cells. FACS, fluorescence-activated cell sorting; FCS, fetal calf serum; GFP, green fluorescent protein; PGK, phosphoglycerate kinase.
Combined transcriptional/posttranscriptional regulation further improves targeting to differentiated myeloid cells
With the aim of maximizing expression in differentiated myeloid cells while minimizing it in HSPC, we combined posttranscriptional, miR-126-based detargeting with a myeloid-specific promoter (MSP) composed of a 1.5 kb minimal promoter sequence from the gp91phox locus fused to the SP146 synthetic enhancer/promoter.23,28 CD34+ HSPC from human BM were transduced with matched doses of a phosphoglycerate kinase (PGK).GFP control LV, the transcriptionally targeted MSP.GFP LV and the dual-targeted MSP.GFP.126T(2) LV (Figure 2a), and promoter activity was compared in liquid culture as described above for BdLV.126T(2), as well as in myelo-erythroid colonies. Average vector copy number (VCN) per cell measured after 2 weeks in culture was comparable for the 3 LVs (Figure 2b), as was the percentage of GFP+ cells in the more differentiated, CD34−CD38+ compartment (Figure 2c). On the contrary, CD34+CD38− cells transduced with the MSP constructs expressed less GFP compared with the PGK LV, indicating that the transcriptional activity of the MSP is decreased in HSPC with respect to PGK. Strikingly, GFP expression was virtually absent in CD34+CD38−CD90+ HSPC when the MSP was combined with the 126T sequence (Figure 2c,d). Transgene expression was quantified as the mean fluorescence intensity (MFI) corrected for transduction level (see Materials and Methods). The MSP and the combined MSP.126T LVs were, respectively, 10- and 100-fold less active in the most primitive CD34+CD38−CD90+ compartment compared with PGK. During myeloid differentiation, transgene expression in MSP- or MSP.126T-transduced cells was similar and surpassed the PGK group 5–10 fold (Figure 2d,e). A similar expression pattern was seen for CD14+ monocyte/macrophage subsets, while the MSP was not active in CD235+ erythroid cells (Figure 2f). To compare the differentiation-stage specific activity of each LV construct, we measured the GFP MFI in myeloid subpopulations in a bulk culture containing both primitive and progressively differentiated cellular elements, and normalized it to the MFI of CD34hiCD11b−CD13− HSPC within the same vector group (Figure 2g). The activity of the PGK promoter peaked in progenitors and progressively decreased during myeloid differentiation to ~50% of its original value, and addition of the 126T(2) element improved this myeloid specificity index from 0.5 to 5. Noteworthy, the MSP had a very favorable expression profile, with a 30-fold induction of transgene expression during myeloid differentiation, and addition of the miR-126 target sequences further improved this regulation index to more than 100-fold. Finally, we tested the performance of the MSP LVs in comparison with PGK in vivo after long-term repopulation of NSG mice with LV-transduced BM HSPC. Analysis of human CD45+ BM cells at 18 weeks post-transplant showed little GFP expression in the CD34+ HSPC compartment, particularly in the most primitive HSC compartment and in CD10+ B cell progenitors. Expression was further reduced to barely detectable levels by adding the 126T(2) element, fully in line with the in vitro culture data (Figure 2h). Expression from the MSP was absent in T cells and similar to that of PGK in CD19+ B lymphocytes. Importantly, we confirmed a robust induction of transgene expression in CD33+ myeloid cells in vivo, surpassing the PGK promoter several fold (Figure 2i). These in vivo results were confirmed in an independent experiment, and transduction of mature human monocytes/macrophages showed that our MSP performed at least equally to PGK in the latter phagocytic cell population (Supplementary Figure S3).
Figure 2.

Transcriptional targeting by the SP146/gp91phox promoter in human BM populations. (a) Vector maps of the myeloid-specific promoter (MSP) based on a minimal gp91phox promoter from the endogenous locus fused to the synthetic SP146 element and the ubiquitous PGK promoter (Ctrl LV) which was used as a reference. (b) CD34+ bone marrow (BM) cells (n = 4, two different donors) were transduced with the vectors indicated in (a), and vector copy number (VCN) was assessed after 14 days of in vitro culture (mean + SEM). (c) Transgene expression during in vitro culture is shown for the two vector groups (mean + SEM). Culture conditions and subpopulations are the same as in Figure 1d. P < 0.001 (two-way analysis of variance (ANOVA)), * indicates significance levels after Bonferroni post-test correction, with the following reference populations: before the slash: MSP versus PGK; after the slash: MSP versus MSP.126T. (d) Representative FACS plots showing GFP expression of MSP- (black line) or Ctrl LV- (gray area) transduced cells in CD34+CD38+CD90+ hematopoietic stem/progenitor cells (HSPC) (top) or CD34−CD13+CD11b+ myeloid progeny (bottom). (e) Mean fluorescence intensity (MFI) (arithmetic mean) of the GFP-positive cells shown in (c). P < 0.001 (two-way ANOVA), and * indicates significance levels after Bonferroni post-test correction. (f) Representative histograms and MFI quantification in the indicated subpopulations recovered from day 14 myeloerythroid colonies (n = 3). Two-way ANOVA: P < 0.0001 (subpopulations), ns (MSP versus MSP.126T). (g) Differentiation-stage-specific activity of PGK, PGK.126T(2), MSP, MSP.126T(2). Indicated subpopulations (see Figure 1b for legend) were identified in a 2-week myeloid differentiation culture of CD34+ bone marrow (BM), and MFI of each subpopulation was internally normalized to the most primitive cell fraction in the culture, i.e., CD34hi13−11b− cells (HSPC). (h,i) In vivo activity of the MSPs. BM CD34+ HSPC were transduced with MSP LV, MSP.LV.126T(2), or PGK Ctrl.LV, and transplanted into NSG mice (n = 4 per group). Representative FACS plots show GFP expression in the indicated subpopulations of human CD45+ cells recovered from the PB of hematochimeric mice at 10 (CD33+, CD19+ cells) or 17 weeks (CD3+ cells) post-transplantation. The bar graph on the right shows GFP expression of the MSP in the indicated subpopulations (green bars, median; d) and the MFI of the GFP-positive cells relative to the mean MFI of the Ctrl LV in the indicated subpopulation (white bars, mean + SEM). The dotted line represents the average %GFP+ cells in the CtrlLV group (left y-axis) and the GFP MFI of the Ctrl LV (right y-axis). FACS, fluorescence-activated cell sorting; PB, peripheral blood; PGK, phosphoglycerate kinase.
Regulated LVs reduce ectopic gp91phox expression in human CD34+ cells from normal donors
We next generated and tested different LVs that expressed human codon-optimized gp91phox cDNA29 and were transcriptionally and/or posttranscriptionally detargeted from HSPC (MSP.gp91_126T(2), MSP.gp91, or PGK.gp91_126T(2), respectively) (Supplementary Figure S1). As a control, we used a ubiquitous cellular promoter (PGK.gp91). Initially, we measured gp91phox expression in BM CD34+ cells from healthy donor (HD) after short-term culture (4 days) and found that gp91phox protein was almost undetectable in CD34+ HSPC (1.31%), while it started to be expressed (15.3%) in CD34− cells generated during in vitro culture (Supplementary Figure S4). Then we compared transgene regulation from PGK.gp91_126T(2), MSP.gp91, and MSP_126T(2) with the constitutive PGK.gp91 vector. We found that HD BM CD34+ cells transduced with the PGK.gp91 vector ectopically expressed gp91phox (Figure 3a; Supplementary Figure S4). In contrast, transduction with PGK.gp91_126T(2), MSP.gp91, and MSP_126T(2) resulted in substantially reduced gp91phox expression in CD34+ cells with respect to PGK-transduced cells (5.9% ± 1.6, 9.5% ± 3.3 and 6.6 ± 1.8 versus 32.7% ± 6.8). Reduced ectopic gp91phox expression was also evident when comparing MFI values, confirming the proficiency of both regulation systems in detargeting HSPC in the CGD setting (Figure 3b). On the other hand, gp91phox was overexpressed in differentiated CD34− cells transduced by all vectors (Supplementary Figure S4).
Figure 3.

Prevention of ectopic expression in human CD34+ cells of healthy donor transduced with regulated vectors. (a) The histograms represent % of gp91phox positive cells (mean ± SEM) within CD34+ cell gates, in four independent experiments. Statistical analysis was performed with one-way analysis of variance (ANOVA) with Bonferroni post-test correction, * indicates P < 0.05, **indicates P < 0.01, and *** indicates P < 0.001. (b) gp91phox-estimated mean fluorescence intensity (MFI) (as reported in Supplementary Methods) of HD CD34+ cells transduced with the indicated vectors. Statistical analysis was performed as in (a). (c) FACS analysis of transgene expression in human progenitor cell subsets: hematopoietic stem cell (CD34+ CD38− CD90+ CD45RA−), MPP (CD34+ CD38− CD90− CD45RA−), MLP (CD34+ CD38− CD90− CD45RA+), GMP (CD34+ CD38+ CD90− CD45RA+), CMP/MEP (CD34+ CD38+ CD90− CD45RA+). The histograms represent the percentage of gp91phox positive cells in the indicated subsets (n = 3, mean ± SEM). The mean vector copy number ± SEM was 1.0 ± 0.2 for MSP.gp91 and 1.1 ± 0.2 for MSP.gp91.1262T. Statistical analysis was performed with one-way ANOVA with Bonferroni post-test correction, * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001, and **** indicates P < 0.0001. FACS, fluorescence-activated cell sorting; HD, healthy donor; PGK, phosphoglycerate kinase.
Finally, we decided to analyze more in detail the heterogeneous CD34+ populations to test the efficiency of MSP-regulated vectors in the most primitive human cell subsets. MSP.gp91_126T(2) was most efficient in detargeting transgene expression and more importantly, the lowest gp91phox expression was observed in the most primitive fraction of HSC and MPP, revealing a significative difference between the MSP.gp91 vector and the combined MSP.gp91_126T(2) LV at a comparable VCN (Figure 3c).
Regulated expression of the gp91phox transgene and biochemical correction in HSPC from X-CGD patients
We then collected BM from two X-CGD patients (Pt1 and Pt2) and transduced CD34+ cells with the therapeutic LVs.30 After short-term culture, all LVs restored gp91phox expression in differentiating cells at levels (range of gp91phox 6–23%) consistent with transduction efficiency (VCN per cell range 0.1–0.5). In addition, X-CGD CD34+ cells transduced with LV.PGK.gp91 showed ectopic expression of gp91phox (7–13%), which was strongly reduced by the regulated vectors (Figure 4a,b).
Figure 4.

Regulated transgene expression with restored gp91phox expression and function in X-linked chronic granulomatous disease (X-CGD) cells. (a,b) X-CGD CD34+ cells from Pt1 and Pt2 were transduced with the indicated vectors at 100 MOI and analyzed for the surface expression of gp91phox by FACS, after 4 days of liquid culture in serum-free medium with SCF, FLT3L, TPO, and IL-3. Vector copy number reported for each sample is calculated on the total population after in vitro culture. In (c) bars show the gp91phox MFI of transduced CD34+ cells relative to the mean fluorescence intensity of HD untransduced cells in CD16+CD11b+ and CD14+CD11b+ gated cells for Pt 1 and Pt 2. (d,e) Nicotinamide adenine dinucleotide phosphate oxidase activity in myeloid (CD11b+) cells from Pt1 and Pt2 measured by DHR assay. The bars show the percentage of Rhodamine123 cells after PMA stimulation. DHR, dihydrorhodamine 123; FACS, fluorescence-activated cell sorting; HD, healthy donor; MOI, multiplicity of infection; PMA, phorbol 12-myristate 13-acetate; SCF, stem cell factor; TPO, thrombopoietin.
To evaluate transgene expression and function in differentiated cells, LV-transduced CD34+ X-CGD patients' cells were differentiated along the myeloid lineage. Gp91phox expression was restored in neutrophils (CD16+CD11b+) and monocytes (CD14+CD11b+) after transduction with therapeutic LV at comparable VCN (Supplementary Figure S5). Restored intracellular gp91phox expression was also confirmed in differentiated CD11b+ cells (Supplementary Figure S6). Expression on a per-cell basis was highest in MSP vector-transduced cells, reaching 50–70% and 120–130% of normal donor levels in granulocytes and monocytes, respectively (Figure 4c). Importantly, NADPH oxidase function was reestablished in Pt1 in 25–40% of cells (Figure 4d), and transduced cells reached on average 45% of HD oxidase activity after phorbol 12-myristate 13-acetate stimulation (data not shown). In Pt2 (Figure 4e), a single integrant of MSP.gp91_126T(2) LV restored NADPH oxidase function after myeloid differentiation, similar to the MSP LV and better than the phosphoglycerate kinase (PGK) vectors.
Correction of gp91phox expression in primary human macrophages from X-CGD patients
To confirm the ability of the combined regulated vector to restore gp91phox expression in human macrophages, we isolated peripheral blood (PB) monocytes from three X-CGD patients and induced them to differentiate in culture in the presence of M-CSF. Macrophages were transduced in the presence of virus-like particles containing Vpx to overcome inhibition of LV transduction.31 Both PGK.gp91_126T(2) and MSP.gp91_126T(2) vectors restored transgene expression in 22–48% of patient cells, with a tendency of the MSP-based vector to induce higher MFI compared with PGK, to ~50% of HD levels (Figure 5a). Transduced macrophages showed restoration of NADPH oxidase activity as assessed by cytochrome C assay, in agreement with gp91phox expression levels (Figure 5b).
Figure 5.

Restored gp91phox expression and function in primary X-linked chronic granulomatous disease macrophages. PB monocytes from three patients were cultured with M-CSF and transduced after 1 week with the indicated vectors in the presence of virus-like particles-VPX. (a) Mean fluorescence intensity and % of gp91phox expression are indicated in cells gated for CD206 and CD14. Untransduced cells and HD macrophages are shown for comparison. (b) Representative NADPH oxidase activity measured by cytochrome c assay is shown. The bars show the superoxide production after PMA-stimulation of patient 1. M-CSF, macrophage colony stimulating factor; PB, peripheral blood; PMA, phorbol 12-myristate 13-acetate; VPX, virion protein X.
Correction of gp91phox expression and NADPH activity in X-CGD mice
To confirm that the regulated vectors effectively restored gp91phox expression and NADPH activity in vivo, we transduced Lineage negative (Lin−) cells, harvested from BM of X-CGD mice, with the therapeutic vectors and transplanted them into irradiated recipients. All GT-treated mice showed engraftment of vector-transduced cells at sacrifice (Table 1 and Figure 6). In the PB, 20–70% of granulocytes and monocytes expressed human gp91phox, in agreement with the VCN of the respective treatment group (which was lowest for PGK and MSP.gp91_126T(2) mice). As previously suggested,23 gp91phox expression on B and T cells was significantly restricted using both MSP-driven vectors (Figure 6a). Next, we investigated correction of NADPH oxidase activity in these cells. As expected, dihydrorhodamine 123 activity was absent (0.4%) in PB neutrophils of mice transplanted with X-CGD BM, while it was restored in mice transplanted with wt BM or gene therapy-treated X-CGD BM to the degree of gp91phox positive cell engraftment (Figure 6b and data not shown). By comparing oxidase activity in corrected granulocytes, we confirmed the superior performance of the MSP-driven vectors (59 and 38% of wt NADPH oxidase activity in the MSP and MSP.126T(2) groups, respectively) compared with PGK (17 and 23% for PGK and PGK.126T(2), respectively) (Figure 6c).
Table 1. Vector copy number of gene therapy-mice calculated at sacrifice in bone marrow, spleen, thymus, and peripheral blood.

Figure 6.

Restored gp91phox expression and biochemical correction in X-linked chronic granulomatous disease (X-CGD) mice treated with gene-corrected Lin-negative cells. X-CGD mice were irradiated at 900 RAD and injected with X-CGD Lin− cells transduced with the indicated regulated vectors. Mice were sacrificed after 20 weeks and analyzed for gp91phox expression in PB and bone marrow (BM); nicotinamide adenine dinucleotide phosphate oxidase function was evaluated in PB granulocytes and monocytes. (a) Expression of gp91phox in PB Granulocytes (CD11b+, Gr1+, CD48+), monocytes (CD11b+, Gr1−, CD48hi), B cells (B220+, CD48+), and T cells (CD3+). Statistical analysis was performed with one-way analysis of variance with Bonferroni post-test correction, * indicates P < 0.05, ** indicates P < 0.01, and *** P < 0.001. (b) Representative plots of DHR assay on PB cells of mice transplanted with X-CGD Lin- or wt Lin- or or transduced X-CGD Lin−. (c) Mean fluorescence intensity of DHR-positive cells in PB granulocytes and monocytes. (d) The presence of the therapeutic gene gp91phox was investigated in different BM progenitor cell subsets. Statistical analysis was performed as in (a). DHR, dihydrorhodamine 123.
Finally, we analyzed gp91phox expression in successive stages of granulocytic differentiation in the BM, comparing the expression levels of the four therapeutic vectors in most primitive HSC, multipotent progenitors with myeloid and lymphoid potential (GMLP), in lineage-committed GMP, and in immature granulocytes (Figure 6d). The PGK vectors showed similar gp91phox expression in all these developmental stages, with no obvious difference between PGK and PGK.126T(2). This is not surprising, given that miR-126 is 10-fold less active in mouse with respect to human HSC,27 and 2 miR-126 targets might not be sufficient to achieve stringent regulation in mice. However, we noted a clear developmental stage-specific upregulation of gp91phox expression from the MSP constructs, which stand out as the superior vectors for CGD gene therapy in this work.
Discussion
The present study describes a novel gene therapy approach for X-CGD which is based on the use of autologous HSPC transduced ex vivo with novel, gp91phox-expressing SIN LVs exploiting two complementary strategies to regulate transgene expression. Our MSP.gp91_126T(2) vector restored up to 50% of normal NADPH oxidase activity in X-CGD mice, as well as in primary X-CGD patient phagocytes (myeloid cells differentiated in vitro from CD34+ cells or macrophages differentiated from PB monocytes). Gp91phox was detected by surface and intracellular expression on primary cells and confirmed by western blot on a human cell line (data not shown). In contrast, expression was nearly absent in CD34+ HSPC. To our best knowledge, this vector most closely mimics gp91phox expression in healthy donors and features the strongest transgene induction during myeloid differentiation reported so far, although a formal side-by-side comparison ex vivo and in vivo with previously described myeloid-specific LV-gp91phox vectors20,22 remains to be done. This stringent level of transgene expression control is based on two cornerstones: posttranscriptional and transcriptional regulation.
Posttranscriptional regulation relies on the HSPC-specific expression of miRNAs with high regulatory potential, which degrade transcripts containing complementary binding sites.26,27 In this work, we confirm substantial activity of miR-126 and miR-130a in human BM HSPC and suggest, in the case of miR-126T, an optimum of two tandem repeats, since more copies reduced expression in differentiated cells. Even though the addition of two miR-130aTs further increased HSPC de-targeting by 30–40%, we decided to proceed with the 126T(2) element due to emerging data on the functional role of miR-126 in hematopoiesis. MiR-126 loss-of-function in HSC is well tolerated32 and can result in benign HSC expansion, without causing exhaustion or transformation.33 On the other hand, ectopic miR-126 expression can contribute to hematologic malignancy.34 It is highly unlikely that expression of the 126T(2) sequence from the promoters used in this study, which have moderate or low activity in CD34+ cells, inhibits the regulation of natural miR-126 targets. Indeed, more than 10 integrations per cell of a vector that overexpresses 8 tandem repeats of an optimized, bulged miR-126 target sequence from the strong SFFV promoter were necessary to produce a loss-of-function phenotype in HSPC.33 In a worst-case scenario, if ex vivo transduction of CD34+ HSPC with the GT constructs generated rare clones which express, due to copy number accumulation and integration site-specific features, the 126T(2) to high enough levels to saturate miR-126, we would expect the resulting phenotype to be benign. This is further supported by the description of a single-nucleotide polymorphism in pri-mir-126, which is fairly prevalent in the general population and reduces miR-126 expression several fold.35
The second cornerstone to direct transgene expression to the differentiated myeloid compartment relies on transcriptional targeting. Several groups have proposed myeloid-specific regulatory elements for CGD gene therapy.20,22,23,28 The MRP8 promoter combined with a chromatin-opening element, A2UCOE, was shown to effectively drive gp91phox expression in myeloid cells.22 However, concerns of disturbing host gene function and driving insertional mutagenesis by the A2UCOE element remain.36 Recently, Santilli et al.20 designed a vector containing a chimeric myeloid-specific promoter which reestablished transgene expression in gp91phox−/− myeloid cells, but limited data were available to support differentiation stage specificity in human CD34+ cells. These studies underline the challenges to find specific, yet sufficiently strong, promoters to allow correction of the CGD disease phenotype. We focused on the SP146.gp91phox MSP, which has shown promise in terms of specificity and efficacy in mice, as well as in an in vitro culture model of human CB.23,28 We provide a detailed characterization of this promoter in human BM cells and in NSG mice, both in vitro and in vivo, document its excellent differentiation stage specificity with robust expression in mature myeloid cells, and show disease correction in granulocytes/monocytes differentiated in vitro from CD34+ BM cells or PB monocytes of CGD patients, and in X-CGD mice in vivo.
The degree of HSPC detargeting in total human CD34+ cells by the MSP promoter was similar to PGK-driven 126T(2). When the transcriptional and posttranscriptional regulations were combined in the same vector using either a GFP reporter or gp91phox, this resulted in the strongest downregulation of the transgene in most primitive human CD34+ cell subsets (CD34+/CD38−/CD90+).
Notably, the MSP provided stronger transgene expression than PGK in myeloid cells derived from primary huCD34+ BM cells, in primary human macrophages as well as in murine myeloid cells differentiated in vivo. Indeed, the performance in relevant primary cells should dictate promoter choice for clinical development and, based on this premise, all tested vectors restored NADPH oxidase activity in patient cells to sufficient levels to confer therapeutic benefit.1,37 This was seen with a VCN between 0.1 and 0.5, compatible with a single integrant per transduced cell. Transduction efficiency can be further enhanced by the use of optimized protocols, increased rounds of transductions, and purified vectors.38
Due to their different mechanisms of action, posttranscriptional and transcriptional regulation, are complementary. Combining both approaches in the context of the LV backbone might have several advantages: expression in HSPC is virtually abrogated, concerns for miRNA saturation are further mitigated due to the very modest activity of the MSP in HSPC. This hypothesis is based on the assumption that HSPC represent a more efficient substrate for malignant transformation than differentiated myeloid cells. Even though this has been documented in experimental models,39 the improved safety of the MSP still needs to be formally demonstrated using insertional mutagenesis models.40,41
The regulated SIN LVs described in this work represent a significant evolution with respect to previous generation retroviral vectors and constitutively expressing LVs. Recent data from clinical trials indicate that the SIN LV platform allows highly efficient in vitro HSPC transduction (>90%) resulting in robust and stable (>2 years) in vivo marking of HSPC with a safe integration profile in children with metachromatic leukodystrophy or Wiskott-Aldrich syndrome.42,43 The regulatory elements included in the vectors described here represent a further evolution of the SIN LV platform by avoiding the exposure of CD34+ cells to the transgene product. This overcomes potential concerns that inappropriate gp91phox expression may interfere with reactive oxygen species homeostasis, which is evolving as an important principle in regulating HSC fate.17,18 Indeed, replication stress induces elevation of intracellular reactive oxygen species levels and the accumulation of persistent DNA damage within human HSCs, as observed in in vivo models of human hematopoiesis and HSC transplanted patients. However, while Stein et al. observed an increase in DNA double-strand breaks post-gene therapy in X-CGD patients, the phenotype could not be reproduced in vitro by overexpression of gp91phox. Furthermore, the patient would not be exposed to the transgene product at the time of transplant infusion and during subsequent weeks, potentially reducing the risk of triggering an antitransgene immune response. This risk has to be considered especially in patients exposed to neutrophil transfusions or with extensive tissue damage due to the conditioning regimen, ongoing infection, and autoimmunity related to the CGD disease and in those receiving conditioning regimens without immunosuppression. Importantly, the vectors employed in this work support robust and stable transgene expression in differentiated myeloid cells which are the natural target population in CGD, and the MSP closely mimics the endogenous expression profile of gp91phox—not surprisingly, since specificity is, at least in part, dictated by its own promoter. The paradigm of regulated vector cassettes that lack expression in HSC but get activated during differentiation is applicable to a broader spectrum of diseases, e.g., lysosomal storage disorders,27 and will help to make HSC gene therapy safer and more efficacious. CGD represents an optimal scenario to clinically test this next generation of regulated vectors.
Materials and Methods
Lentiviral vectors. Bidirectional miRNA reporter vectors were described previously.25 A codon-optimized gp91phox sequence29 was cloned into the BamHI-SalI sites of the various LV containing different promoters. The PGK and SFFV backbones were previously described by our laboratory,27 while the MSP was obtained from Barde et al.23 miRNA target sequences (miRT) were designed and cloned as described19,25,44 For the LV production and purification, see Supplementary Methods and ref. 45.
Isolation, transduction, and culture of human HSPC. Human CD34+ cells were purified from BM of X-CGD patients using the CD34 MicroBeads kit (Miltenyi Bergisch Gladbach, Germany). BM samples were obtained after informed consent following the Declaration of Helsinki standard ethics procedure with approval of the Children's Hospital Bambino Gesù Ethical Committee, while patients were undergoing general anesthesia for unrelated diagnostic procedures. Human BM or CB CD34+ cells from healthy donors were purchased from Lonza (Basel, Switzerland). Culture conditions and transduction procedures are described in Supplementary Methods.
NSG and X-CGD model. NOD.Cg-Prkdcscid Il2rgtm1Wjil/SzJ (NSG) and B6.129S6− Cybbtm1Din/J mice, Ly5.2 (X-CGD) mice were purchased from the Jackson Laboratory (Charles River Italia, Lecco, Italy) and maintained in specific pathogen-free conditions at the Charles River Animal Facility (Calco, Italy). All animal work was approved by the Animal Care and Use Committee of the San Raffaele Hospital (IACUC 455 and 476) and communicated to the Ministry of Health and local authorities according to Italian law. Eight-to-10-week-old mice (male or female) were sublethally irradiated (210 cGy for NSG and 900 cGy split dose for X-CGD) and transplanted with 500,000 transduced CD34+ BM HSPC or transduced Lin− from X-CGD mice. Cell engraftment was measured in the peripheral blood starting at 7–8 weeks post-transplant.
Flow cytometry and functional and molecular analyses. Antibody staining and flow cytometry are described in Supplementary Methods. Functional oxidase activity assays were performed by the “Burst test kit” (Orpegen Pharma, Heidelberg, Germany) following the manufacturer's instructions. Superoxide release was assessed by a cytochrome c reduction assay.46
Statistical analysis. Calculation of comparative transgene expression levels is described in the Supplementary Methods. Unless otherwise indicated, graphs show the mean ± SEM. For pairwise comparisons, we used a Student's t-test if n > 6 and a Mann–Whitney test if n ≤ 6. If more than two groups were to be compared, we used an analysis of variance test with Bonferroni posttest. Significance levels are as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
SUPPLEMENTARY MATERIAL Figure S1. Schematic diagram of the lentiviral vectors used in this study. Figure S2. Optimization of miRNA target sequences to de-targeted the CD34+ HSPC compartment. Figure S3. Performance of the SP146/gp91phox MSP promoter in human primary cells. Figure S4. Regulated vectors reduce gp91phox ectopic expression in HD CD34+ cells gp91phox. Figure S5. Restored gp91phox expression in monocytes and neutrophils differentiated in vitro. Figure S6. Restored intracellular gp91phox expression in differentiated X-CGD CD34+ cells. Methods
Acknowledgments
We thank Giulia Schira, Nicola Bocchini, Tiziana Plati, and Antonella Isgrò for technical help. The SIV3+ packaging construct was kindly provided by A. Cimarelli (ENS. Lyon, France). This work was supported by grants from Fondazione Roma (Stem Cells based approaches to monogenic diseases) to A.A., MIUR (Progetto PRIN 2008), Progetto Giovani Ricercatori to A.F.; European Commission (E-rare project EURO-CGD to A.A., M.G., P.R.; CELL-PID HEALTH-F5-2010–261387 to A.A., M.G. and P.R.; GA 222878 PERSIST to A.A., L.N., D.T.; ERC, Advanced Grant 249845 TARGETING GENE THERAPY to L.N.); Fondazione Telethon (TIGET core grant to A.A., L.N., and B.G.). M.C., G.F., and V.C. performed the experiments with gp91phox encoding vectors in myeloid cell lines and human primary cells, carried out immunological, molecular, and functional readouts, analyzed the data, and wrote the paper. E.Z. performed in vivo and in vitro experiments with human primary cells, interpreted and analyzed the data concerning the GFP reporter vectors, and wrote the paper. S.S. performed experiments in X-CGD mice and participated in study design and vector production. M.M. and R.J.H. participated in the experiments in X-CGD mice. G.D.M. contributed to molecular analysis and genetic studies. L.S.S. produced the LV. E.G. performed cell sorting. A.K.R., F.B., D.T., and M.G. provided critical reagents for the study. P.R. and A.F. followed X-CGD patients and provided biological samples. L.N. participated in the study design, data interpretation, and paper revision. B.G. provided the vectors; conceived, performed, and supervised the studies with the GFP reporter vectors; analyzed the data; and wrote the paper. A.A. conceived and supervised the studies with gp91phox vectors, analyzed the data, and wrote the paper. All authors critically revised the manuscript. The authors declare that they do not have any conflict of interest.
Supplementary Material
Schematic diagram of the lentiviral vectors used in this study.
Optimization of miRNA target sequences to de-targeted the CD34+ HSPC compartment.
Performance of the SP146/gp91phox MSP promoter in human primary cells.
Regulated vectors reduce gp91phox ectopic expression in HD CD34+ cells gp91phox.
Restored gp91phox expression in monocytes and neutrophils differentiated in vitro.
Restored intracellular gp91phox expression in differentiated X-CGD CD34+ cells.
References
- Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol. 2010;38:3–10. doi: 10.1007/s12016-009-8136-z. [DOI] [PubMed] [Google Scholar]
- Seger RA. Chronic granulomatous disease: recent advances in pathophysiology and treatment. Neth J Med. 2010;68:334–340. [PubMed] [Google Scholar]
- Di Matteo G, Giordani L, Finocchi A, Ventura A, Chiriaco M, Blancato J, et al. IPINET (Italian Network for Primary Immunodeficiencies) Molecular characterization of a large cohort of patients with Chronic Granulomatous Disease and identification of novel CYBB mutations: an Italian multicenter study. Mol Immunol. 2009;46:1935–1941. doi: 10.1016/j.molimm.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Roos D, Kuhns DB, Maddalena A, Bustamante J, Kannengiesser C, de Boer M, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update) Blood Cells Mol Dis. 2010;44:291–299. doi: 10.1016/j.bcmd.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger RA. Modern management of chronic granulomatous disease. Br J Haematol. 2008;140:255–266. doi: 10.1111/j.1365-2141.2007.06880.x. [DOI] [PubMed] [Google Scholar]
- Arruda VR, Favaro P, Finn JD. Strategies to modulate immune responses: a new frontier for gene therapy. Mol Ther. 2009;17:1492–1503. doi: 10.1038/mt.2009.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez CA, Shah S, Shearer WT, Rosenblatt HM, Paul ME, Chinen J, et al. Excellent survival after sibling or unrelated donor stem cell transplantation for chronic granulomatous disease. J Allergy Clin Immunol. 2012;129:176–183. doi: 10.1016/j.jaci.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti A, Bacchetta R, Seger R, Villa A, Cavazzana-Calvo M. Gene therapy for primary immunodeficiencies: Part 2. Curr Opin Immunol. 2012;24:585–591. doi: 10.1016/j.coi.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation; European Society for Immunodeficiency Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better. J Allergy Clin Immunol. 2010;126:602–10.e1. doi: 10.1016/j.jaci.2010.06.015. [DOI] [PubMed] [Google Scholar]
- Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383:436–448. doi: 10.1016/S0140-6736(13)62069-3. [DOI] [PubMed] [Google Scholar]
- Kang EM, Choi U, Theobald N, Linton G, Long Priel DA, Kuhns D, et al. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115:783–791. doi: 10.1182/blood-2009-05-222760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Bartholomae CC, Paruzynski A, Arens A, Kim S, Yu SS, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol Ther. 2011;19:2092–2101. doi: 10.1038/mt.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Wu C, Dunbar CE. Stem cell gene therapy: the risks of insertional mutagenesis and approaches to minimize genotoxicity. Front Med. 2011;5:356–371. doi: 10.1007/s11684-011-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Yahata T, Takanashi T, Muguruma Y, Ibrahim AA, Matsuzawa H, Uno T, et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood. 2011;118:2941–2950. doi: 10.1182/blood-2011-01-330050. [DOI] [PubMed] [Google Scholar]
- Grez M, Reichenbach J, Schwäble J, Seger R, Dinauer MC, Thrasher AJ. Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther. 2011;19:28–35. doi: 10.1038/mt.2010.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301–315. doi: 10.1038/nrg2985. [DOI] [PubMed] [Google Scholar]
- Santilli G, Almarza E, Brendel C, Choi U, Beilin C, Blundell MP, et al. Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther. 2011;19:122–132. doi: 10.1038/mt.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel C, Hänseler W, Wohlgensinger V, Bianchi M, Tokmak S, Chen-Wichmann L, et al. Human miR223 promoter as a novel myelo-specific promoter for chronic granulomatous disease gene therapy. Hum Gene Ther Methods. 2013;24:151–159. doi: 10.1089/hgtb.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel C, Müller-Kuller U, Schultze-Strasser S, Stein S, Chen-Wichmann L, Krattenmacher A, et al. Physiological regulation of transgene expression by a lentiviral vector containing the A2UCOE linked to a myeloid promoter. Gene Ther. 2012;19:1018–1029. doi: 10.1038/gt.2011.167. [DOI] [PubMed] [Google Scholar]
- Barde I, Laurenti E, Verp S, Wiznerowicz M, Offner S, Viornery A, et al. Lineage- and stage-restricted lentiviral vectors for the gene therapy of chronic granulomatous disease. Gene Ther. 2011;18:1087–1097. doi: 10.1038/gt.2011.65. [DOI] [PubMed] [Google Scholar]
- Brown BD, Gentner B, Cantore A, Colleoni S, Amendola M, Zingale A, et al. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol. 2007;25:1457–1467. doi: 10.1038/nbt1372. [DOI] [PubMed] [Google Scholar]
- Brown BD, Venneri MA, Zingale A, Sergi Sergi L, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12:585–591. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- Gentner B, Naldini L. Exploiting microRNA regulation for genetic engineering. Tissue Antigens. 2012;80:393–403. doi: 10.1111/tan.12002. [DOI] [PubMed] [Google Scholar]
- Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A, et al. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci Transl Med. 2010;2:58ra84. doi: 10.1126/scitranslmed.3001522. [DOI] [PubMed] [Google Scholar]
- He W, Qiang M, Ma W, Valente AJ, Quinones MP, Wang W, et al. Development of a synthetic promoter for macrophage gene therapy. Hum Gene Ther. 2006;17:949–959. doi: 10.1089/hum.2006.17.949. [DOI] [PubMed] [Google Scholar]
- Moreno-Carranza B, Gentsch M, Stein S, Schambach A, Santilli G, Rudolf E, et al. Transgene optimization significantly improves SIN vector titers, gp91phox expression and reconstitution of superoxide production in X-CGD cells. Gene Ther. 2009;16:111–118. doi: 10.1038/gt.2008.143. [DOI] [PubMed] [Google Scholar]
- Berger G, Durand S, Goujon C, Nguyen XN, Cordeil S, Darlix JL, et al. A simple, versatile and efficient method to genetically modify human monocyte-derived dendritic cells with HIV-1-derived lentiviral vectors. Nat Protoc. 2011;6:806–816. doi: 10.1038/nprot.2011.327. [DOI] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechman ER, Gentner B, van Galen P, Giustacchini A, Saini M, Boccalatte FE, et al. Attenuation of miR-126 activity expands HSC in vivo without exhaustion. Cell Stem Cell. 2012;11:799–811. doi: 10.1016/j.stem.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnprasopwat R, Ha D, Toyoshima T, Lodish H, Tojo A, Kotani A. Alteration of processing induced by a single nucleotide polymorphism in pri-miR-126. Biochem Biophys Res Commun. 2010;399:117–122. doi: 10.1016/j.bbrc.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Frost AR, Blundell MP, Bales O, Antoniou MN, Thrasher AJ. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation-mediated silencing of lentiviral vectors. Mol Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björgvinsdóttir H, Ding C, Pech N, Gifford MA, Li LL, Dinauer MC. Retroviral-mediated gene transfer of gp91phox into bone marrow cells rescues defect in host defense against Aspergillus fumigatus in murine X-linked chronic granulomatous disease. Blood. 1997;89:41–48. [PubMed] [Google Scholar]
- Scaramuzza S, Biasco L, Ripamonti A, Castiello MC, Loperfido M, Draghici E, et al. Preclinical safety and efficacy of human CD34(+) cells transduced with lentiviral vector for the treatment of Wiskott-Aldrich syndrome. Mol Ther. 2013;21:175–184. doi: 10.1038/mt.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova OS, Schiedlmeier B, Brugman MH, Stahlhut M, Bartels S, Li Z, et al. Cell-intrinsic and vector-related properties cooperate to determine the incidence and consequences of insertional mutagenesis. Mol Ther. 2009;17:1537–1547. doi: 10.1038/mt.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Cesana D. Genotoxicity assay for gene therapy vectors in tumor prone Cdkn2a-/- mice. Methods Enzymol. 2012;507:171–185. doi: 10.1016/B978-0-12-386509-0.00009-0. [DOI] [PubMed] [Google Scholar]
- Modlich U, Bohne J, Schmidt M, von Kalle C, Knöss S, Schambach A, et al. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner B, Schira G, Giustacchini A, Amendola M, Brown BD, Ponzoni M, et al. Stable knockdown of microRNA in vivo by lentiviral vectors. Nat Methods. 2009;6:63–66. doi: 10.1038/nmeth.1277. [DOI] [PubMed] [Google Scholar]
- Follenzi A, Naldini L. HIV-based vectors. Preparation and use. Methods Mol Med. 2002;69:259–274. [PubMed] [Google Scholar]
- Mayo LA, Curnutte JT. Kinetic microplate assay for superoxide production by neutrophils and other phagocytic cells. Methods Enzymol. 1990;186:567–575. doi: 10.1016/0076-6879(90)86151-k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic diagram of the lentiviral vectors used in this study.
Optimization of miRNA target sequences to de-targeted the CD34+ HSPC compartment.
Performance of the SP146/gp91phox MSP promoter in human primary cells.
Regulated vectors reduce gp91phox ectopic expression in HD CD34+ cells gp91phox.
Restored gp91phox expression in monocytes and neutrophils differentiated in vitro.
Restored intracellular gp91phox expression in differentiated X-CGD CD34+ cells.